Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum

1
Department of Pathology, Dalhousie University, Halifax, NS B3H 4G7, Canada
2
Department of Microbiology and Immunology, Dalhousie University, Halifax, NS B3H 4G7, Canada
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(4), 101; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040101
Submission received: 14 February 2018
/
Revised: 23 March 2018
/
Accepted: 27 March 2018
/
Published: 1 April 2018
(This article belongs to the Special Issue Epigenetic Influence on Cancer Metastasis and/or Treatment Resistance)
Abstract
:Aberrant epigenetic modifications are an early event in carcinogenesis, with the epigenetic landscape continuing to change during tumor progression and metastasis—these observations suggest that specific epigenetic modifications could be used as diagnostic and prognostic biomarkers for many cancer types. DNA methylation, post-translational histone modifications, and non-coding RNAs are all dysregulated in cancer and are detectable to various degrees in liquid biopsies such as sputum, urine, stool, and blood. Here, we will focus on the application of liquid biopsies, as opposed to tissue biopsies, because of their potential as non-invasive diagnostic tools and possible use in monitoring therapy response and progression to metastatic disease. This includes a discussion of septin-9 (SEPT9) DNA hypermethylation for detecting colorectal cancer, which is by far the most developed epigenetic biomarker assay. Despite their potential as prognostic and diagnostic biomarkers, technical issues such as inconsistent methodology between studies, overall low yield of epigenetic material in samples, and the need for improved histone and non-coding RNA purification methods are limiting the use of epigenetic biomarkers. Once these technical limitations are overcome, epigenetic biomarkers could be used to monitor cancer development, disease progression, therapeutic response, and recurrence across the entire cancer care continuum.
1. Introduction
Cancer has been referred to as “cellular chaos”. This is an appropriate description for a disease which is characterized by uncontrolled cell proliferation and avoiding the host’s strategies to eliminate aberrant cells. Part of the chaotic nature of cancer cells is that though all cancers share certain hallmark traits, the driving forces and resulting phenotype of the aberrant cells can vary greatly [1]. While the role of genetic mutations as drivers of carcinogenesis has been firmly established, epigenetic modifications have more recently been proposed as important drivers of cancer [2].
The term epigenotype was first coined by C.H. Waddington in 1942 to describe the heritable alterations in gene expression which affect phenotype and do not change the DNA sequence itself [3]. Epigenetic modifications are key regulators of gene expression and also contribute to genomic stability/chromatin structure. Regardless of whether aberrant epigenetic modifications are required for carcinogenesis, certain modifications are consistently dysregulated among cancers. This presents an opportunity to use these modifications as biomarkers for screening, detection, prediction of therapeutic response, and relapse surveillance.
1.1. DNA Methylation
The nucleotide alphabet has been expanded beyond ATGC with the discovery of modified bases, the best-characterized of which is 5-methylcytosine (5mC) [4]. In 5mC, a methyl moiety, donated by S-adenosylmethionine (SAM), is added to the 5′ position of a cytosine residue in CpG dinucleotides [5]. The maintenance and de novo generation of 5mC is mediated by DNA methyltransferases DNMT1/3A/3B. Genomic regions with high concentration of CpGs are known as CpG islands and seem to have an important role in gene expression regulation. Approximately 40–60% of human genes have CpG islands in the promoter region; and when these islands acquire 5mC, transcription of the gene is inhibited. Recently, 5mC-mediated transcriptional repression was also observed in genes without promoter CpG islands [6,7,8]. A prototypical cancer phenotype consists of a globally hypomethylated genome (which disrupts genomic stability) concurrent with promoter-specific hypermethylation (which silences tumor suppressor genes) [2,9,10].
Several methods exist for assessing DNA methylation at a global or CpG site-specific level. Global methylation levels can be determined via liquid chromatography–electrospray ionization–tandem mass spectrometry, luminometric methylation assay, or using methylation of repetitive sequences like long interspersed nuclear element (LINE) as a proxy of global methylation [11,12,13]. Site-specific methylation assays are more prevalent in clinical biomarker studies and can use either genome-wide discovery/screening approaches (e.g., Illumina beadchip, reduced representation or whole genome bisulfite sequencing, or methylated DNA immunoprecipitation sequencing), or can be used to investigate a single region (e.g., pyrosequencing or methylation-specific polymerase chain reaction (MS-PCR)) [14,15]. The most commonly used assays by far are MS-PCR and the Illumina HumanMethylation bead kits [16,17]. The Illumina kits are typically used for genome-wide searches for methylation biomarkers, while a clinical assay will likely resemble MS-PCR, which assays a single CpG island.
1.2. Post-Translational Histone Modifications
The human genome consists of approximately 3 billion base pairs and is able to fit within a cell due to the tightly regulated process of DNA compaction, the first stage of which is based around the nucleosome. The nucleosome is a core unit of chromatin consisting of an octamer of four histone proteins (H2A, H2B, H3, and H4) with approximately 147 bp of DNA wrapped twice around the complex [18]. An amino acid tail extends from each histone, and it is post-translational modifications to these tails which affect histone–DNA interactions and nuclear architecture [19]. Over 60 distinct histone modifications exist, though most cancer-related research focuses on acetylation (mediated by histone deacetylases (HDACs) and histone acetyltransferases (HATs)) and methylation (mediated by several protein lysine methyl-transferases like polycomb repression complex) [20].
