
Application of LC-MS/MS MRM to Determine Staphylococcal Enterotoxins (SEB and SEA) in Milk

 ,
,
Abstract
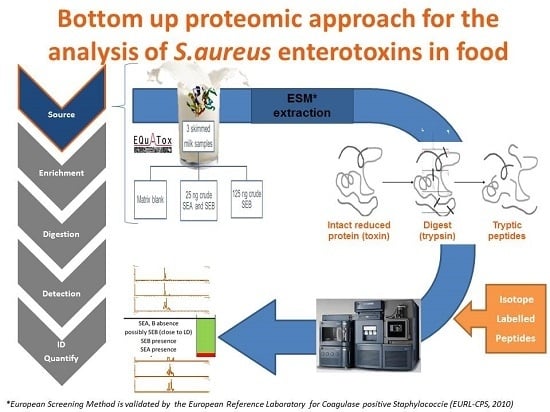
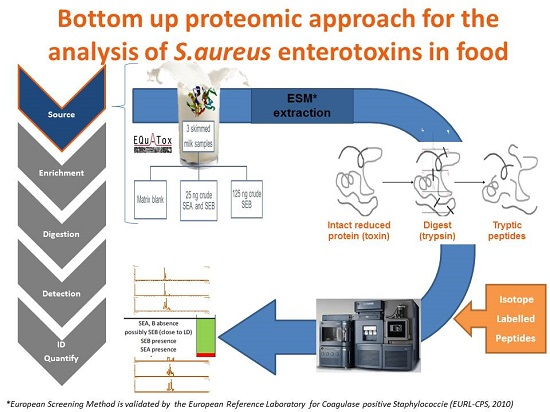
:
1. Introduction
2. Results and Discussion
2.1. In Silico Selection of SEB Peptides
2.2. In Silico Selection of SEA Peptides
2.3. Toxin Detection
3. Experimental Section
3.1. Samples
3.2. Sample Preparation
3.3. Stock Solutions and Reagents
3.4. In Silico Selection of Peptides
3.5. Internal Standards (IS)
3.6. LC-MS/MS Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rajkovic, A. Staphylococcus: Food poisoning. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 133–139. [Google Scholar]
- Hennekinne, J.A.; de Buyser, M.L.; Dragacci, S. Staphylococcus aureus and its food poisoning toxins: Characterization and outbreak investigation. Fed. Eur. Microbiol. Soc. Microbiol. Rev. 2012, 36, 815–836. [Google Scholar]
- Grumann, D.; Nübel, U.; Bröker, B.M. Staphylococcus aureus toxins—Their functions and genetics. Infect. Genet. Evol. 2014, 21, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Le Loir, Y.; Baron, F.; Gautier, M. Staphylococcus aureus and food poisoning. Genet. Mol. Res. 2003, 2, 63–76. [Google Scholar] [PubMed]
- Marta, D.; Wallin-Carlquist, N.; Schelin, J.; Borch, E.; Radstrom, F. Extended staphylococcal enterotoxin D expression in ham products. Food Microbiol. 2011, 28, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Wallin-Carlquist, N.; Marta, D.; Borch, E.; Radström, P. Prolonged expression and production of Staphylococcus aureus enterotoxin A in processed pork meat. Int. J. Food Microbiol. 2010, 141, S69–S74. [Google Scholar] [CrossRef] [PubMed]
- Principato, M.; Qian, B.F. Staphylococcal enterotoxins in the etiopathogenesis of mucosal autoimmunity within the gastrointestinal tract. Toxins 2014, 6, 1471–1489. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Shimomura, Y.; Murayama, S.Y.; Yagi, J.; Ubukata, K.; Kirikae, T.; Miyoshi-Akiyama, T. Evolutionary paths of streptococcal and staphylococcal superantigens. BMC Genom. 2012, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Evenson, M.L.; Hinds, M.W.; Bernstein, R.S.; Bergdoll, M.S. Estimation of human dose of staphylococcal enterotoxin A from a large outbreak of staphylococcal food poisoning involving chocolate milk. Int. J. Food Microbiol. 1988, 7, 311–316. [Google Scholar] [CrossRef]
- Asao, T.; Kumeda, Y.; Kawai, T.; Shibata, T.; Oda, H.; Haruki, K.; Nakazawa, H.; Kozaki, S. An extensive outbreak of staphylococcal food poisoning due to low-fat milk in Japan: Estimation of enterotoxin A in the incriminated milk and powdered skim milk. Epidemiol. Infect. 2003, 130, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Schelin, J.; Wallin-Carlquist, N.; Cohn, M.T.; Lindqvist, R.; Barker, G.C. The formation of Staphylococcus aureus enterotoxin in food environments and advances in risk assessment. Virulence 2011, 2, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.D.; Moon, B.Y.; Park, J.H.; Chang, H.I.; Kim, W.J. Expression of enterotoxin genes in Staphylococcus aureus isolates based on mRNA analysis. J. Microbiol. Biotechnol. 2007, 17, 461–467. [Google Scholar] [PubMed]
