
Recent Advances and Future Challenges in Modified Mycotoxin Analysis: Why HRMS Has Become a Key Instrument in Food Contaminant Research


Abstract
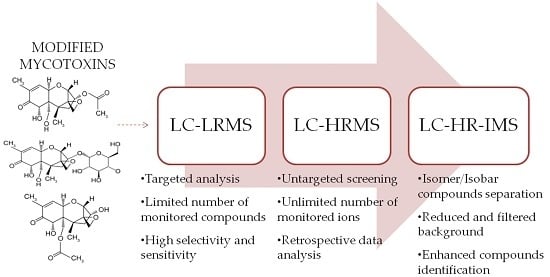
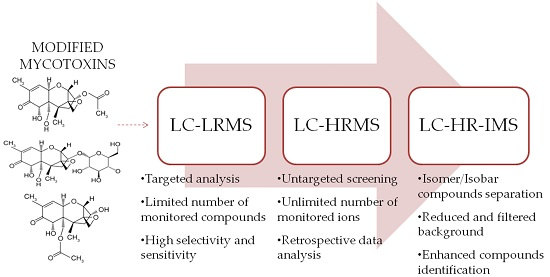
:
1. Introduction
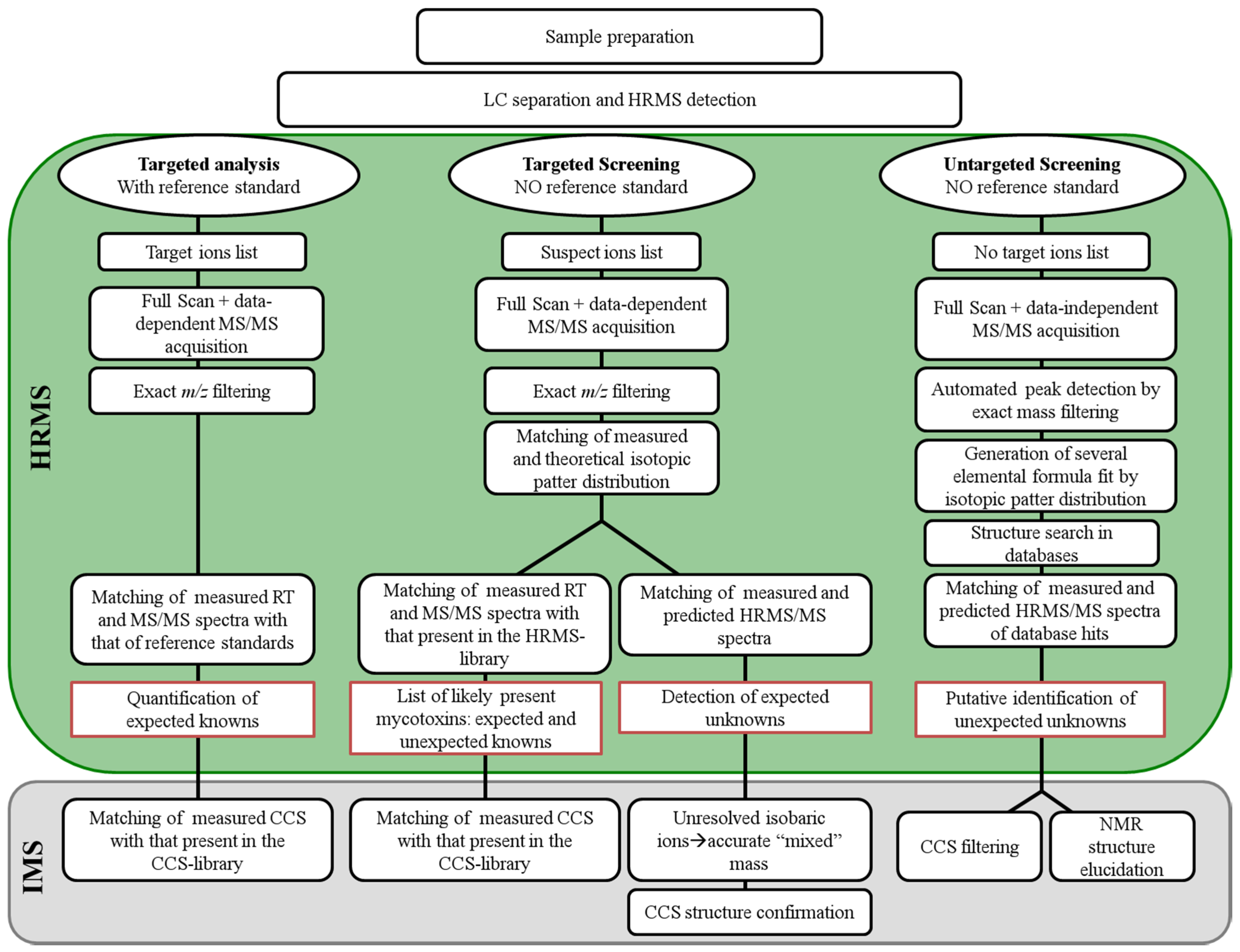
2. From Targeted LC-MS/MS Determination to Untargeted HR-MS Analysis
3. Use of HRMS Methods for Targeted Quantification of Natural Toxins
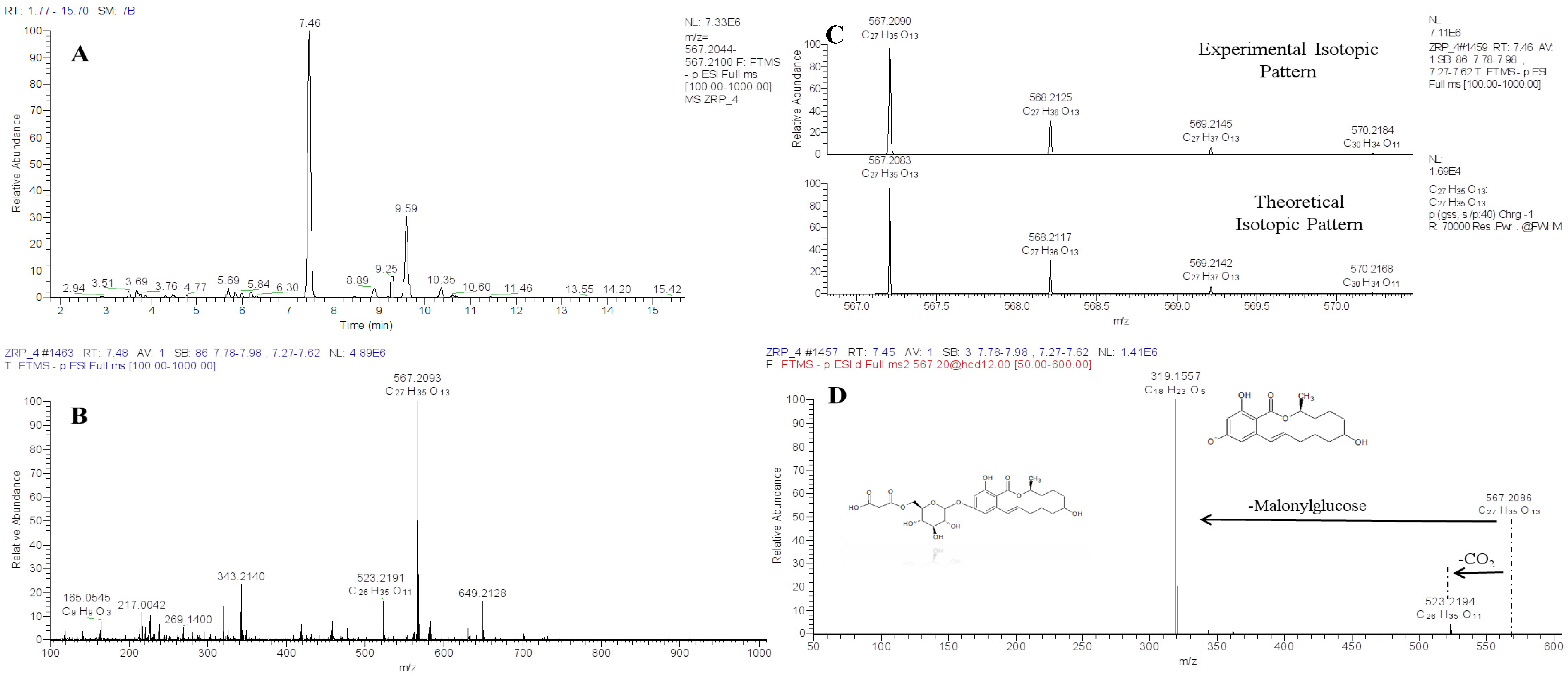
4. Use of HRMS Non-Targeted Screening Methods for Natural Toxins
5. Use of HRMS Methods for Structural Identification of Unknowns
6. Advantages and Challenges of HRMS in Modified Mycotoxin Analysis
7. The Potential Benefits of Ion Mobility Mass Spectrometry in Mycotoxin Analysis
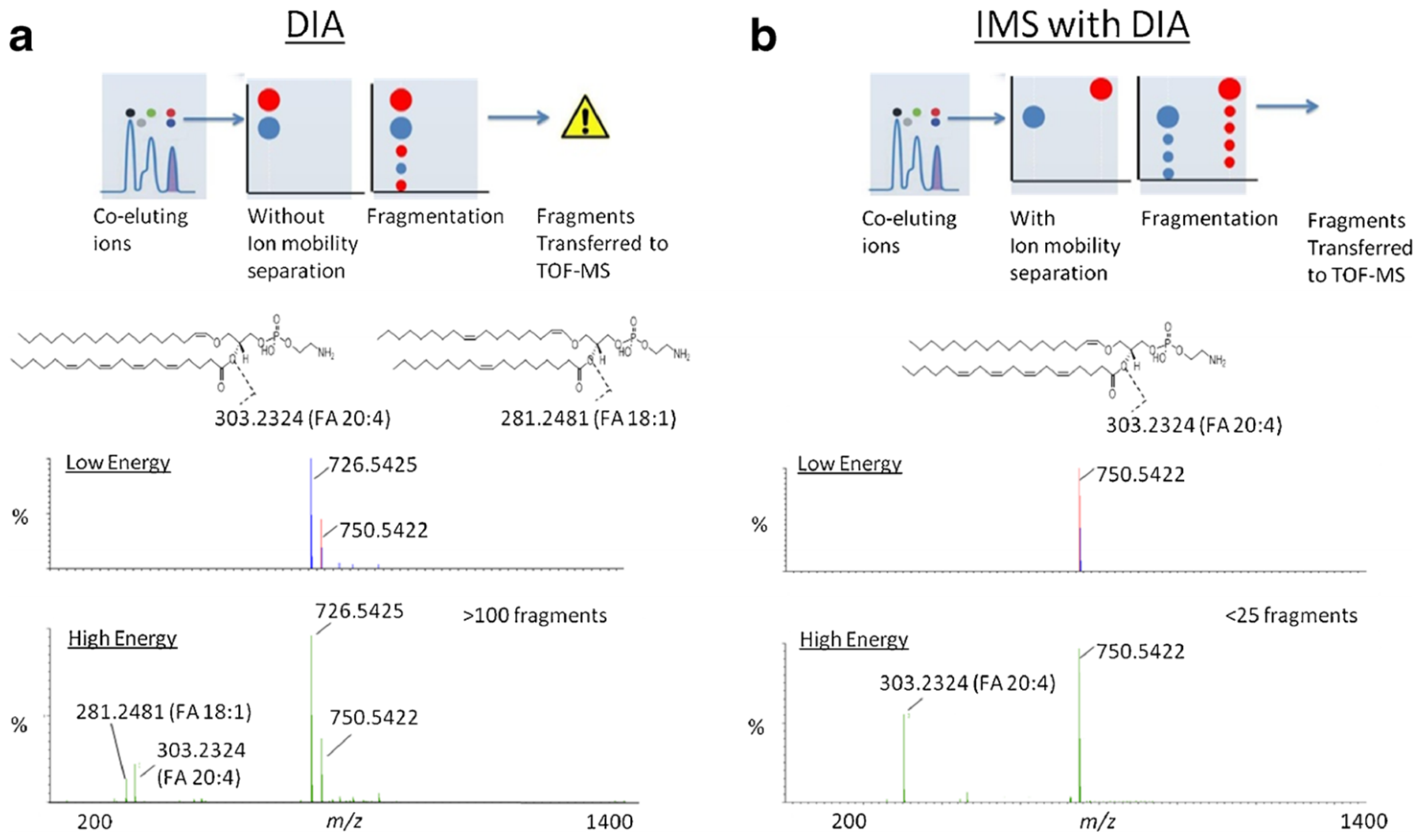
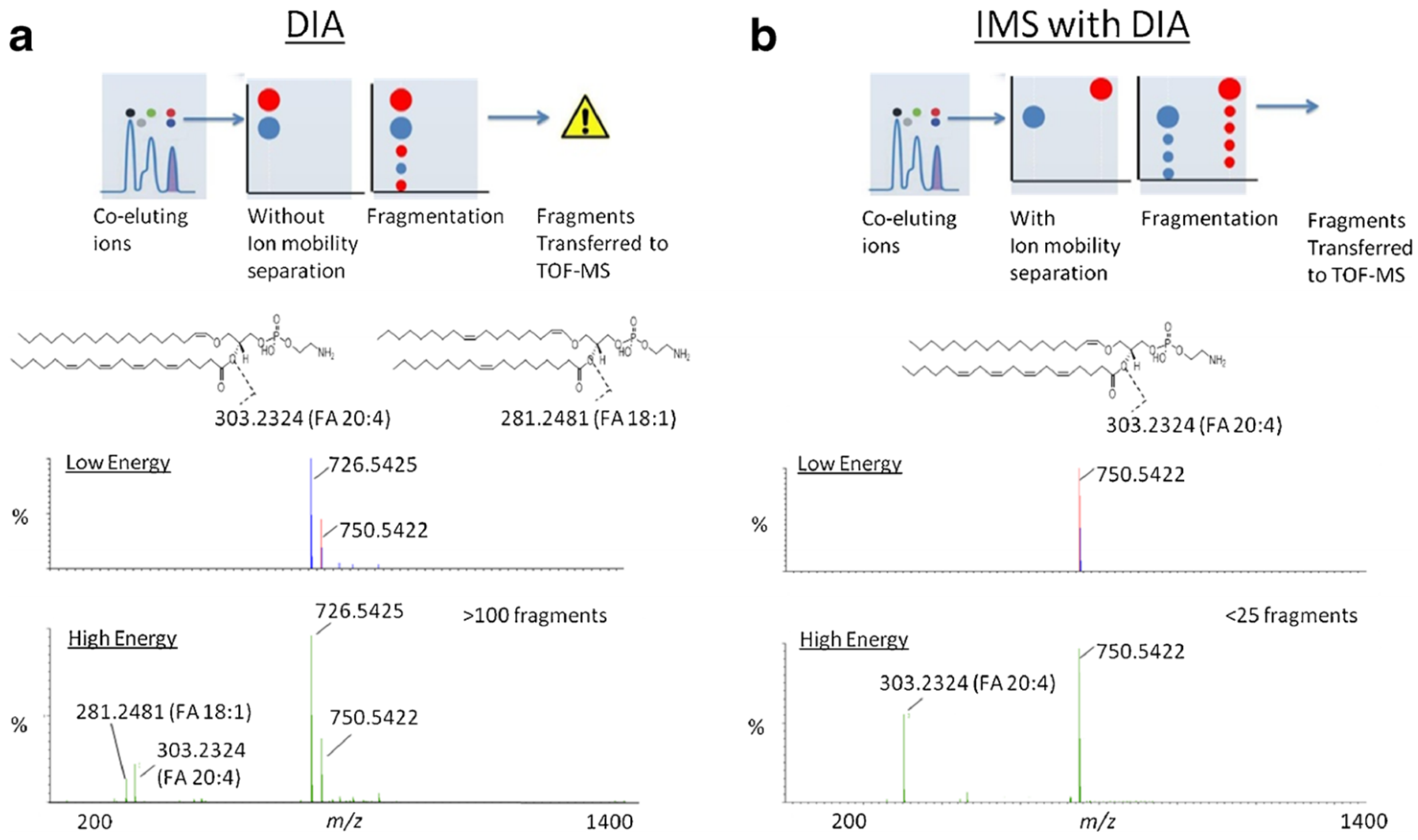
7.1. Peak Capacity and Signal-to-Noise Ratio Improvement
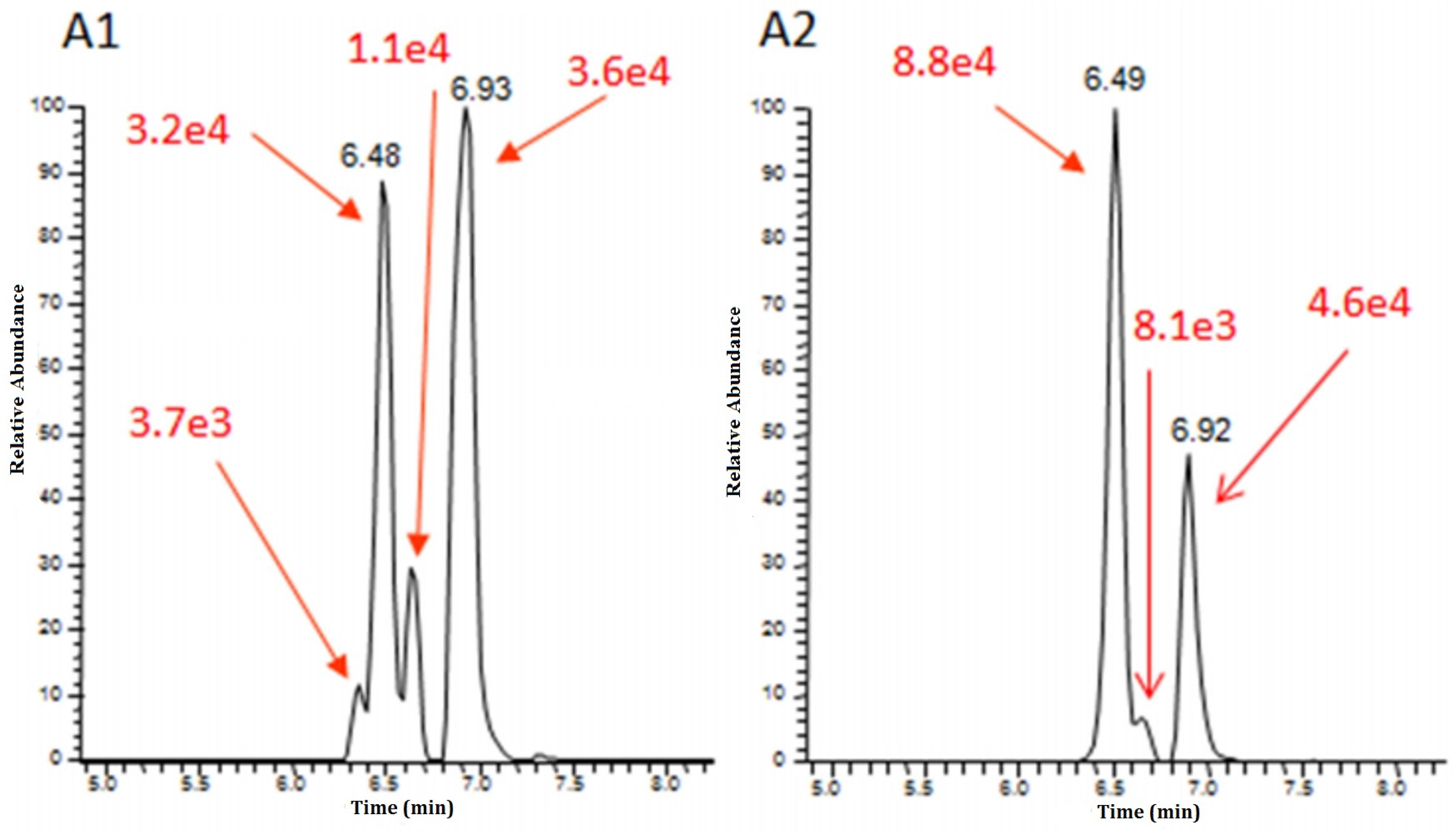
7.2. LC-IMS-MS Enables the Separation of Isobar Molecules
7.3. CCS Value: A New Unambiguous Molecular Descriptor
8. Conclusions and Future Trends
Author Contributions
Conflicts of Interest
References
- Battilani, P.; Toscano, P.; Van der Fels-Klerx, H.J.; Moretti, A.; Camardo Leggieri, M.; Brera, C.; Rortais, A.; Goumperis, T.; Robinson, T. Aflatoxin B1 contamination in maize in Europe increases due to climate change. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Dall’Asta, C.; Berthiller, F. Masked Mycotoxins in Food: Formation, Occurrence and Toxicological Relevance, 1st ed.; Royal Society of Chemistry: Cambridge, UK, 2016; Volume 24. [Google Scholar]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA J. 2014, 12. [Google Scholar] [CrossRef] [Green Version]
- Rychlik, M.; Humpf, H.U.; Marko, D.; Danicke, S.; Mally, A.; Berthiller, F.; Klaffke, H.; Lorenz, N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boevre, M.; Jacxsens, L.; Lachat, C.; Eeckhout, M.; Di Mavungu, J.D.; Audenaert, K.; Maene, P.; Haesaert, G.; Kolsteren, P.; De Meulenaer, B.; et al. Human exposure to mycotoxins and their masked forms through cereal-based foods in Belgium. Toxicol. Lett. 2013, 218, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, C.; Lazzaro, I.; Galaverna, G.; Battilani, P.; Dall’Asta, C. Fatty acid esters of fumonisins: First evidence of their presence in maize. Food Addit. Contam. Part A 2013, 30, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Dall’Erta, A.; Cirlini, M.; Dall’Asta, M.; Del Rio, D.; Galaverna, G.; Dall’Asta, C. Masked mycotoxins are efficiently hydrolyzed by human colonic microbiota releasing their aglycones. Chem. Res. Toxicol. 2013, 26, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Gareis, M.; Bauer, J.; Thiem, J.; Plank, G.; Grabley, S.; Gedek, B. Cleavage of zearalenone-glycoside, a “masked” mycotoxin, during digestion in swine. Zentralbl Veterinarmed B 1990, 37, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Comprehensive comparison of liquid chromatography selectivity as provided bytwo types of liquid chromatography detectors (high resolution mass spectrometry and tandem mass spectrometry): “Where is the crossover point”. Anal. Chim. Acta 2010, 673, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Mol, H.G.J.; Van Dam, R.C.J.; Zomer, P.; Mulder, P.P.J. Screening of plant toxins in food, feed and botanicals using full-scan high-resolution (Orbitrap) mass spectrometry. Food Addit. Contam. 2011, 28, 1405–1423. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Domínguez, G.; Romero-González, R.; Arrebola, F.J.; Garrido Frenich, A. Multi-class determination of pesticides and mycotoxins in isoflavones supplements obtained from soy by liquid chromatography coupled to Orbitrap high resolution mass spectrometry. Food Control 2015, 59, 218–224. [Google Scholar] [CrossRef]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Sulyok, M.; Beltrán, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography–tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chrom. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprikova, Z.; Slavikova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Cirlini, M.; Dall’Asta, C.; Galaverna, G. Hyphenated chromatographic techniques for structural characterization and determination of masked mycotoxins. J. Chromatog. A 2012, 1255, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Brera, C.; Crews, C.; Iha, M.H.; Kraska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; et al. Development in mycotoxin analysis: An update for 2014–2015. World Mycotoxin J. 2016, 9, 5–29. [Google Scholar] [CrossRef]
- Regulation No 882/2004/EC of 29 April 2004 of the European Parliament and of the Council on Official Controls Performed to Ensure the Verification of Compliance with Feed and Food Law, Animal Health and Animal Welfare Rules. Available online: http://data.europa.eu/eli/reg/2004/882/oj (accessed on 18 November 2016).
- ISO/IEC 17025:2005 General Requirements for the Competence of Testing and Calibration Laboratories. Available online: http://www.iso.org/iso/catalogue_detail?csnumber=39883 (accessed on 18 November 2016).
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis, IUPAC Technical report. Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- Walravens, J.; Mikula, H.; Rychlik, M.; Asam, S.; Ediage, E.M.; Di Mavungu, J.D.; Van Landschoot, A.; Vanhaecke, L.; De Saeger, S. Development and validation of an ultra-high-performance liquid chromatography tandem mass spectrometric method for the simultaneous determination of free and conjugated Alternaria toxins in cereal-based foodstuffs. J. Chromatog. A. 2014, 1372, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Dall’Asta, C.; Schuhmacher, R.; Lemmens, M.; Adam, G.; Krska, R. Masked Mycotoxins: Determination of a Deoxynivalenol Glucoside in Artificially and Naturally Contaminated Wheat by Liquid Chromatography—Tandem Mass Spectrometry. J. Agric. Food Chem. 2005, 53, 3421–3425. [Google Scholar] [CrossRef] [PubMed]
- Vend, O.; Berthiller, F.; Crews, C.; Krska, R. Simultaneous determination of deoxynivalenol, zearalenone, and their major masked metabolites in cereal-based food by LC-MS-MS. Anal. Bioanal. Chem. 2009, 395, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A. The current role of high-resolution mass spectrometry in food analysis. Anal. Bioanal. Chem. 2012, 403, 1233–1249. [Google Scholar] [CrossRef] [PubMed]
- Krauss, M.; Singer, H.; Hollender, J. LC-high resolution MS in environmental analysis: From target screening to the identification of unknowns. Anal. Bional. Chem. 2010, 397, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.; Asam, S. Stable isotope dilution assays in mycotoxin analysis. Anal. Bioanal. Chem. 2008, 390, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Asam, S.; Rychlik, M. Quantitation of type B-trichothecene mycotoxins in foods and feeds by a multiple stable isotope dilution assay. Eur. Food Res. Technol. 2007, 224, 769–783. [Google Scholar] [CrossRef]
