
Do the A Subunits Contribute to the Differences in the Toxicity of Shiga Toxin 1 and Shiga Toxin 2?

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure
3. Catalytic Activity and Cytotoxicity of Stx1 and Stx2
3.1. Differences in Cytotoxicity
3.2. Differences in Catalytic Activity
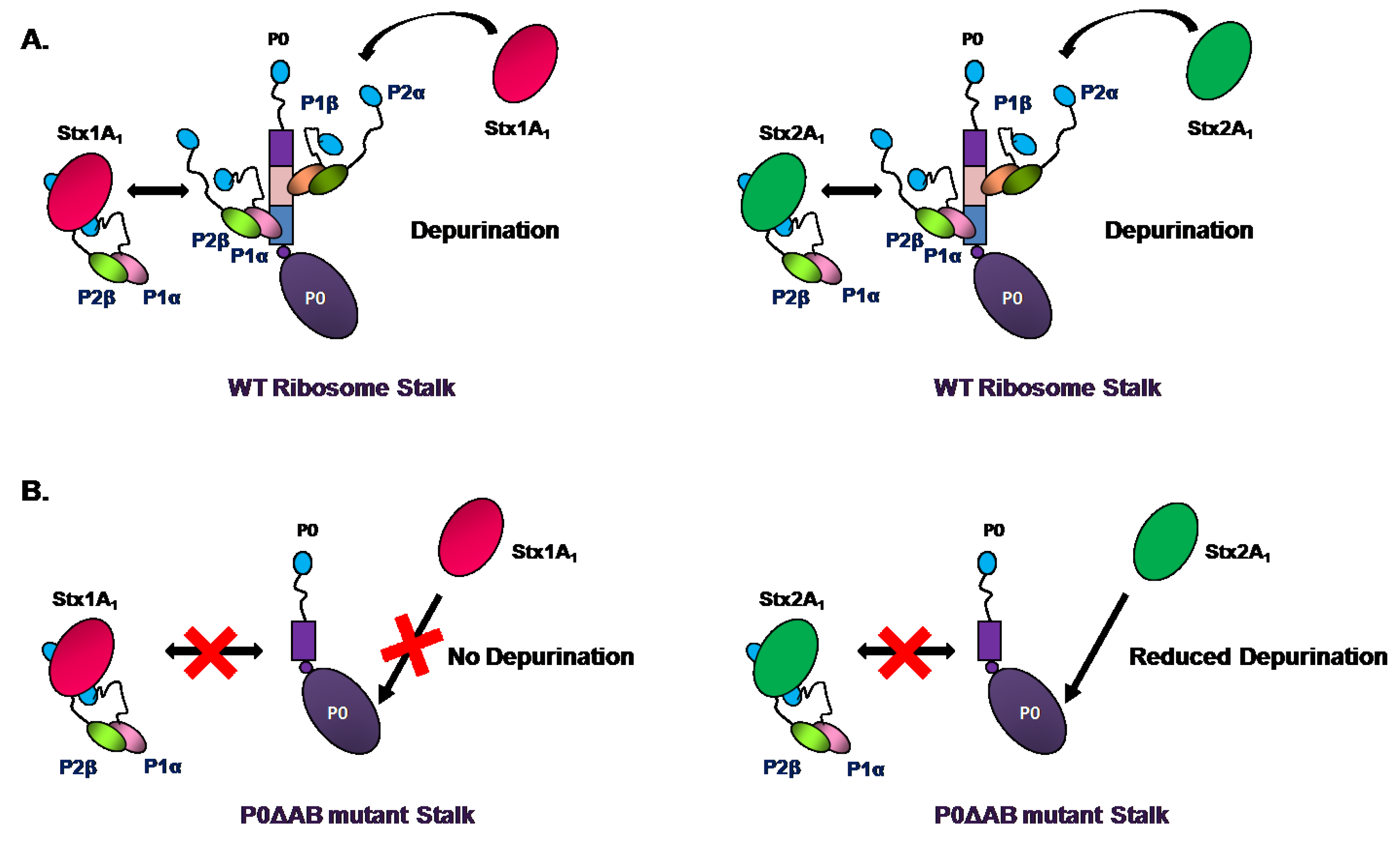
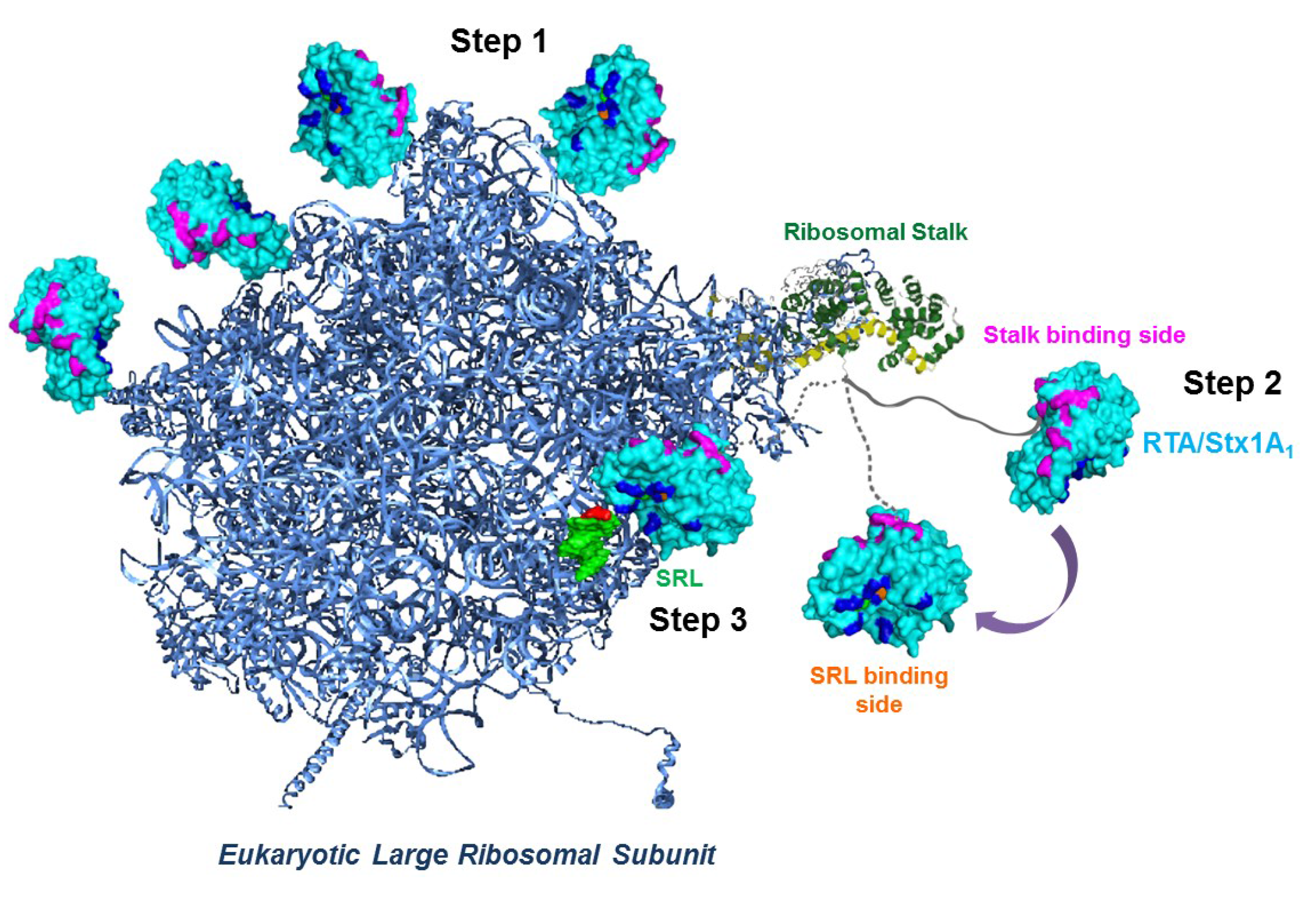
4. Ribosome Interactions


5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boerlin, P.; McEwen, S.; Boerlin-Petzold, F.; Wilson, J.; Johnson, R.; Gyles, C. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Siegler, R.; Oakes, R. Hemolytic uremic syndrome; pathogenesis, treatment, and outcome. Curr. Opin. Pediatr. 2005, 17, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A. Epidemic profile of Shiga-toxin-producing Escherichia coli O104: H4 outbreak in Germany. New Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.; O’Brien, A. Overview and historical perspectives. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Karch, H.; Denamur, E.; Dobrindt, U.; Finlay, B.B.; Hengge, R.; Johannes, L.; Ron, E.Z.; Tønjum, T.; Sansonetti, P.J.; Vicente, M. The enemy within us: Lessons from the 2011 European Escherichia coli O104: H4 outbreak. EMBO Mol. Med. 2012, 4, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Köck, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [PubMed]
- Pickering, L.; Obrig, T.; Stapleton, F. Hemolytic-uremic syndrome and enterohemorrhagic Escherichia coli. Pediatr. Infect. Dis. J. 1994, 13, 459. [Google Scholar] [CrossRef] [PubMed]
- Manning, S.D.; Motiwala, A.S.; Springman, A.C.; Qi, W.; Lacher, D.W.; Ouellette, L.M.; Mladonicky, J.M.; Somsel, P.; Rudrik, J.T.; Dietrich, S.E. Variation in virulence among clades of Escherichia coli O157: H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA 2008, 105, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Bergan, J.; Lingelem, A.B.D.; Simm, R.; Skotland, T.; Sandvig, K. Shiga toxins. Toxicon 2012, 60, 1085–1107. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F. Ribosome-inactivating proteins: From toxins to useful proteins. Toxicon 2013, 67, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.; Boston, R.S. Ribosome-inactivating proteins: A plant perspective. Annu. Rev. Plant Biol. 2001, 52, 785–816. [Google Scholar] [CrossRef]
- Zhabokritsky, A.; Kutky, M.; Burns, L.; Karran, R.; Hudak, K. RNA toxins: Mediators of stress adaptation and pathogen defense. Wiley Interdiscip. Rev. RNA 2010, 2, 890–903. [Google Scholar] [CrossRef]
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar] [CrossRef] [PubMed]