- Duquenne, M.; Fleurot, I.; Aigle, M.; Darrigo, C.; Borezée-Durant, E.; Derzelle, S.; Bouix, M.; Deperrois-Lafarge, V.; Delacroix-Buchet, A. Tool for quantification of staphylococcal enterotoxin gene expression in cheese. Appl. Environ. Microbiol. 2010, 76, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Hennekinne, J.A.; Guillier, F.; Perelle, S.; de Buyser, M.L.; Dragacci, S.; Krys, S.; Lombard, B. Intralaboratory validation according to the EN ISO 16 140 Standard of the Vidas SET2 detection kit for use in official controls of staphylococcal enterotoxins in milk products. J. Appl. Microbiol. 2007, 102, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Rajkovic, A.; Moualij, B.E.; Uyttendaele, M.; Brolet, P.; Zorzi, W.; Heinen, E.; Foubert, E.; Debevere, J. Immunoquantitative real-time PCR for detection and quantification of Staphylococcus aureus enterotoxin B in foods. Appl. Environ. Microbiol. 2006, 72, 6593–6599. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, H.J.; Mathisen, T.; Lovseth, A.; Omoe, K.; Qvale, K.S.; Loncarevic, S. An outbreak of staphylococcal food poisoning caused by enterotoxin H in mashed potato made with raw milk. Fed. Eur. Microbiol. Soc. Microbiol. Lett. 2005, 252, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Adrait, A.; Lebert, D.; Trauchessec, M.; Dupuis, A.; Louwagie, M.; Masselon, C.; Jaquinod, M.; Chevalier, B.; Vandenesch, F.; Garin, J.; et al. Development of a Protein Standard Absolute Quantification (PSAQ (TM)) assay for the quantification of Staphylococcus aureus enterotoxin A in serum. J. Proteom. 2012, 75, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.H.; Shefcheck, K.J.; Williams, T.L.; Musser, S.M. Detection, confirmation, and quantification of staphylococcal enterotoxin B in food matrixes using liquid chromatography—Mass spectrometry. Anal. Chem. 2006, 78, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, A.; Hennekinne, J.A.; Garin, J.; Brun, V. Protein Standard Absolute Quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.D.; Letellier, A.; Beaudry, F. Analysis of Staphylococcus enterotoxin B using differential isotopic tags and liquid chromatography quadrupole ion trap mass spectrometry. Biomed. Chromatogr. 2012, 26, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, I.; Soler, C.; Mañes, J.; Soriano, J.M. Rapid whole protein quantitation of staphylococcal enterotoxins A and B by liquid chromatography/mass spectrometry. J. Chromatogr. 2012, 1238, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, I.; Soler, C.; Manes, J.; Soriano, J.M. Analysis of staphylococcal enterotoxin A in milk by matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.; van Dijl, J.M.; Hecker, M.; Becher, D. The Staphylococcus aureus proteome. Int. J. Med. Microbiol. 2014, 304, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Muratovic, A.Z.; Hagstrom, T.; Rosen, J.; Granelli, K.; Hellenas, K.E. Quantitative analysis of staphylococcal enterotoxins A and B in food matrices using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS). Toxins 2015, 7, 3637–3656. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Yu, Y.Y.; Schaffner, D.W.; Chen, S.G.; Ye, X.Q.; Liu, D.H. Farm to consumption risk assessment for Staphylococcus aureus and staphylococcal enterotoxins in fluid milk in China. Food Control 2016, 59, 636–643. [Google Scholar] [CrossRef]
- Cosijns, S.; Andjelkovic, M.; de Cremer, K.; van Loco, J.; Rajkovic, A. Identification and quantification of staphyloccocus aureus using online SPE-UPLC-MSMS. In Proceedings of the 23rd International ICFMH Symposium FoodMicro 2012, Istanbul, Turkey, 3–7 September 2012.