- Habler, K.; Rychlik, M. Multi-mycotoxin stable isotope dilution LC-MS/MS method for Fusarium toxins in cereals. Anal. Bioanal. Chem. 2016, 408, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Habler, K.; Frank, O.; Rychlik, M. Chemical synthesis of deoxynivalenol-3-β-D-[13C6]-glucoside and application in stable isotope dilution assay. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Glauner, T.; Köppen, R.; Mayer, K.; Sulyok, M.; Schuhmacher, R.; Krska, R.; Berthiller, F. Stable isotope dilution assay for the accurate determination of mycotoxins in maize by UHPLC-MS/MS. Anal. Bional. Chem. 2012, 402, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Veprikova, Z.; Godula, M. Multi-analyte high performance liquid chromatography coupled to high resolution tandem mass spectrometry method for control of pesticide residues, mycotoxins, and pyrrolizidine alkaloids. Anal. Chim. Acta 2015, 863, 29–40. [Google Scholar] [CrossRef] [PubMed]
- León, N.; Pastor, A.; Yusà, V. Target analysis and retrospective screening of veterinary drugs, ergot alkaloids, plant toxins and other undesirable substances in feed using liquid chromatography–high resolution mass spectrometry. Talanta 2016, 149, 43–52. [Google Scholar] [CrossRef] [PubMed]
- De Boevre, M.; Vanheule, A.; Audenaert, K.; Bekaert, B.; Di Mavungu, J.D.; Werbrouck, S.; Haesaert, G.; De Saeger, S. Detached leaf in vitro model for masked mycotoxin biosynthesis and subsequent analysis of unknown conjugates. World Mycotoxin J. 2014, 7, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Rochat, B. From targeted quantification to untargeted metabolomics: Why LC-high-resolution-MS will become a key instrument in clinical labs. Trend Analyt Chem. 2016, 84, 151–164. [Google Scholar] [CrossRef]
- Senyuva, H.Z.; Gökmen, V.; Sarikaya, E.A. Future perspectives in Orbitrap™-high-resolution mass spectrometry in food analysis: A review. Food Addit. Contam. Part A 2015, 32, 1568–1606. [Google Scholar] [CrossRef] [PubMed]
- Ates, E.; Godula, M.; Stroka, J.; Senyuva, H. Screening of plant and fungal metabolites in wheat, maize and animal feed using automated on-line clean-up coupled to high resolution mass spectrometry. Food Chem. 2014, 142, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Kovalsky, M.P.; Schweiger, W.; Hametner, C.; Stuckler, R.; Muehlbauer, G.J.; Varga, E.; Krska, R.; Berthiller, F.; Adam, G. Zearalenone-16-O-glucoside: A new masked mycotoxin. J. Agric. Food Chem. 2014, 62, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Vaclavikova, M.; Lacina, O.; Vaclavik, L.; Hajslova, J. Deoxynivalenol oligoglycosides: New “masked” fuarium toxins occurring in malt, beer, and breadstuff. J. Agric. Food Chem. 2012, 60, 9280–9291. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Foglia, P.; Guarino, C.; Marzioni, F.; Nazzari, M.; Samperi, R.; Laganà, A. Aflatoxin M1 determination in cheese by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2006, 1135, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Sahin, H.Z.; Celik, M.; Kotay, S.; Kabak, B. Aflatoxins in dairy cow feed, raw milk and milk products from Turkey. Food Addit. Contam. Part B 2016, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Veprikova, Z.; Zachariasova, M.; Dzuman, Z.; Zachariasova, A.; Fenclova, M.; Slavikova, P.; Vaclavikova, M.; Mastovska, K.; Hengst, D.; Hajslova, J. Mycotoxins in Plant-Based Dietary Supplements: Hidden Health Risk for Consumers. J. Agric. Food Chem. 2015, 63, 6633–6643. [Google Scholar] [CrossRef] [PubMed]