- May, K.L.; Yan, Q.; Tumer, N.E. Targeting ricin to the ribosome. Toxicon 2013, 69, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Clementi, N.; Chirkova, A.; Puffer, B.; Micura, R.; Polacek, N. Atomic mutagenesis reveals A2660 of 23S ribosomal RNA as key to EF-G GTPase activation. Nat. Chem. Biol. 2010, 6, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Khade, P.K.; Sanbonmatsu, K.Y.; Joseph, S. Functional role of the sarcin-ricin loop of the 23S rRNA in the elongation cycle of protein synthesis. J. Mol. Biol. 2012, 419, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Domashevskiy, A.V.; Goss, D.J. Pokeweed antiviral protein, a ribosome inactivating protein: Activity, inhibition and prospects. Toxins 2015, 7, 274–298. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.K.; Wong, E.C.; Lee, K.-M.; Wong, K.-B. Structures of eukaryotic ribosomal stalk proteins and its complex with trichosanthin, and their implications in recruiting ribosome-inactivating proteins to the ribosomes. Toxins 2015, 7, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Di, R.; Tumer, N.E. Pokeweed antiviral protein: Its cytotoxicity mechanism and applications in plant disease resistance. Toxins 2015, 7, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.J.; Dodd, J.E.; Hautbergue, G.M. Ribosome-inactivating proteins: Potent poisons and molecular tools. Virulence 2013, 4, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Parikh, B.; Tumer, N. Antiviral activity of ribosome inactivating proteins in medicine. Mini Rev. Med. Chem. 2004, 4, 523. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Souady, J.; Hülsewig, M.; Drmić-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S. Shiga toxin receptor Gb3Cer/CD77: Tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS ONE 2009, 4, e6813. [Google Scholar] [CrossRef] [PubMed]
- Maak, M.; Nitsche, U.; Keller, L.; Wolf, P.; Sarr, M.; Thiebaud, M.; Rosenberg, R.; Langer, R.; Kleeff, J.; Friess, H. Tumor-specific targeting of pancreatic cancer with Shiga toxin B-subunit. Mol. Cancer Ther. 2011, 10, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Römer, W. Shiga toxins—From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar] [PubMed]
- Strockbine, N.A.; Jackson, M.; Sung, L.; Holmes, R.; O’Brien, A.D. Cloning and sequencing of the genes for Shiga toxin from Shigella dysenteriae type 1. J. Bacteriol. 1988, 170, 1116–1122. [Google Scholar] [PubMed]
- Jackson, M.P.; Neill, R.J.; O’Brien, A.D.; Holmes, R.K.; Newland, J.W. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol. Lett. 1987, 44, 109–114. [Google Scholar] [CrossRef]
- Strockbine, N.A.; Marques, L.; Newland, J.W.; Smith, H.W.; Holmes, R.K.; O’brien, A.D. Two toxin-converting phages from Escherichia coli O157: H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 1986, 53, 135–140. [Google Scholar] [PubMed]
- Calderwood, S.B.; Auclair, F.; Donohue-Rolfe, A.; Keusch, G.T.; Mekalanos, J.J. Nucleotide sequence of the Shiga-like toxin genes of Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 4364–4368. [Google Scholar] [CrossRef] [PubMed]
- Karch, H.; Tarr, P.; Bielaszewska, M. Enterohaemorrhagic Escherichia coli in human medicine. Int. J. Med. Microbiol. 2005, 295, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Tyrrell, G.J.; Brunton, J.L.; Read, R.J. Crystal structure of the cell-binding B oligomer of Verotoxin-1 from E. coli. Nature 1992, 355, 748–750. [Google Scholar] [CrossRef]
