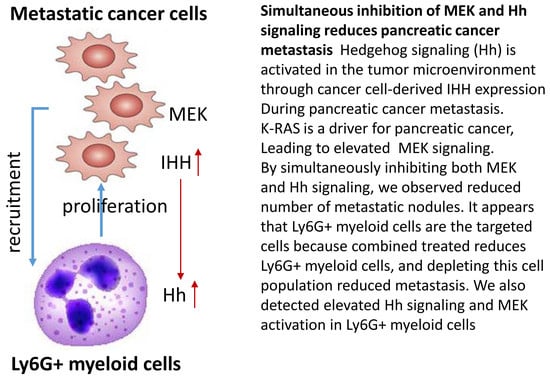
Simultaneous Inhibition of MEK and Hh Signaling Reduces Pancreatic Cancer Metastasis

Abstract
:
1. Introduction
2. Results
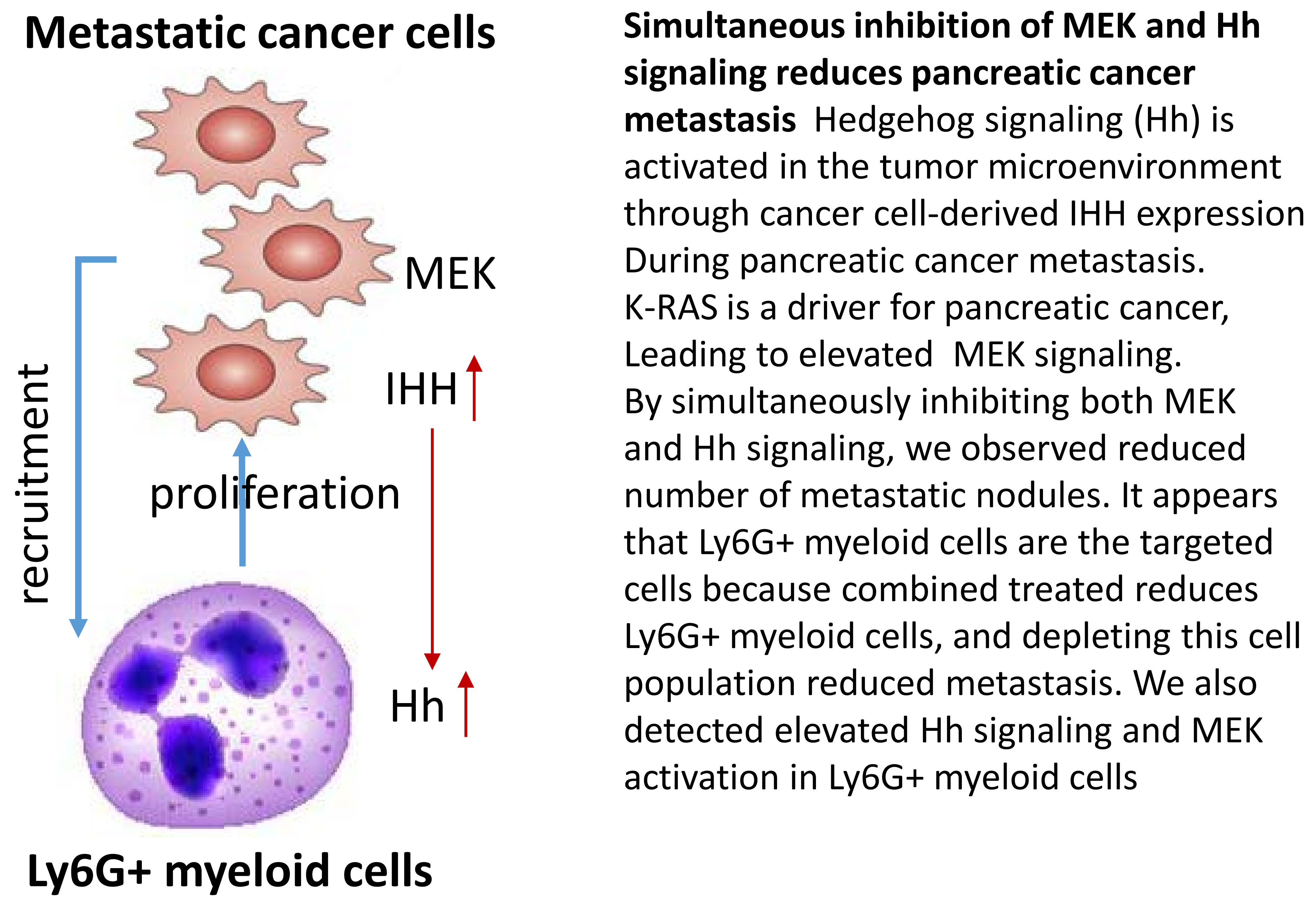
2.1. Effects of Hh Signaling on Metastatic Niche Gene Expression
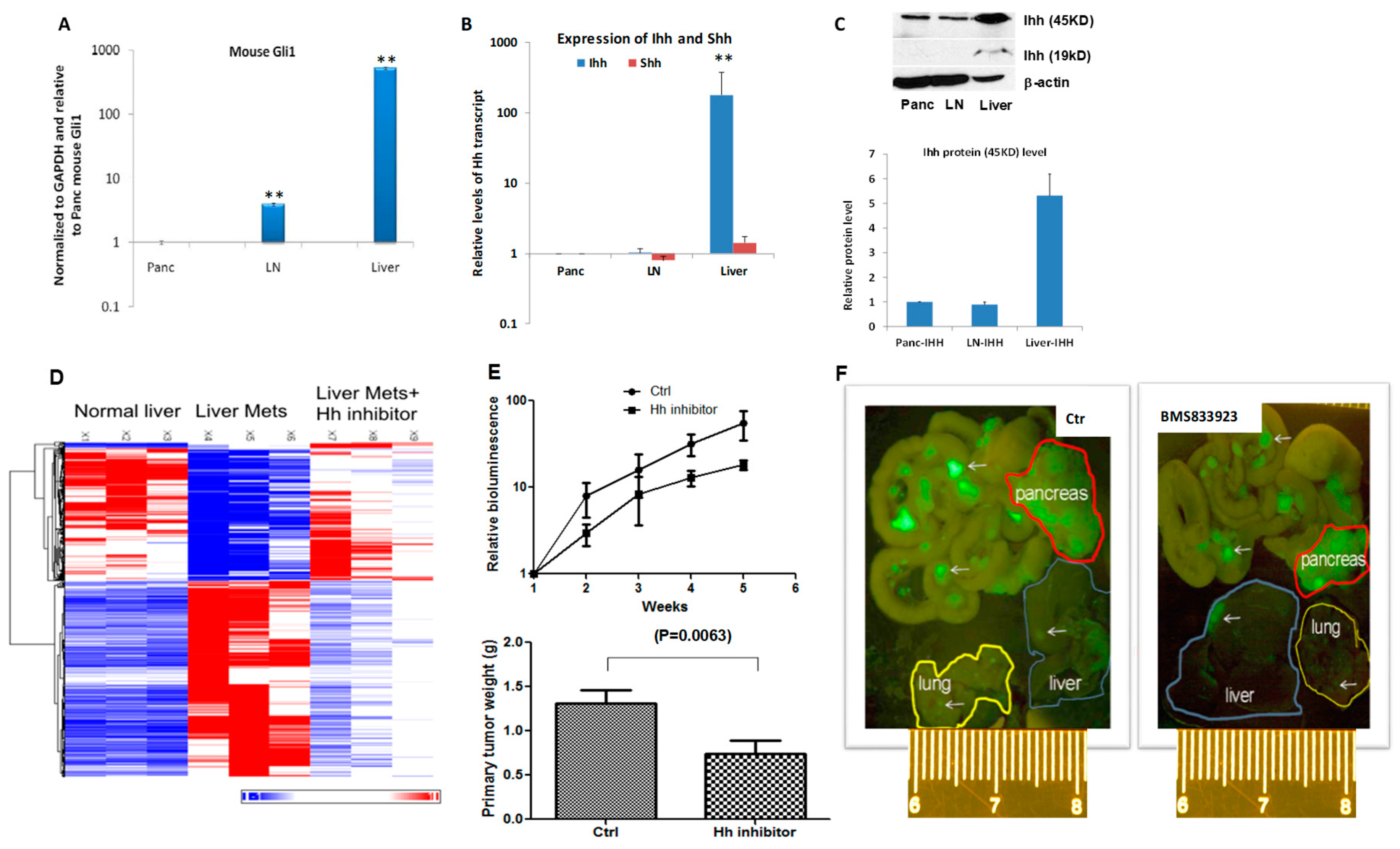
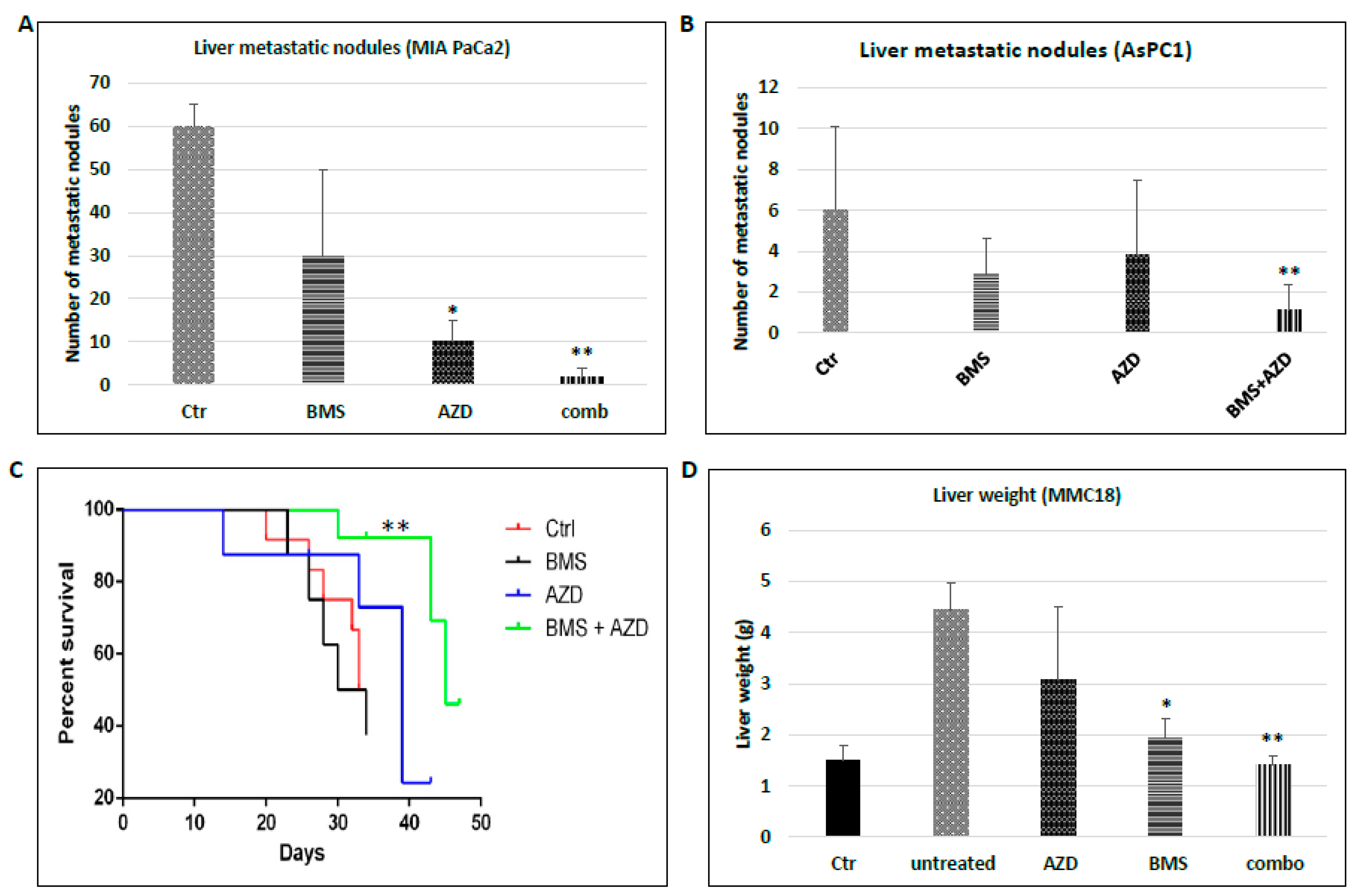
2.2. Suppression of MEK/ERK and Hh Signaling Reduces Pancreatic Cancer Metastasis
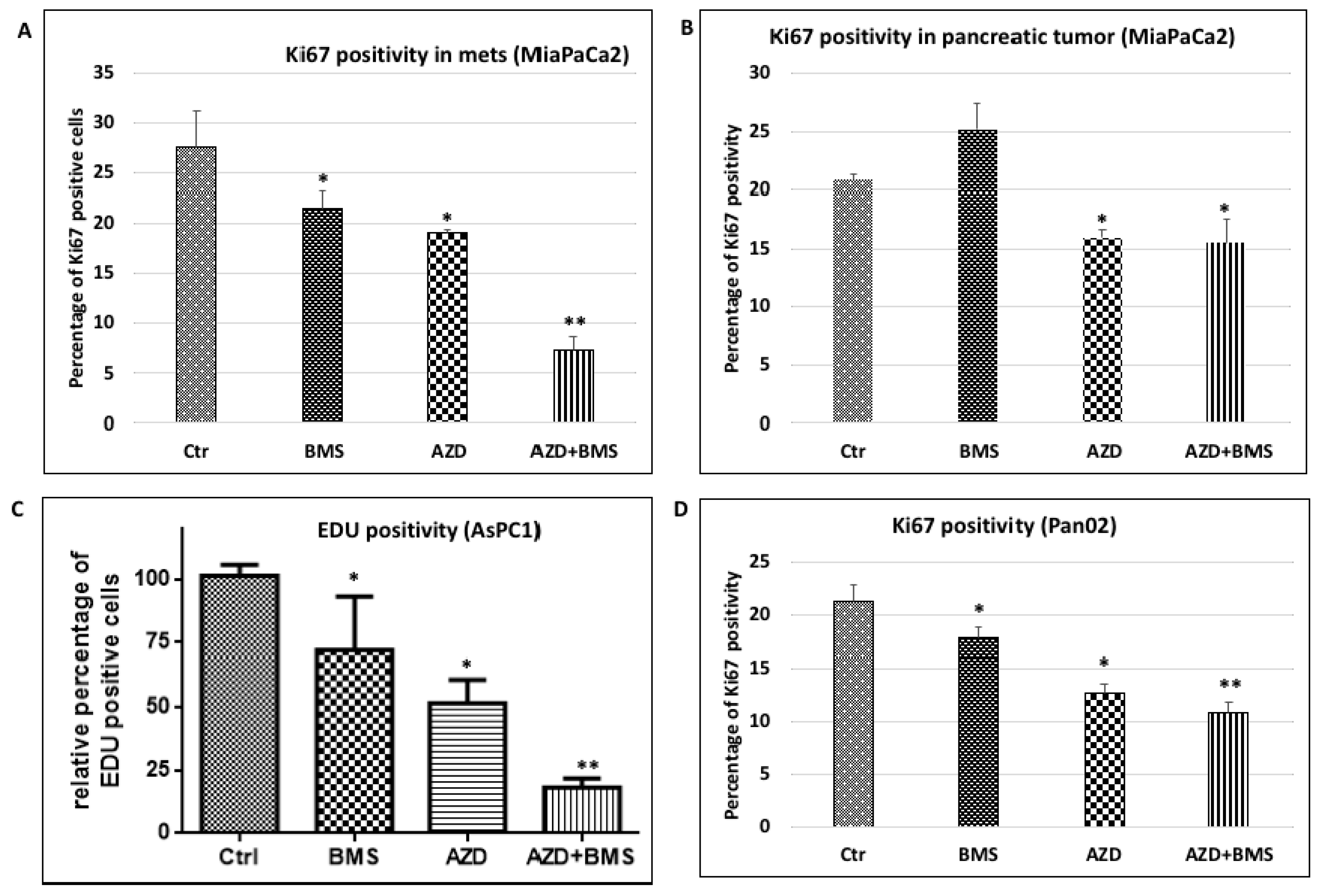
2.3. Effects of MEK Inhibitor AZD6244 and Smoothened Inhibitor BMS833923 on Cell Proliferation
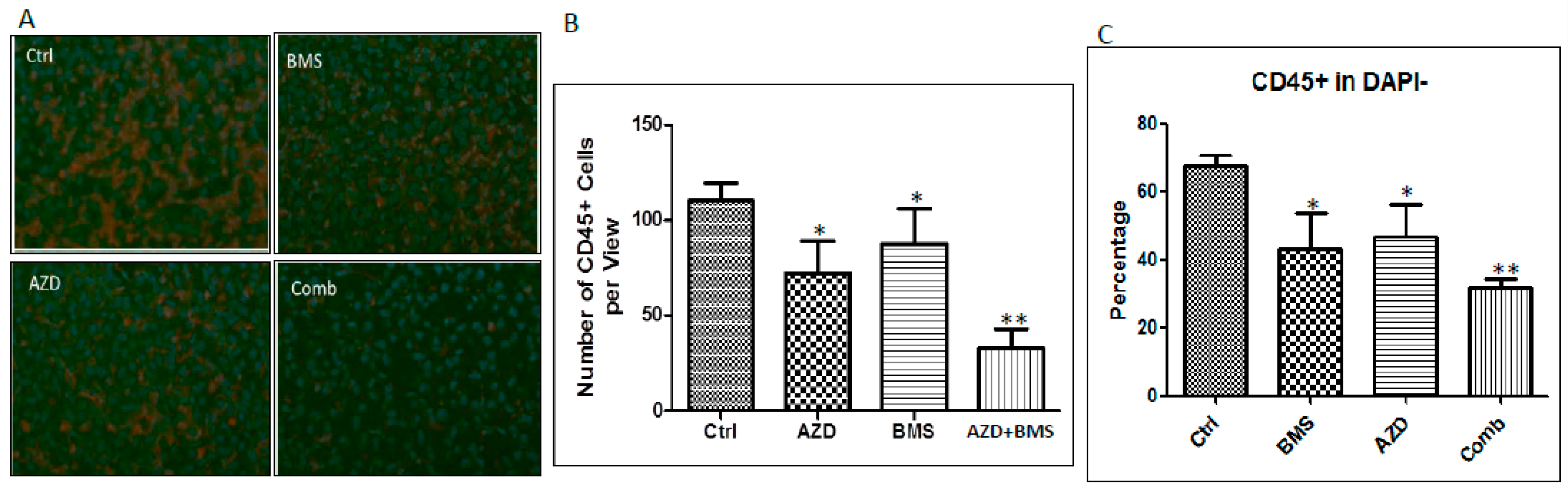
2.4. Effects of MEK Inhibitor AZD6244 and Smoothened Inhibitor BMS833923 on Cell Population at the Metastatic Niche
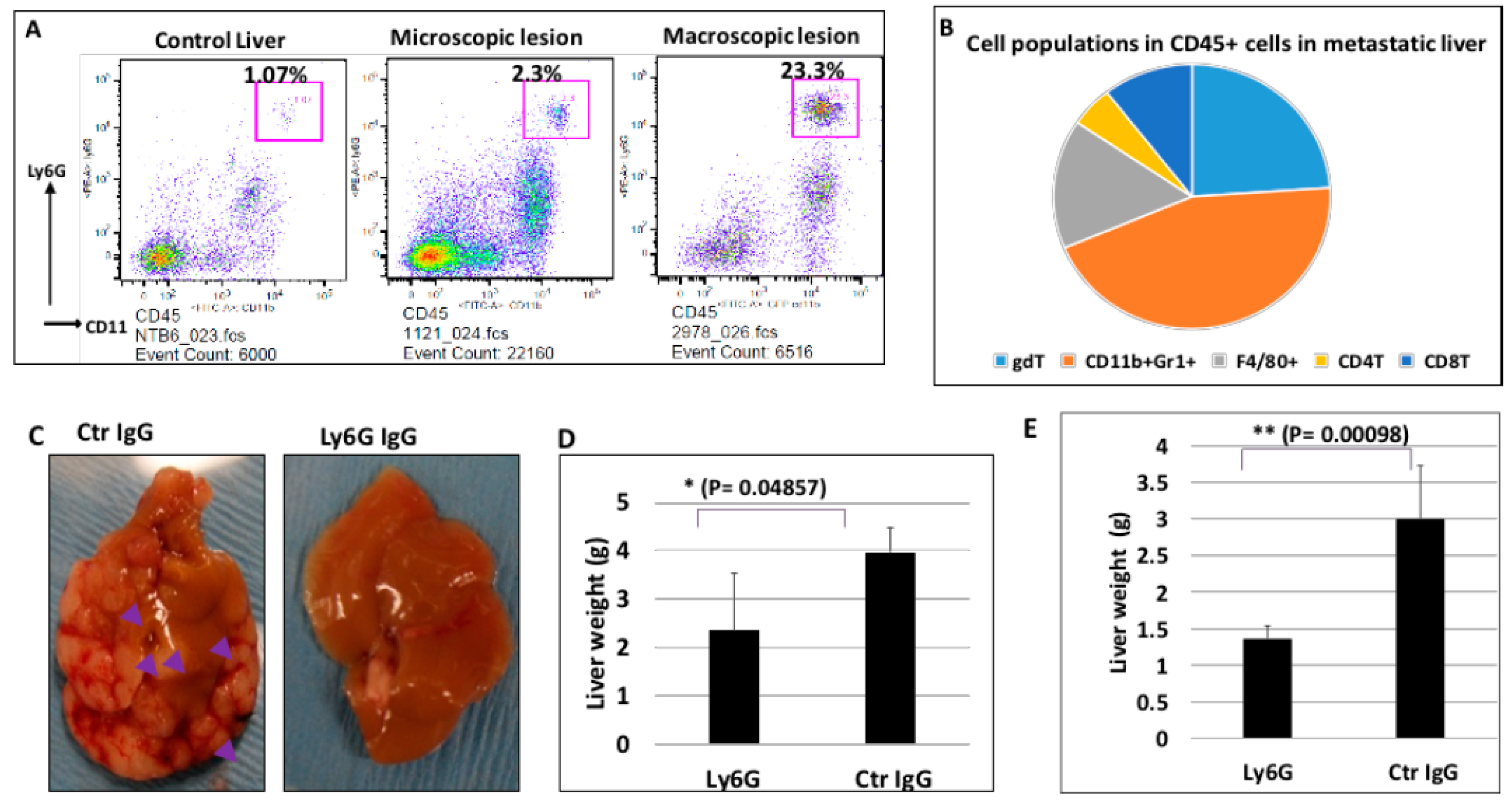
2.5. The Significance of Ly6G+CD11b+ Cells for Liver Metastasis of Pancreatic Cancer
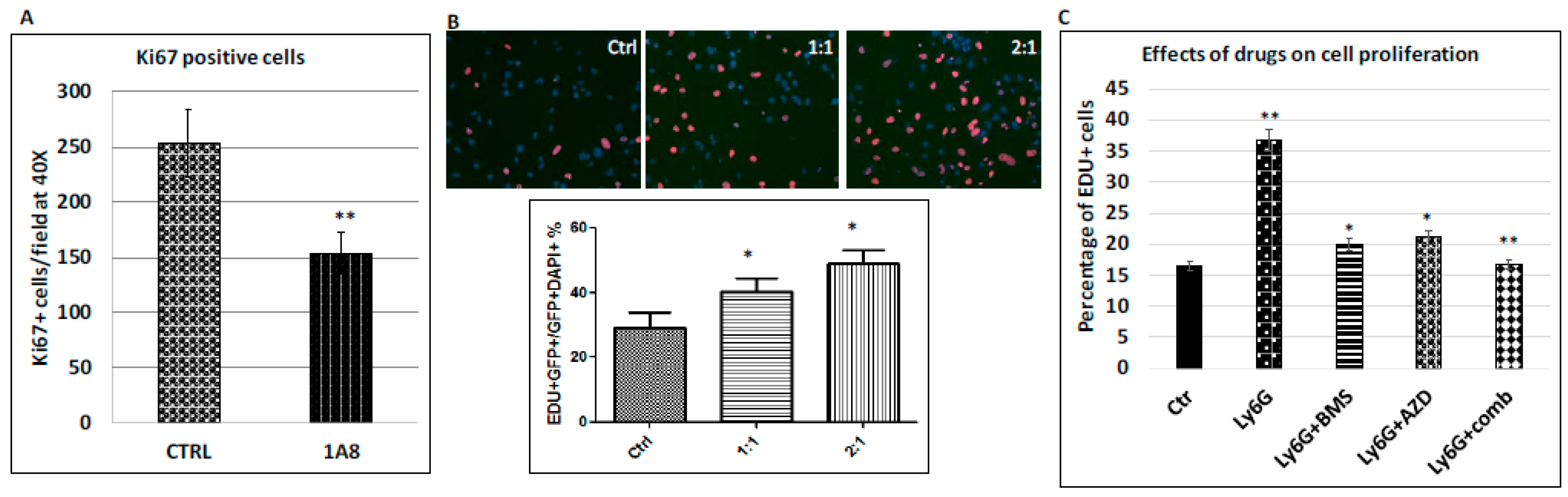
2.6. Stimulation of Cancer Cell Proliferation by Ly6G+CD11b+ Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines
4.3. Animals
4.4. Orthotopic Mouse Model of Pancreatic Cancer Metastasis
4.5. Depletion of Ly6G+ Cells
4.6. Quantification of Metastatic Burden
4.7. Single Cell Isolation