- Frisvad, J.C.; Larsen, T.O.; de Vries, R.; Meijer, M.; Houbraken, J.; Cabañes, F.J.; Ehrlich, K.; Samson, R.A. Secondary metabolite profiling, growth profiles and other tools for species recognition and important Aspergillus mycotoxins. Stud. Mycol. 2007, 59, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T.; Visconti, A.; Haidukowski, M.; Pascale, M. Identification and characterization of new Fusariummasked mycotoxins, T2 and HT2 glycosyl derivatives, in naturally contaminated wheat and oats by liquid chromatography–high-resolution mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 47, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Bueschl, C.; Lemmens, M.; Michlmayr, H.; Malachova, A.; Koutnik, A.; Maloku, I.; Berthiller, F.; Adam, G.; Krska, R.; et al. Biotransformation of the mycotoxin deoxynivalenol in fusarium resistant and susceptible near isogenic wheat lines. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Ohmichi, K.; Sakamoto, S.; Sago, Y.; Kushiro, M.; Nagashima, H.; Yoshida, M.; Nakajima, T. Detection of a new Fusarium masked mycotoxin in wheat grain by high-resolution LC–OrbitrapTM MS. Food Addit. Contam. 2011, 28, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Kruse Fæste, C.; Uhlig, S. In vitro phase I metabolism of the depsipeptide enniatin B. Anal. Bioanal. Chem. 2011, 400, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Werner, U.; Suliok, M.; Kraska, R.; Hauser, M.T.; Schuhmacher, R. Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) determination of phase II metabolites of the mycotoxin zearalenone in the model plant Arabidopsis thaliana. Food Addit. Contam. 2006, 23, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Meng-Reiterer, J.; Varga, E.; Nathanail, A.V.; Bueschl, C.; Rechthaler, J.; McCormick, S.P.; Michlmayr, H.; Malachová, A.; Fruhmann, P.; Adam, G.; et al. Tracing the metabolism of HT-2 toxin and T-2 toxin in barley by isotope-assisted untargeted screening and quantitative LC-HRMS analysis. Anal. Bioanal. Chem. 2015, 407, 8019–8033. [Google Scholar] [CrossRef] [PubMed]
- Fruhmann, P.; Mikula, H.; Wiesenberger, G.; Varga, E.; Lumpi, D.; Stöger, B.; Häubl, G.; Lemmens, M.; Berthiller, F.; Krska, R.; et al. Isolation and Structure Elucidation of Pentahydroxyscirpene, a Trichothecene Fusarium Mycotoxin. J. Nat. Prod. 2014, 77, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Kostelanska, M.; Dzuman, Z.; Malachova, A.; Capouchova, I.; Prokinova, E.; Skerikova, A.; Hajslova, J. Effects of Milling and Baking Technologies on Levels of Deoxynivalenol and its Masked Form Deoxynivalenol-3-Glucoside. J. Agric. Food Chem. 2011, 59, 9303–9312. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal. Rev. 2010, 2, 23–60. [Google Scholar] [CrossRef] [PubMed]
- Zedda, M.; Zwiener, C. Is nontarget screening of emerging contaminants by LC-HRMS successful? A plea for compound libraries and computer tools. Anal. Bioanal. Chem. 2012, 403, 2493–2502. [Google Scholar] [CrossRef] [PubMed]
- Nathanail, A.V.; Varga, E.; Meng-Reiterer, J.; Bueschl, C.; Michlmayr, H.; Malachova, A.; Fruhmann, P.; Jestoi, M.; Peltonen, K.; Adam, G.; et al. Metabolism of the Fusarium Mycotoxins T-2 Toxin and HT-2 Toxin in Wheat. J. Agric. Food Chem. 2015, 63, 7862–7872. [Google Scholar] [CrossRef] [PubMed]
- Dall’Asta, C.; Berthiller, F.; Schuhmacher, R.; Adam, G.; Lemmens, M.; Krska, R. DON-glycosides: Characterisation of synthesis products and screening for their occurrence in DON treated wheat samples. Mycotoxin Res. 2005, 21, 123–127. [Google Scholar] [CrossRef] [PubMed]