- Fraser, M.; Chernaia, M.; Kozlov, Y.; James, M. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat. Struct. Biol. 1994, 1, 59. [Google Scholar] [CrossRef]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157: H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of the Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998, 37, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Sato, T.; Kawakami, S.; Ohta, T.; Noda, M.; Hamabata, T. Receptor affinity, stability and binding mode of Shiga toxins are determinants of toxicity. Microb. Pathog. 2007, 43, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Samuel, J.; Perera, L.; Ward, S.; O'brien, A.; Ginsburg, V.; Krivan, H. Comparison of the glycolipid receptor specificities of Shiga-like toxin type II and Shiga-like toxin type II variants. Infect. Immun. 1990, 58, 611–618. [Google Scholar] [PubMed]
- DeGrandis, S.; Law, H.; Brunton, J.; Gyles, C.; Lingwood, C. Globotetraosylceramide is recognized by the pig edema disease toxin. J. Biol. Chem. 1989, 264, 12520–12525. [Google Scholar] [PubMed]
- Nakajima, H.; Kiyokawa, N.; Katagiri, Y.U.; Taguchi, T.; Suzuki, T.; Sekino, T.; Mimori, K.; Ebata, T.; Saito, M.; Nakao, H. Kinetic analysis of binding between Shiga toxin and receptor glycolipid Gb3Cer by surface plasmon resonance. J. Biol. Chem. 2001, 276, 42915–42922. [Google Scholar] [CrossRef] [PubMed]
- Müthing, J.; Meisen, I.; Zhang, W.; Bielaszewska, M.; Mormann, M.; Bauerfeind, R.; Schmidt, M.A.; Friedrich, A.W.; Karch, H. Promiscuous Shiga toxin 2e and its intimate relationship to Forssman. Glycobiology 2012, 22, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.M.; Yin, J.; Kitov, P.I.; Mulvey, G.; Griener, T.P.; James, M.N.; Armstrong, G.; Bundle, D.R. The crystal structure of Shiga toxin type 2 with bound disaccharide guides the design of a heterobifunctional toxin inhibitor. J. Biol. Chem. 2014, 289, 885–894. [Google Scholar] [CrossRef] [PubMed]
- van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.E.; Cherney, M.M.; Marcato, P.; Mulvey, G.L.; Armstrong, G.D.; James, M.N. Binding of adenine to Stx2, the protein toxin from Escherichia coli O157: H7. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Arfilli, V.; Carnicelli, D.; Rocchi, L.; Ricci, F.; Pagliaro, P.; Tazzari, P.; Brigotti, M. Shiga toxin 1 and ricin A chain bind to human polymorphonuclear leucocytes through a common receptor. Biochem. J. 2010, 432, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E. Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Delivery into cells: Lessons learned from plant and bacterial toxins. Gene Ther. 2005, 12, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.L.; Leibowitz, C.S.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga Toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins 2012, 4, 1261. [Google Scholar] [CrossRef]
- Tesh, V.L.; Burris, J.; Owens, J.; Gordon, V.; Wadolkowski, E.; O’brien, A.; Samuel, J. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [PubMed]