- Martinez-Bartolome, S.; Deutsch, E.W.; Binz, P.A.; Jones, A.R.; Eisenacher, M.; Mayer, G.; Campos, A.; Canals, F.; Bech-Serra, J.J.; Carrascal, M.; et al. Guidelines for reporting quantitative mass spectrometry based experiments in proteomics. J. Proteom. 2013, 95, 84–88. [Google Scholar] [CrossRef] [PubMed]
- European Commission. 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Off. J. Eur. Comm. L 2002, L221, 8–36. [Google Scholar]
- Solodoukhina, D. Food safety and bioterrorism from public health perspective. In Advances in Food Protection; Hefnawy, M., Ed.; NATO Science for Peace and Security Series A: Chemistry and Biology; Springer: Doordrecht, The Netherlands, 2011; pp. 17–25. [Google Scholar]
- Nedelkov, D.; Rasooly, A.; Nelson, R.W. Multitoxin biosensor-mass spectrometry analysis: A new approach for rapid, real-time, sensitive analysis of staphylococcal toxins in food. Int. J. Food Microbiol. 2000, 60, 1–13. [Google Scholar] [CrossRef]
- Nedelkov, D.; Nelson, R.W. Detection of Staphylococcal enterotoxin B via biomolecular interaction analysis mass spectrometry. Appl. Environ. Microbiol. 2003, 69, 5212–5215. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Kuster, B.; Mortensen, P.; Andersen, J.S.; Mann, M. Mass spectrometry allows direct identification of proteins in large genomes. Proteomics 2001, 1, 641–650. [Google Scholar] [CrossRef]
- Steen, H.; Mann, M. The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Peptide Calculator. Available online: http://www.bachem.com/service-support/peptide-calculator/ (accessed on 25 November 2015).




{kind=link}
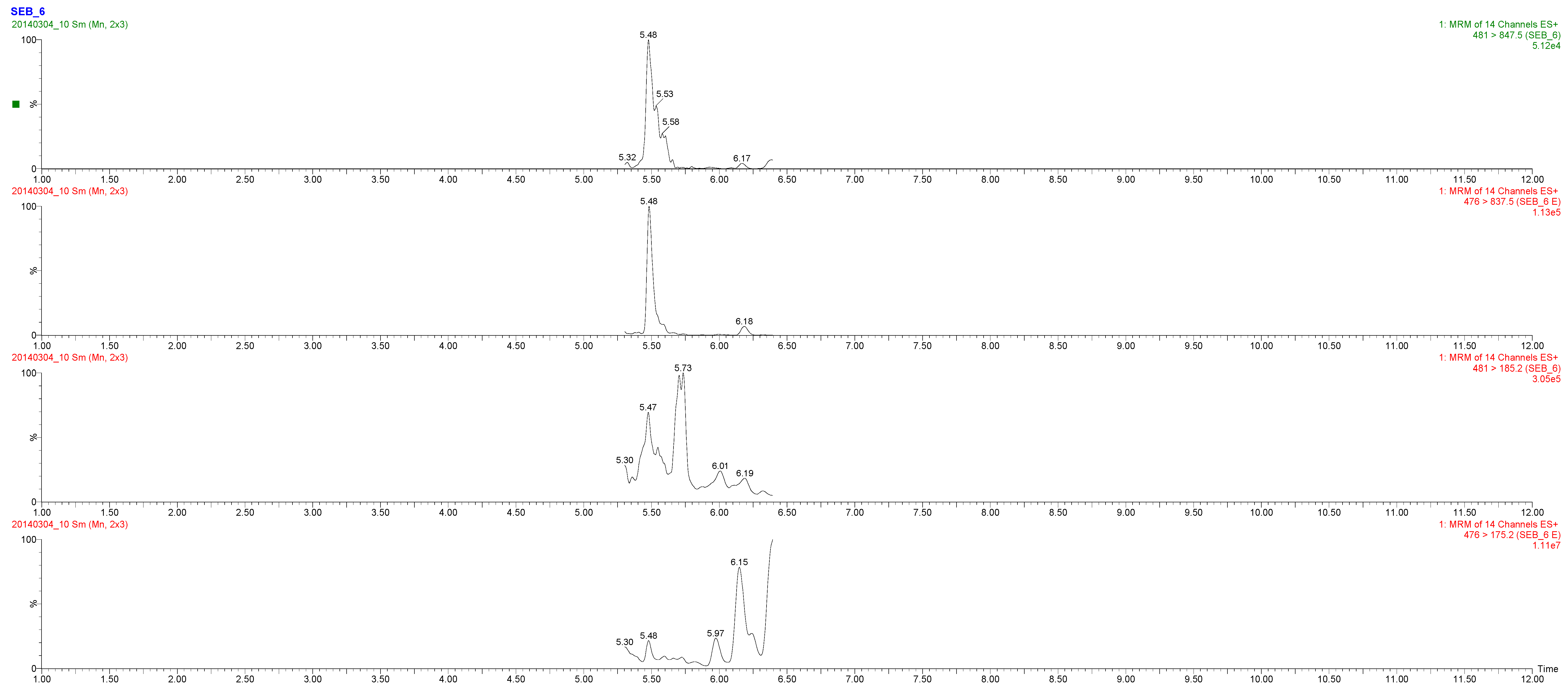{kind=link}
{kind=link}
{kind=link}
| Toxin | Peptide Sequence | Peptide Study Code * | Position | C-End | Peptide Mass | Q1 > Q2 m/z ** | Cone Voltage (V) | Collision Energy (eV) |
|---|---|---|---|---|---|---|---|---|
| SEA | YNLYNSDVFDGK | SEA-2 | 191–202 | K | 1434.654 | 722.0 > 474.40 | 25 | 33 |
| 722.0 > 391.30 | 25 | 23 | ||||||
| 722.0 > 278.20 | 25 | 23 | ||||||
| 722.0 > 212.20 | 25 | 23 | ||||||