- De Boevre, M.; Ediage, E.N.; Van Poucke, C.; De Sager, S. Untargeted analysis of modified mycotoxins using high-resolution mass spectrometry. In Masked Mycotoxins in Food: Formation, Occurrence and Toxicological Relevance, 1st ed.; Dall’Asta, C., Berthiller, F., Eds.; Royal Society of Chemistry: Cambridge, UK, 2016; Volume 24, pp. 50–72. [Google Scholar]
- Keller, B.O.; Sui, J.; Young, A.B.; Whittal, R.M. Interferences and contaminants encountered in modern mass spectrometry. Anal. Chim. Acta 2008, 627, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Kluger, B.; Lemmens, M.; Adam, G.; Wiesenberger, G.; Maschietto, V.; Marocco, A.; Strauss, J.; Bodi, S.; Thallinger, G.G.; et al. A novel stable isotope labelling assisted workflow for improved untargeted LC–HRMS based metabolomics research. Metabolomics 2014, 10, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Bueschl, C.; Lemmens, M.; Berthiller, F.; Häubl, G.; Jaunecker, G.; Adam, G.; Krska, R.; Schuhmacher, R. Stable isotopic labelling-assisted untargeted metabolic profiling reveals novel conjugates of the mycotoxin deoxynivalenol in wheat. Anal. Bioanal. Chem. 2013, 405, 5031–5036. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Kluger, B.; Berthiller, F.; Lirk, G.; Winkler, S.; Krska, R.; Schuhmacher, R. MetExtract: A new software tool for the automated comprehensive extraction of metabolite-derived LC/MS signals in metabolomics research. Bioinformatics 2012, 28, 736–738. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.P.; Kato, T.; Maragos, C.M.; Busman, M.; Lattanzio, V.M.T.; Galaverna, G.; Dall-Asta, C.; Crich, D.; Price, N.P.J.; Kurtzman, C.P. Anomericity of T-2 Toxin-glucoside: Masked Mycotoxin in Cereal Crops. J. Agric. Food Chem. 2015, 63, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Cumeras, R.; Figueras, E.; Davis, C.E.; Baumbach, J.I.; Gràcia, I. Review on ion mobility spectrometry. Part 1: Current instrumentation. Analyst 2015, 140, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Kafle, G.K.; Khot, L.R.; Sankaran, S.; Bahlol, H.Y.; Tufariello, J.A.; Hill, H.H. State of ion mobility spectrometry and applications in agriculture: A review. Eng. Agric. Environ. Food 2016, 9, 346–357. [Google Scholar] [CrossRef]
- Ibrahim, Y.M.; Baker, E.S.; Danielson, W.F.; Norheim, R.V.; Prior, D.C.; Anderson, G.A.; Belov, M.E.; Smith, R.D. Development of a New Ion Mobility (Quadrupole) Time-of-Flight Mass Spectrometer. Int. J. Mass Spectrum. 2015, 377, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Giles, K. Travelling wave ion mobility. Int. J. Ion Mobil. Spectrom. 2013, 16, 1–3. [Google Scholar] [CrossRef]
- Shvartsburg, A.A.; Smith, R.D. Fundamentals of traveling wave ion mobility spectrometry. Anal. Chem. 2008, 80, 9689–9699. [Google Scholar] [CrossRef] [PubMed]
- Shvartsburg, A.A. Differential Ion Mobility Spectrometry: Nonlinear Ion Transport and Fundamentals of FAIMS, 1st ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Michelmann, K.; Silveira, J.A.; Ridgeway, M.E.; Park, M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.R.; Debord, J.D.; Ridgeway, M.E.; Kaplan, D.A.; Park, M.A.; Fernandez-Lima, F. Ion dynamics in a trapped ion mobility spectrometer. Analyst 2014, 139, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lima, F.; Kaplan, D.A.; Suetering, J.; Park, M.A. Gas-phase separation using a trapped ion mobility spectrometer. Int. J. Ion Mobil. Spectrom. 2011, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lapthorn, C.; Pullen, F.; Chowdhry, B.Z. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules: Separating and assigning structures to ions. Mass Spectrom. Rev. 2013, 32, 43–71. [Google Scholar] [CrossRef] [PubMed]