- Siegler, R.L.; Obrig, T.G.; Pysher, T.J.; Tesh, V.L.; Denkers, N.D.; Taylor, F.B. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr. Nephrol. 2003, 18, 92–96. [Google Scholar] [PubMed]
- Wadolkowski, E.; Sung, L.; Burris, J.; Samuel, J.; O’brien, A. Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect. Immun. 1990, 58, 3959–3965. [Google Scholar] [PubMed]
- Stearns-Kurosawa, D.; Collins, V.; Freeman, S.; Tesh, V.L.; Kurosawa, S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga Toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect. Immun. 2010, 78, 2497. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Oh, S.-Y.; Cherla, R.P.; Lee, M.-S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli Shiga toxins in non-human primate models of hemolytic uremic syndrome. Am. J. Pathol. 2013, 182, 1227. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, A.; Betz, J.; Meisen, I.; Kemper, B.; Karch, H.; Müthing, J. Facing glycosphingolipid-Shiga toxin interaction: Dire straits for endothelial cells of the human vasculature. Cell. Mol. Life Sci. 2013, 70, 425–457. [Google Scholar] [CrossRef] [PubMed]
- Louise, C.B.; Obrig, T.G. Specific interaction of Escherichia coli 0157: H7-derived Shiga-like toxin II with human renal endothelial cells. J. Infect. Dis. 1995, 172, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, A.; Bielaszewska, M.; Kemper, B.; Langehanenberg, P.; von Bally, G.; Reichelt, R.; Mulac, D.; Humpf, H.-U.; Friedrich, A.W.; Kim, K.S. Differential cytotoxic actions of Shiga toxin 1 and Shiga toxin 2 on microvascular and macrovascular endothelial cells. Thromb. Haemost. 2011, 105, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Fuller, C.A.; Pellino, C.A.; Flagler, M.J.; Strasser, J.E.; Weiss, A.A. Shiga toxin subtypes display dramatic differences in potency. Infect. Immun. 2011, 79, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Flagler, M.J.; Mahajan, S.S.; Kulkarni, A.A.; Iyer, S.S.; Weiss, A.A. Comparison of binding platforms yields insights into receptor binding differences between Shiga toxins 1 and 2. Biochemistry 2010, 49, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Head, S.; Karmali, M.; Lingwood, C. Preparation of VT1 and VT2 hybrid toxins from their purified dissociated subunits. Evidence for B subunit modulation of a subunit function. J. Biol. Chem. 1991, 266, 3617–3621. [Google Scholar]
- Lingwood, C.A. Role of Verotoxin receptors in pathogenesis. Trends Microbiol. 1996, 4, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor globotetraosylceramide. Infect. Immun. 2010, 78, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Rutjes, N.W.; Binnington, B.A.; Smith, C.R.; Maloney, M.D.; Lingwood, C.A. Differential tissue targeting and pathogenesis of Verotoxins 1 and 2 in the mouse animal model. Kidney Int. 2002, 62, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Karve, S.S.; Weiss, A.A. Glycolipid binding preferences of Shiga toxin variants. PLoS One 2014, 9, e101173. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yutsudo, T.; Hirayama, T.; Takeda, Y. Isolation and some properties of A and B subunits of Vero toxin 2 and in vitro formation of hybrid toxins between subunits of Vero toxin 1 and Vero toxin 2 from Escherichia coli O157: H7. Microb. Pathog. 1988, 5, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.L.; Jackson, M.P.; Perera, L.P.; Holmes, R.K.; O’Brien, A.D. In vivo formation of hybrid toxins comprising Shiga toxin and the Shiga-like toxins and role of the B subunit in localization and cytotoxic activity. Infect. Immun. 1989, 57, 3743–3750. [Google Scholar] [PubMed]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.J.; O’Brien, A.D. Comparisons of native Shiga Toxins (Stxs) Type 1 and 2 with chimeric toxins indicate that the source of the binding subunit dictates degree of toxicity. PLoS ONE 2014, 9, e93463. [Google Scholar] [CrossRef] [PubMed]
- Hovde, C.; Calderwood, S.; Mekalanos, J.; Collier, R. Evidence that glutamic acid 167 is an active-site residue of Shiga-like toxin I. Proc. Natl. Acad. Sci. USA 1988, 85, 2568. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Deresiewicz, R.; Calderwood, S. Mutational analysis of the Shiga toxin and Shiga-like toxin II enzymatic subunits. J. Bacteriol. 1990, 172, 3346. [Google Scholar] [PubMed]
- Yamasaki, S.; Furutani, M.; Ito, K.; Igarashi, K.; Nishibuchi, M.; Takeda, Y. Importance of arginine at position 170 of the A subunit of Verotoxin 1 produced by enterohemorrhagic Escherichia coli for toxin activity. Microb. Pathog. 1991, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Di, R.; Kyu, E.; Shete, V.; Saidasan, H.; Kahn, P.; Tumer, N. Identification of amino acids critical for the cytotoxicity of Shiga toxin 1 and 2 in Saccharomyces cerevisiae. Toxicon 2011, 57, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Deresiewicz, R.L.; Austin, P.R.; Hovde, C.J. The role of tyrosine-114 in the enzymatic activity of the Shiga-like toxin I A-chain. Mol. Genet. Genomics 1993, 241, 467–473. [Google Scholar]
- Skinner, L.; Jackson, M. Investigation of ribosome binding by the Shiga toxin A1 subunit, using competition and site-directed mutagenesis. J. Bacteriol. 1997, 179, 1368–1374. [Google Scholar] [PubMed]
- McCluskey, A.; Bolewska-Pedyczak, E.; Jarvik, N.; Chen, G.; Sidhu, S.; Johannes, L. Charged and hydrophobic surfaces on the A chain of Shiga-Like Toxin 1 recognize the C-terminal domain of ribosomal stalk proteins. PLoS ONE 2012, 7, e31191. [Google Scholar] [CrossRef] [PubMed]
- Brigotti, M.; Carnicelli, D.; Alvergna, P.; Mazzaracchio, R.; Sperti, S.; Montanaro, L. The RNA-N-glycosidase activity of Shiga-like toxin I: Kinetic parameters of the native and activated toxin. Toxicon 1997, 35, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Chiou, J.-C.; Li, X.-P.; Remacha, M.; Ballesta, J.P.; Tumer, N.E. Shiga toxin 1 is more dependent on the P proteins of the ribosomal stalk for depurination activity than Shiga toxin 2. Int. J. Biochem. Cell. Biol. 2011, 43, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.; Kahn, J.N.; Chiou, J.; Tumer, N.E. Development of a quantitative RT-PCR assay to examine the kinetics of ribosome depurination by ribosome inactivating proteins using Saccharomyces cerevisiae as a model. RNA 2011, 17, 201–210. [Google Scholar] [CrossRef] [PubMed]