The presence of these modifications forms a histone “code” that can affect transcriptional activity of the associated DNA sequence via directly impacting DNA wrapping or through recruiting enzyme complexes to wrap the DNA [21]. Histone modifications which generally indicate areas of active transcription include H3K4me (methylation of histone 3 at lysine position 4), H3K36me, H3KAc, H4K16Ac, and H3KAc; while H3K27me and H3K20me are associated with gene repression [21,22,23,24]. Cancer cells display silencing histone modifications on tumor-suppressive genes and encourage active transcription histone modifications on oncogenes [25]. Global histone profiling revealed a general loss of H4K16Ac and H4K20me3 (trimethylation) in a variety of cancers [25,26]. As we improve our understanding of the role histone modifications play in cancer biology, histone-based biomarkers could potentially be used in the cancer care continuum [27].
Dysregulation of histone variants has also been observed in cancer. These proteins are functionally distinct from the canonical replication-coupled core histones and endow special properties to chromatin. For example, H2A.Bbd incorporation results in nucleosomes containing 118–130 bp; this less compact chromatin is potentially more transcriptionally active [28]. Histone variants can also be post-translationally modified; thus the many combinations of canonical and variant histones (with their associated post-translational modifications) form a “variant network” to epigenetically alter chromatin structure and transcription [29]. Expression or mutation of specific histone variants are prognostic biomarkers, as in the case of H2A.Z in melanoma and breast where overexpression confers a poor prognosis [30,31], macroH2A expression which is lost during anal carcinoma progression [32]; or mutant H3., which is a common driving event in pediatric brain tumors [33,34].
Methods for quantifying histone modifications fall into two broad categories: methods designed to elucidate histone modifications affecting specific nucleosomes/loci and methods to determine global levels of a histone modification in a sample. Chromatin immunoprecipitation (ChIP) using antibodies specific to the histone modification of interest followed by sequencing of the DNA associated with the isolated histone modification (ChIP-Seq) is the most commonly used approach to investigate specific histone–DNA interactions [21,35]. Global levels can be determined using relatively simple Western blot or enzyme-linked immunosorbent assay (ELISA)-based methods [36].
1.3. Non-Coding RNAs
With the discovery that only approximately 3% of transcribed RNAs were subsequently translated into proteins, there was a surge of interest in the role of the non-coding RNA transcriptome [37]. There are several types of non-coding RNAs, such as small nucleolar RNAs (snoRNAs), and short interfering RNAs (siRNAs); but microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have been the most extensively characterized in cancer [38,39]. As their names suggest, miRNAs are small (18–20 nucleotides) while long non-coding RNAs are significantly longer (200–100,000 nucleotides). While these RNA species are divergent in their size and how they are post-transcriptionally processed, they share a common feature: a single miRNA or lncRNA is able to affect multiple genes/proteins [40,41]. Thus, deregulation of a single miRNA or lncRNA can influence many pathways and alter downstream processes such as apoptosis, proliferation, differentiation, etc. and act as either oncogene or tumor suppressor [42]. Many studies have described differential miRNA/lncRNA expression profiles between normal and cancerous human cells [43].
MicroRNAs repress protein production by binding to the 3′ untranslated region (3′-UTR) of their target messenger RNA (mRNA); this miRNA–mRNA duplex is both actively degraded and also prevents translation initiation [41]. While miRNAs are canonically repressive, lncRNA functions are more diverse. There are four archetypes of lncRNA function (decoy, activator, guide, or scaffold) which help dictate the interactions between transcription factors or chromatin modifier complexes; ultimately changing either the transcription of an mRNA or participating in post-transcriptional regulation of mRNA maturation processes [40].
Techniques that were originally developed to quantify mRNA have been adapted for quantification of miRNAs and lncRNAs. For detecting a broad range of non-coding RNAs, RNA sequencing (RNA-Seq) or microarrays are effective; however care should be taken with sample preparation and data analysis to ensure an accurate depiction of miRNAs/lncRNAs [44,45,46,47]. Secondary validation is necessary, often using real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) techniques to detect single non-coding RNAs of interest [48].
2. Risk Factors and Screening Strategies
Many factors influence cancer etiology including age, body mass index, physical activity, alcohol intake, smoking, environmental exposures, and family history [49,50,51,52,53]. Identifying and efficiently screening individuals who have the highest risk of developing cancer should improve the rate of early-stage diagnosis and translate to a direct survival benefit for the patient. Such a risk-stratified screening strategy could include epigenetic considerations as it is possible to identify and screen high-risk individuals with a pre-cancerous epigenotype. The success of these screening programs is dependent on tests that are: sensitive, low-cost, and relatively non-invasive. Here, minimally-invasive tests for epigenetic markers that could be used to screen high-risk individuals will be described.