- McCooeye, M.; Kolakowski, B.; Boison, J.; Mester, Z. Evaluation of high-field asymmetric waveform ion mobility spectrometry for the analysis of the mycotoxin zearalenone. Anal. Chim. Acta 2008, 627, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Karpas, Z. Applications of ion mobility spectrometry (IMS) in the field of foodomics. Food. Res. Int. 2013, 54, 1146–1151. [Google Scholar] [CrossRef]
- Beach, D.G.; Melanson, J.E.; Purves, R.W. Analysis of paralytic shellfish toxins using high-field asymmetric waveform ion mobility spectrometry with liquid chromatography-mass spectrometry. Anal. Bional. Chem. 2015, 407, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Nielen, M.W.F.; Van Beek, T.A. Macroscopic and microscopic spatially-resolved analysis of food contaminants and constituents using laser-ablation electrospray ionization mass spectrometry imaging. Anal. Bional. Chem. 2014, 406, 6805–6815. [Google Scholar] [CrossRef] [PubMed]
- Goscinny, S.; Joly, L.; De Pauw, E.; Hanot, V.; Eppe, G. Travelling-wave ion mobility time-of-flight mass spectrometry as an alternative strategy for screening of multi-class pesticides in fruits and vegetables. J. Chromatogr. A 2015, 1405, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.B.; Smagghe, G.; Coelus, S.; Adriaenssens, D.; De Winter, K.; Desmet, T.; Raes, K.; Van Camp, J. Collision cross section prediction of deprotonated phenolics in a travelling-wave ion mobility spectrometer using molecular descriptors and chemometrics. Anal. Chim. Acta 2016, 924, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Sheibani, A.; Tabrizchi, M.; Ghaziaskar, H. Determination of aflatoxins B1 and b2 using ion mobility spectrometry. Talanta 2007, 75, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Khalesi, M.; Sheikh-Zeinoddin, M.; Tabrizchi, M. Determination of ochratoxin A in licorice root using inverse ion mobility spectrometry. Talanta 2011, 83, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Fenclová, M.; Lacina, O.; Zachariášová, M.; Hajšlová, J. Application of ion-mobility Q-TOF LC/MS platform in masked mycotoxins research. In Proceedings of the Recent Advances in Food Analysis, Prague, Czech Republic, 3–6 November 2015; Pulkrabová, J., Tomaniová, M., Nielen, M., Hajšlová, J., Eds.; UCT Prague Press: Prague, Czech Republic, 2015; p. 122. [Google Scholar]
- Stead, S.; Joumier, J.M.; McCullagh, M.; Busman, M.; McCormick, S.; Maragos, C. Using ion mobility mass spectrometry and collision cross section areas to elucidate the α and β epimeric forms of glycosilated T-2 and HT-2 toxins. In Proceedings of the Recent Advances in Food Analysis, Prague, Czech Republic, 3–6 November 2015; Pulkrabová, J., Tomaniová, M., Nielen, M., Hajšlová, J., Eds.; UCT Prague Press: Prague, Czech Republic, 2015; p. 338. [Google Scholar]
- Ruotolo, B.T.; Gillig, K.J.; Stone, E.G.; Russell, D.H. Peak capacity of ion mobility mass spectrometry: Separation of peptides in helium buffer gas. J. Chromatogr. B 2002, 782, 385–392. [Google Scholar] [CrossRef]