- May, K.L.; Li, X.P.; Martinez-Azorin, F.; Ballesta, J.P.; Grela, P.; Tchorzewski, M.; Tumer, N.E. The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J. 2012, 279, 3925–3936. [Google Scholar] [CrossRef] [PubMed]
- Sturm, M.B.; Schramm, V.L. Detecting ricin: Sensitive luminescent assay for ricin A-chain ribosome depurination kinetics. Anal. Chem. 2009, 81, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Kahn, P.C.; Kahn, J.N.; Grela, P.; Tumer, N.E. Arginine residues on the opposite side of the active site stimulate the catalysis of ribosome depurination by ricin A chain by interacting with the P-protein stalk. J. Biol. Chem. 2013, 288, 30270–30284. [Google Scholar] [CrossRef] [PubMed]
- Chiou, J.C.; Li, X.P.; Remacha, M.; Ballesta, J.P.; Tumer, N.E. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol. 2008, 70, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Chiou, J.-C.; Remacha, M.; Ballesta, J.P.; Tumer, N.E. A two-step binding model proposed for the electrostatic interactions of ricin a chain with ribosomes. Biochemistry 2009, 48, 3853–3863. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157: H7 and of Shiga toxin on eukaryotic ribosomes. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Poon, G.M.; Bolewska-Pedyczak, E.; Srikumar, T.; Jeram, S.M.; Raught, B.; Gariépy, J. The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol. 2008, 378, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Vater, C.A.; Bartle, L.M.; Leszyk, J.D.; Lambert, J.M.; Goldmacher, V.S. Ricin A chain can be chemically cross-linked to the mammalian ribosomal proteins L9 and L10e. J. Biol. Chem. 1995, 270, 12933–12940. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Hung, F.S.J.; Chan, D.S.B.; Shaw, P.C. Trichosanthin interacts with acidic ribosomal proteins P0 and P1 and mitotic checkpoint protein MAD2B. Eur. J. Biochem. 2001, 268, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.; Chu, L.-O.; Lee, K.-M.; Too, P.H.; Ma, K.-W.; Sze, K.-H.; Zhu, G.; Shaw, P.-C.; Wong, K.-B. Interaction between trichosanthin, a ribosome-inactivating protein, and the ribosomal stalk protein P2 by chemical shift perturbation and mutagenesis analyses. Nucleic Acids Res. 2007, 35, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Too, P.H.-M.; Ma, M.K.-W.; Mak, A.N.-S.; Wong, Y.-T.; Tung, C.K.-C.; Zhu, G.; Au, S.W.-N.; Wong, K.-B.; Shaw, P.-C. The C-terminal fragment of the ribosomal P protein complexed to trichosanthin reveals the interaction between the ribosome-inactivating protein and the ribosome. Nucleic Acids Res. 2009, 37, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-P.; Grela, P.; Krokowski, D.; Tchórzewski, M.; Tumer, N.E. Pentameric organization of the ribosomal stalk accelerates recruitment of ricin a chain to the ribosome for depurination. J. Biol. Chem. 2010, 285, 41463–41471. [Google Scholar] [CrossRef] [PubMed]