4.8. Immunohistochemistry (IHC) and Immunofluorescence (IF)
4.9. Flow Cytometry
4.10. Cell Proliferation Assay
4.11. RNA Extraction, RT-PCR and Real-Time PCR
4.12. RNA-seq, Sequence Alignment, Differential Expression Analyses
4.13. Western Blot Analysis
4.14. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; De La Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- McCormick, F. K-Ras protein as a drug target. J. Mol. Med. 2016, 94, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Z.; Li, Z.; Wang, X.; Kang, Y.; Yuan, Y.; Niu, J.; Wang, H.; Chatterjee, D.; Fleming, J.B.; Li, M.; et al. Deciphering the mechanisms of tumorigenesis in human pancreatic ductal epithelial cells. Clin. Cancer Res. 2013, 19, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Sahin, F.; Iacobuzio-Donahue, C.A.; Garcia-Carracedo, D.; Wang, W.M.; Kuo, C.Y.; Chen, D.; Arking, D.E.; Lowy, A.M.; Hruban, R.H.; et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2011, 2, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.; Liu, H.; Su, G.H.; Zhang, X.; Chin-Sinex, H.; Hanenberg, H.; Mendonca, M.S.; Shannon, H.E.; Chiorean, E.G.; Xie, J. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Mol. Cancer Ther. 2013, 12, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Abbruzzese, J.L.; Huang, S.; Fidler, I.J.; Xiong, Q.; Xie, K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin. Cancer Res. 1999, 5, 3711–3721. [Google Scholar] [PubMed]
- Bruns, C.J.; Shinohara, H.; Harbison, M.T.; Davis, D.W.; Nelkin, G.; Killion, J.J.; McConkey, D.J.; Dong, Z.; Fidler, I.J. Therapy of human pancreatic carcinoma implants by irinotecan and the oral immunomodulator JBT 3002 is associated with enhanced expression of inducible nitric oxide synthase in tumor-infiltrating macrophages. Cancer Res. 2000, 60, 2–7. [Google Scholar] [PubMed]
- Bruns, C.J.; Solorzano, C.C.; Harbison, M.T.; Ozawa, S.; Tsan, R.; Fan, D.; Abbruzzese, J.; Traxler, P.; Buchdunger, E.; Radinsky, R.; et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000, 60, 2926–2935. [Google Scholar] [PubMed]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsson, B.; Rushfeldt, C.; Seljelid, R.; Smedsrod, B. Inhibition of establishment and growth of mouse liver metastases after treatment with interferon gamma and beta-1,3-D-glucan. Hepatology 1998, 27, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Little, E.C.; Wang, C.; Watson, P.M.; Watson, D.K.; Cole, D.J.; Camp, E.R. Novel immunocompetent murine models representing advanced local and metastatic pancreatic cancer. J. Surg. Res. 2012, 176, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, H.; Visbal, A.P.; Obeid, N.F.; Ta, A.Q.; Faruki, A.A.; Wu, M.F.; Hilsenbeck, S.G.; Shaw, C.A.; Yu, P.; Plummer, N.W.; et al. An essential role for Galpha(i2) in Smoothened-stimulated epithelial cell proliferation in the mammary gland. Sci. Signal 2015, 8, ra92. [Google Scholar] [CrossRef] [PubMed]
- Bosco-Clement, G.; Zhang, F.; Chen, Z.; Zhou, H.M.; Li, H.; Mikami, I.; Hirata, T.; Yagui-Beltran, A.; Lui, N.; Do, H.T.; et al. Targeting Gli transcription activation by small molecule suppresses tumor growth. Oncogene 2014, 33, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Grausam, K.B.; Wang, J.; Lun, M.P.; Ohli, J.; Lidov, H.G.; Calicchio, M.L.; Zeng, E.; Salisbury, J.L.; Wechsler-Reya, R.J.; et al. Sonic Hedgehog promotes proliferation of Notch-dependent monociliated choroid plexus tumour cells. Nat. Cell Biol. 2016, 18, 418–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, N. Understanding synergy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E237–E253. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Chornoguz, O.; Ecker, C. Regulating the suppressors: Apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol. Immunother. 2012, 61, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peranzoni, E.; Zilio, S.; Marigo, I.; Dolcetti, L.; Zanovello, P.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 2010, 22, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.F. A niche role for cancer exosomes in metastasis. Nat. Cell Biol. 2015, 17, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, C.C.; Baker, C.H.; Tsan, R.; Traxler, P.; Cohen, P.; Buchdunger, E.; Killion, J.J.; Fidler, I.J. Optimization for the blockade of epidermal growth factor receptor signaling for therapy of human pancreatic carcinoma. Clin. Cancer Res. 2001, 7, 2563–2572. [Google Scholar] [PubMed]
- Nagrath, S.; Jack, R.M.; Sahai, V.; Simeone, D.M. Opportunities and Challenges for Pancreatic Circulating Tumor Cells. Gastroenterology 2016, 151, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Feigin, M.E.; Tuveson, D.A. Challenges and Opportunities in Modeling Pancreatic Cancer. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuveson, D.A.; Neoptolemos, J.P. Understanding metastasis in pancreatic cancer: A call for new clinical approaches. Cell 2012, 148, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Maitra, A. Pancreatic Cancer Genomics 2.0: Profiling Metastases. Cancer Cell 2017, 31, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Maitra, A.; Wang, H. The emerging roles of F-box proteins in pancreatic tumorigenesis. Semin. Cancer Biol. 2016, 36, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Serge, A.; Lavaut, M.N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, T.J.; Brown, K.M.; LaFleur, B.; Peterson, K.; Lawlor, C.; Chen, Y.; Packer, R.J.; Cogen, P.; Stephan, D.A. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat. Genet. 2001, 29, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Aszterbaum, M.; Zhang, X.; Bonifas, J.M.; Zachary, C.; Epstein, E.; McCormick, F. A role of PDGFRalpha in basal cell carcinoma proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 9255–9259. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Yokoi, K.; Bucana, C.D.; Tsan, R.; Killion, J.J.; Evans, D.B.; Fidler, I.J. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin. Cancer Res. 2003, 9, 6534–6544. [Google Scholar] [PubMed]