- Ruotolo, B.T.; McLean, J.A.; Gillig, K.J.; Russell, D.H. Peak capacity of ion mobility mass spectrometry: The utility of varying drift gas polarizability for the separation of tryptic peptides. J. Mass Spectrom. 2004, 39, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.T.; Saraji, M.; Yousefi, S. Negative electrospray ionization ion mobility spectrometry combined with microextraction in packed syringe for direct analysis of phenoxyacid herbicides in environmental water. J. Chromatogr. A 2012, 1249, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, J.; Negreira, N.; Berntssen, M.H.G. Ion mobility-derived collision cross section as an additional identification point for multi-residue screening of pesticides in fish feed. Anal. Chem. 2016, 88, 11169–11177. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.T.; Khayamian, T.; Shaer, V.; Zarei, N. Determination of veterinary drug residues in chicken meat using corona discharge ion mobility spectrometry. Anal. Chim. Acta 2007, 581, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Kliman, M.; Claude, E.; Geromanos, S.; Astarita, G. Applications of ion-mobility mass spectrometry for lipid analysis. Anal. Bional. Chem. 2015, 407, 4995–5007. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Tsybulskyy, D.; Lucioli, J.; Laffitte, J.; Callu, P.; Lyazhri, F.; Grosjean, F.; Bracarense, A.P.; Kolf-Clauw, M.; Oswald, I.P. Toxicity of deoxynivalenol and its acetylated derivatives on the intestine: Differential effects on morphology, barrier function, tight junction proteins, and mitogen-activated protein kinases. Toxicol. Sci. 2012, 130, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Monbaliu, S.; Van Poucke, C.; Van Peteghem, C.; Van Poucke, K.; Heungens, K.; De Saeger, S. Development of a multi-mycotoxin liquid chromatography/tandem mass spectrometry method for sweet pepper analysis. Rapid Commun. Mass Spectrom. 2009, 23, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Biancardi, A.; Gasparini, M.; Dall’Asta, C.; Marchelli, R. A rapid multiresidual determination of type A and type B trichothecenes in wheat flour by HPLC-ESIMS. Food. Addit. Contam. 2005, 22, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, I.; Bush, M.F.; Robinson, C.V.; Beaumont, C.; Richardson, K.; Kim, H.; Kim, H.I. Structural characterization of drug-like compounds by ion mobility mass spectrometry: Comparison of theirethical and experimentally derived nitrogen collision cross sections. Anal. Chem. 2012, 84, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Fenn, L.S.; Kliman, M.; Mahsut, A.; Zhao, S.R.; McLean, J.A. Characterizing ion mobility-mass spectrometry conformation space for the analysis of complex biological samples. Anal. Bioanal. Chem. 2009, 394, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.F.; Campuzano, I.D.; Robinson, C.V. Ion mobility mass spectrometry of peptide ions: Effects of drift gas and calibration strategies. Anal. Chem. 2012, 84, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, M.; Cleland, G.; Hanot, V.; Stead, S.; Williams, J.; Goscinny, S. Collision Cross Section a New Identification Point for a “Catch All” Non Targeted Screening Approach; Waters Application Note; Waters and the Scientific Institute of Public Health: Brussels, Belgium, 2014; Available online: http://www.waters.com/waters/library.htm?lid=134803219&locale=it_IT (accessed on 29 November 2016).
- Commission Decision No. 2002/657/EC of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Available online: http://data.europa.eu/eli/dec/2002/657/oj (accessed on 18 November 2016).