- Bargis-Surgey, P.; Lavergne, J.-P.; Gonzalo, P.; Vard, C.; Filhol-Cochet, O.; Reboud, J.-P. Interaction of elongation factor eEF-2 with ribosomal P proteins. Eur. J. Biochem. 1999, 262, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Grela, P.; Sawa-Makarska, J.; Gordiyenko, Y.; Robinson, C.; Grankowski, N.; Tchórzewski, M. Structural properties of the human acidic ribosomal P proteins forming the P1-P2 heterocomplex. J. Biochem. 2008, 143, 169. [Google Scholar] [CrossRef] [PubMed]
- Wool, I.; Chan, Y.; Glück, A.; Suzuki, K. The primary structure of rat ribosomal proteins P0, P1, and P2 and a proposal for a uniform nomenclature for mammalian and yeast ribosomal proteins. Biochimie 1991, 73, 861. [Google Scholar] [CrossRef] [PubMed]
- Planta, R.J.; Mager, W.H. The list of cytoplasmic ribosomal proteins of Saccharomyces cerevisiae. Yeast 1998, 14, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Tchórzewski, M.; Boguszewska, A.; Dukowski, P.; Grankowski, N. Oligomerization properties of the acidic ribosomal P-proteins from Saccharomyces cerevisiae: Effect of P1A protein phosphorylation on the formation of the P1A-P2B hetero-complex. BBA Clin. 2000, 1499, 63. [Google Scholar]
- Krokowski, D.; Boguszewska, A.; Abramczyk, D.; Liljas, A.; Tchórzewski, M.; Grankowski, N. Yeast ribosomal P0 protein has two separate binding sites for P1/P2 proteins. Mol. Microbiol. 2006, 60, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Hagiya, A.; Naganuma, T.; Maki, Y.; Ohta, J.; Tohkairin, Y.; Shimizu, T.; Nomura, T.; Hachimori, A.; Uchiumi, T. A mode of assembly of P0, P1 and P2 proteins at the GTPase-associated center in animal ribosome: In vitro analyses with P0 truncation mutants. J. Biol. Chem. 2005, 280, 39193–39199. [Google Scholar] [CrossRef] [PubMed]
- Grela, P.; Krokowski, D.; Gordiyenko, Y.; Krowarsch, D.; Robinson, C.V.; Otlewski, J.; Grankowski, N.; Tchórzewski, M. Biophysical properties of the eukaryotic ribosomal stalk. Biochemistry 2010, 49, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Ballesta, J.P.; Remacha, M. The large ribosomal subunit stalk as a regulatory element of the eukaryotic translational machinery. Prog. Nucleic Acid Res. Mol. Biol. 1996, 55, 157–193. [Google Scholar] [PubMed]
- Gonzalo, P.; Reboud, J.P. The puzzling lateral flexible stalk of the ribosome. Biol. Cell 2003, 95, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, M.; Kothe, U.; Schlünzen, F.; Fischer, N.; Harms, J.M.; Tonevitsky, A.G.; Stark, H.; Rodnina, M.V.; Wahl, M.C. Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 2005, 121, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Maki, Y.; Hashimoto, T.; Zhou, M.; Naganuma, T.; Ohta, J.; Nomura, T.; Robinson, C.V.; Uchiumi, T. Three binding sites for stalk protein dimers are generally present in ribosomes from archaeal organism. J. Biol. Chem. 2007, 282, 32827–32833. [Google Scholar] [CrossRef] [PubMed]