- Kitadai, Y.; Sasaki, T.; Kuwai, T.; Nakamura, T.; Bucana, C.D.; Hamilton, S.R.; Fidler, I.J. Expression of activated platelet-derived growth factor receptor in stromal cells of human colon carcinomas is associated with metastatic potential. Int. J. Cancer 2006, 119, 2567–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuge, R.; Kitadai, Y.; Shinagawa, K.; Onoyama, M.; Tanaka, S.; Yasui, W.; Chayama, K. mTOR and PDGF pathway blockade inhibits liver metastasis of colorectal cancer by modulating the tumor microenvironment. Am. J. Pathol. 2015, 185, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, K.P.; Park, S.J.; Park, J.H.; Jang, Y.S.; Choi, S.Y.; Jung, J.G.; Jo, K.; Park, D.Y.; Yoon, J.H.; et al. TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial-mesenchymal transition. Oncogene 2008, 27, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Gu, D.; Liu, H.; Yang, L.; Zhang, X.; Yoder, M.C.; Kaplan, M.H.; Xie, J. Defective TGF-beta signaling in bone marrow-derived cells prevents hedgehog-induced skin tumors. Cancer Res. 2014, 74, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Breese, M.R.; Liu, Y. NGSUtils: A software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 2013, 29, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human SHH | HS00179843 |
| Human IHH | HS01081800 |
| Mouse Gli1 | Mm00494654 |
| Mouse Ptch1 | Mm00436026 |
| Human GLI1 | Hs01110766 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, D.; Lin, H.; Zhang, X.; Fan, Q.; Chen, S.; Shahda, S.; Liu, Y.; Sun, J.; Xie, J. Simultaneous Inhibition of MEK and Hh Signaling Reduces Pancreatic Cancer Metastasis. Cancers 2018, 10, 403. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10110403
Gu D, Lin H, Zhang X, Fan Q, Chen S, Shahda S, Liu Y, Sun J, Xie J. Simultaneous Inhibition of MEK and Hh Signaling Reduces Pancreatic Cancer Metastasis. Cancers. 2018; 10(11):403. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10110403
Chicago/Turabian StyleGu, Dongsheng, Hai Lin, Xiaoli Zhang, Qipeng Fan, Shaoxiong Chen, Safi Shahda, Yunlong Liu, Jie Sun, and Jingwu Xie. 2018. "Simultaneous Inhibition of MEK and Hh Signaling Reduces Pancreatic Cancer Metastasis" Cancers 10, no. 11: 403. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10110403