- Paglia, G.; Angel, P.; Williams, J.P.; Richardson, K.; Olivos, H.J.; Thompson, J.W.; Menikarachchi, L.; Lai, S.; Walsh, C.; Moseley, A.; et al. Ion Mobility-Derived Collision Cross Section As an Additional Measure for Lipid Fingerprinting and Identification. Anal. Chem. 2015, 87, 1137–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hines, K.M.; May, J.C.; McLean, J.A.; Xu, L. Evaluation of Collision Cross Section Calibrants for Structural Analysis of Lipids by Traveling Wave Ion Mobility-Mass Spectrometry. Anal. Chem. 2016, 88, 7329–7336. [Google Scholar] [CrossRef] [PubMed]


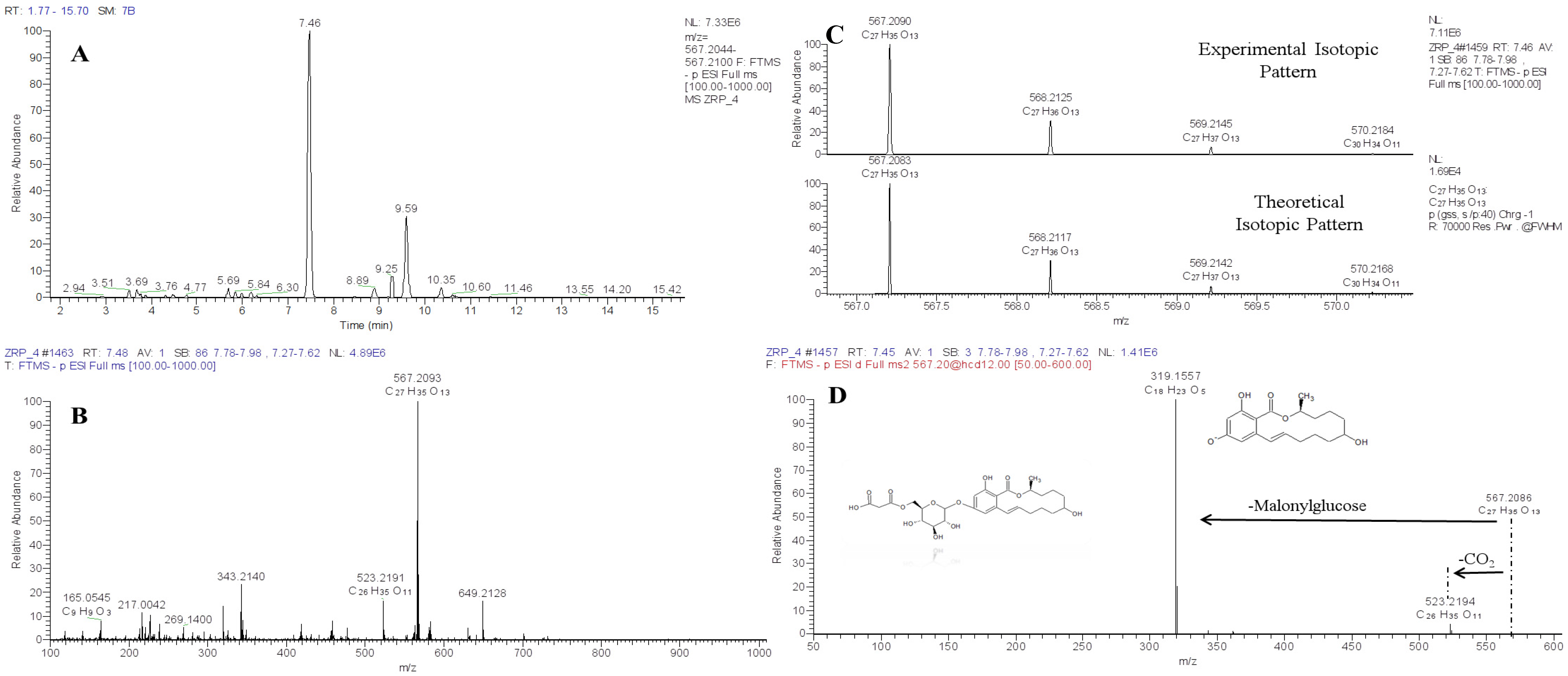

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Known/Unknown Categories | Modified Mycotoxin | Matrix | MS Equipment | Identification Based on | Analytical Standard |
|---|---|---|---|---|---|
| Expected knowns | Aflatoxin M1 | Cheese [39] | Q-Trap | authentic standards | commercially available |
| DON3Glc | Wheat and maize [22] | Q-Trap | authentic standards | commercially available | |
| 3/15Ac-DON | Wheat [23] | Q-Trap | authentic standards | commercially available | |
| Unexpected knowns | Aflatoxin M1 | Feed [40] | QqQ | authentic standards | commercially available |
| enniatins, alternaria toxins, T-2/HT-2 toxins | Dietary supplements [41] | Q-Trap | authentic standards | commercially available | |
| FB2 | Culture media [42] | QqQ | authentic standards | commercially available | |
| Expected unknowns | T2-Glc | Wheat and oats [43] | LTQ Orbitrap | HRMS | in-house synthesized |
| 15Ac-DON-Glc | Wheat [44] | LTQ Orbitrap XL | authentic standards | in-house synthesized | |
| DON-oligoglycoside | Malt and Beer [38] | Exactive Orbitrap | HRMS | n.a. | |
| NIV-Glc | Wheat [45] | LTQ Orbitrap | HRMS/MS | n.a. | |
| Desmethyl Enn B1 | Human liver [46] | Q-Tof | HRMS/MS | n.a. | |
| ZEN-MalGlc | Wheat [47] | Q-trap | MS/MS | n.a. | |
| Unexpected unknowns | Feruloyl-T2 | Barley [48] | Exactive Plus Orbitrap | HRMS/MS | n.a. |
| DON-2H-glutathione | Wheat [44] | LTQ Orbitrap XL | HRMS/MS | n.a. | |
| Pentahydroxyscirpene (PHS) | Barley [49] | Q-Tof | MS/MS | in-house synthesized | |
| DON-3-Glc lactone | Wheat [50] | Exactive Orbitrap | HRMS | n.a. |
| Modification | Mass Change (Da) | Molecular Formula Change |
|---|---|---|
| Hydrogenation | 2.0151 | H2 |
| Hydroxylation | 15.9944 | O |
| Methylation | 14.0151 | CH2 |
| Acetylation | 42.0100 | C2H2O |
| Glycine | 57.0209 | C2H3NO |
| Sulfate | 79.9563 | SO3 |
| Sulfonation | 102.9460 | SO3Na |
| Ferulic acid | 176.0468 | C10H8O3 |
| Cysteine | 119.0036 | C3H5NO2S |
| Acetyl-cysteine | 161.0141 | C5H7NO3S |
| Glucose | 162.0523 | C6H10O5 |
| Cysteine-glycine | 176.0250 | C5H8N2O3S |
| Glucuronic acid | 176.0315 | C6H8O6 |
| Acetyl-glucoside | 203.0550 | C8H11O6 |
| Malonyl glucoside | 248.0527 | C9H12O8 |
| Glutathione | 305.0682 | C10H15N5O6S |
| Di-glucoside | 324.1051 | C12H20O10 |
| Malonyl di-glucoside | 410.1055 | C15H22O13 |
| Tri-glucoside | 486.1579 | C18H30O15 |
| Di malonyl-di glucoside | 497.1137 | C18H25O16 |
| Tetra-glucoside | 648.2107 | C24H40O20 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righetti, L.; Paglia, G.; Galaverna, G.; Dall’Asta, C. Recent Advances and Future Challenges in Modified Mycotoxin Analysis: Why HRMS Has Become a Key Instrument in Food Contaminant Research. Toxins 2016, 8, 361. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8120361
Righetti L, Paglia G, Galaverna G, Dall’Asta C. Recent Advances and Future Challenges in Modified Mycotoxin Analysis: Why HRMS Has Become a Key Instrument in Food Contaminant Research. Toxins. 2016; 8(12):361. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8120361
Chicago/Turabian StyleRighetti, Laura, Giuseppe Paglia, Gianni Galaverna, and Chiara Dall’Asta. 2016. "Recent Advances and Future Challenges in Modified Mycotoxin Analysis: Why HRMS Has Become a Key Instrument in Food Contaminant Research" Toxins 8, no. 12: 361. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8120361