- Ayub, M.J.; Smulski, C.R.; Ma, K.-W.; Levin, M.J.; Shaw, P.-C.; Wong, K.-B. The C-terminal end of P proteins mediates ribosome inactivation by trichosanthin but does not affect the pokeweed antiviral protein activity. Biochem. Biophys. Res. Commun. 2008, 369, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Hudak, K.A.; Dinman, J.D.; Tumer, N.E. Pokeweed antiviral protein accesses ribosomes by binding to L3. J. Biol. Chem. 1999, 274, 3859–3864. [Google Scholar] [CrossRef] [PubMed]
- Lapadula, W.J.; Sanchez-Puerta, M.; Juri Ayub, M. Convergent evolution led ribosome inactivating proteins to interact with ribosomal stalk. Toxicon 2012, 59, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Lapadula, W.J.; Puerta, M.V.S.; Ayub, M.J. Revising the taxonomic distribution, origin and evolution of ribosome inactivating protein genes. PLoS ONE 2013, 8, e72825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernado, P.; Modig, K.; Grela, P.; Svergun, D.I.; Tchorzewski, M.; Pons, M.; Akke, M. Structure and dynamics of ribosomal protein L12: An ensemble model based on SAXS and NMR relaxation. Biophys. J. 2010, 98, 2374–2382. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Yusa, K.; Chu, L.-O.; Yu, C.W.-H.; Oono, M.; Miyoshi, T.; Ito, K.; Shaw, P.-C.; Wong, K.-B.; Uchiumi, T. Solution structure of human P1 P2 heterodimer provides insights into the role of eukaryotic stalk in recruiting the ribosome-inactivating protein trichosanthin to the ribosome. Nucleic Acids Res. 2013, 41, 8776–8787. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Melton-Celsa, A.R.; Sinclair, J.F.; Carvalho, H.M.; Robinson, C.M.; O’Brien, A.D. Monoclonal antibody 11E10, which neutralizes Shiga toxin type 2 (Stx2), recognizes three regions on the Stx2 A subunit, blocks the enzymatic action of the toxin in vitro, and alters the overall cellular distribution of the toxin. Infect. Immun. 2009, 77, 2730–2740. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.-J.; Zeng, X.-Y.; Guo, X.-L.; Shi, Z.-Y.; Feng, Z.-Q.; Wang, H. Monoclonal antibody S2C4 neutralizes the toxicity of Shiga toxin 2 and its variants. Prog. Biochem. Biophys. 2009, 36, 736–742. [Google Scholar] [CrossRef]
- Olson, M.A.; Cuff, L. Free energy determinants of binding the rRNA substrate and small ligands to ricin A-chain. Biophys. J. 1999, 76, 28–39. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basu, D.; Tumer, N.E. Do the A Subunits Contribute to the Differences in the Toxicity of Shiga Toxin 1 and Shiga Toxin 2? Toxins 2015, 7, 1467-1485. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7051467
Basu D, Tumer NE. Do the A Subunits Contribute to the Differences in the Toxicity of Shiga Toxin 1 and Shiga Toxin 2? Toxins. 2015; 7(5):1467-1485. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7051467
Chicago/Turabian StyleBasu, Debaleena, and Nilgun E. Tumer. 2015. "Do the A Subunits Contribute to the Differences in the Toxicity of Shiga Toxin 1 and Shiga Toxin 2?" Toxins 7, no. 5: 1467-1485. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7051467