| SELQGTALGNLK | SEA-4 | 40–51 | K | 1230.669 | 620.0 > 330.20 | 25 | 22 | |
| 620.0 > 217.20 | 25 | 20 | ||||||
| SEB | VLYDDNHVSAINVK | SEB-2 | 53–66 | K | 1586.817 | 798.2 > 692.30 | 25 | 15 |
| 798.2 > 213.30 | 25 | 15 | ||||||
| 798.2 > 185.20 | 25 | 31 | ||||||
| 532.6 > 185.20 | 25 | 31 | ||||||
| VTAQELDYLTR | SEB-5 | 182-192 | R | 1308.679 | 660.0 > 562.50 | 25 | 30 | |
| 660.0 > 919.60 | 25 | 20 | ||||||
| 660.0 > 790.60 | 25 | 20 | ||||||
| 660.0 > 677.50 | 25 | 20 | ||||||
| LGNYDNVR | SEB-6 | 85-92 | R | 950.469 | 481.0 > 847.50 | 25 | 15 | |
| 481.0 > 398.40 | 25 | 21 | ||||||
| 481.0 > 185.20 | 25 | 15 |
| Sample (Matrix) | Test Portion Used (mL or g) | SEA Observed | SEB Observed | Peptide Observed (Study Code) | Actual Toxin Presencse * |
|---|---|---|---|---|---|
| B1 buffer sample spiked at | 100 µL | - | - | no | 0.5 ng SEB/g |
| B2 buffer sample spiked at | 100 µL | - | - | no | 2 ng SEB/g |
| M1 (milk) | 1 mL extract | - | - | no | No |
| M2 (milk) | 1 mL extract | - | ± | SEB-6 | 5 ng SEB/g |
| M3 (milk) | 1 mL extract | - | + | SEB-2 | 25 ng SEB/g |
| SEB-5 | |||||
| SEB-6 | |||||
| M4 (milk) | 1 mL extract | + | - | SEA-4 | 10 ng SEA/g |
| SEA-2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andjelkovic, M.; Tsilia, V.; Rajkovic, A.; De Cremer, K.; Van Loco, J. Application of LC-MS/MS MRM to Determine Staphylococcal Enterotoxins (SEB and SEA) in Milk. Toxins 2016, 8, 118. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8040118
Andjelkovic M, Tsilia V, Rajkovic A, De Cremer K, Van Loco J. Application of LC-MS/MS MRM to Determine Staphylococcal Enterotoxins (SEB and SEA) in Milk. Toxins. 2016; 8(4):118. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8040118
Chicago/Turabian StyleAndjelkovic, Mirjana, Varvara Tsilia, Andreja Rajkovic, Koen De Cremer, and Joris Van Loco. 2016. "Application of LC-MS/MS MRM to Determine Staphylococcal Enterotoxins (SEB and SEA) in Milk" Toxins 8, no. 4: 118. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8040118