2.1. Breast Cancer
In addition to regular mammograms, in several clinical experiments women at high-risk for developing breast cancer underwent ductal lavage, periareolar fine-needle aspiration, or a blood test. These high-risk women could be BRCA1/2 mutation carriers, they could have a family history of the disease, or they may have had a previously identified hyperplasia or ductal carcinoma in situ [54,55].
When ductal lavage was performed on asymptomatic BRCA1/2 mutation carriers, the cells within the lavage fluid of 8/19 women showed promoter hypermethylation of at least one of four genes (BRCA1, BRCA2, ERα, and RARβ2) that are often hypermethylated in breast cancer [56]. While none of the women subsequently developed breast cancer at the time of publication, 2/8 women with hypermethylation markers also presented ductal cell atypia in the lavage. In periareolar fine-needle aspirations from 86 high-risk women, there was actually no association between hypermethylation of three commonly hypermethylated breast cancer genes (p16INK4a/ARF, BRCA1, or BRCA2) and cellular atypia [57]. Strikingly, however, p16INK4a hypermethylation was significantly associated with hypermethylation of BRCA1, BRCA2, ERα, and RARβ2 which is a candidate pre-cancerous hypermethylation profile. This suggests that promoter hypermethylation of candidate genes may be detectable before morphological changes and could be used to identify women who should be closely monitored for breast cancer.
Blood samples are relatively easy to obtain, and while there are no approved serum- or blood-based markers for breast cancer screening, this is still an active area of research [58]. In a genome-wide methylation assessment of blood samples from high-risk women, 250 CpG sites were hypomethylated in individuals who went on to develop breast cancer [59]. Such a DNA methylation risk score could be used to intensify monitoring of certain women, once validated in independent cohorts. One study did use two independent cohorts to generate such a DNA methylation risk score for BRCA1/2 mutation carriers. This signature was composed of 1722 CpG sites, and was relatively successful at predicting a breast cancer diagnosis within 12 years of the blood draw [60]. An early attempt to create a similar risk score with a serum-based model using 20 miRNAs was able to identify 31.7% of the women who developed breast cancer within 18 months of the blood draw [61].
2.2. Nasopharyngeal Carcinoma
Nasal endoscopy is the gold standard for detecting nasopharyngeal carcinoma; however this invasive procedure is not practical for population-level screening [62]. Nasopharyngeal swab collection is a low-cost and minimally-invasive alternative that has been used to screen high-risk individuals (family member of nasopharyngeal carcinoma patient) in a study by Yang et al. [63]. Promisingly, 66/96 patient swabs but 0/43 healthy family member swabs showed promoter hypermethylation in Ras association domain-containing protein 1 (RASSF1). This illustrates a low-cost way to monitor high-risk family members for nasopharyngeal carcinoma [64].
2.3. Lung Cancer
The current screening protocols for individuals at high-risk of developing lung cancer are based around serial computer tomography (CT) scans. Additional screening approaches are being investigated to supplement the use of CT imaging such as sputum analysis for DNA or atypical cells [65]. In a high-risk population with a cigarette smoking history of >30 pack years, hypermethylation of ≥3 genes (within a panel of p16, MGMT, DAPK, RASSF1A, PAX5β, and GATA5) in a sputum sample was associated with a 6.5-fold increase in the risk of developing lung cancer [66]. This suggests that further stratifying the high-risk smoking population could identify an extremely vulnerable population that would require intense monitoring.
3. Diagnostic Biomarkers
3.1. Need for Liquid Biopsies
Surgical resection or biopsy specimens are the gold standard for definitive cancer diagnosis, and this is unlikely to change in the near future. For example, core biopsy has a 93% sensitivity in the detection of breast cancer, and these samples can be used for subsequent immunohistochemical assessment to inform prognosis and therapy [67]. However, tissue biopsies are not always available; either due to anatomical location of the tumor making it difficult to safely biopsy, or because the risk of post-biopsy infection is unacceptable [68,69]. Therefore, liquid biopsies of blood, urine, sputum, or other bodily fluids are used as a less invasive way to detect biomarkers (Figure 1).
An ideal biomarker for cancer detection should be easily and inexpensively measurable and should identify early-stage disease. It is essential that detection methods are sensitive to early-stage disease; as it is no coincidence that cancer types typically diagnosed at later stages (e.g., glioblastoma, pancreatic) also have the worst prognoses [70,71,72,73]. For epigenetic biomarkers to be suitable in liquid biopsy cancer detection, epigenetic dysregulation must occur early in carcinogenesis and the fluid epigenome should be reflective of the tumor epigenome [74].
3.2. Detecting Circulating Nucleotides and Nucleosomes
The presence of elevated cell-free DNA in serum of cancer patients was first observed 40 years ago [75], and is thought to be the result of ongoing release from apoptotic or necrotic cancer cells in the primary tumor [76]. Circulating free DNA (circ-DNA) in plasma, urine, and other bodily fluids is typically at a very low concentration and these double-stranded fragments are often of the length associated with nucleosomes (approximately 147 bp ± 20 bp linker DNA) with a portion of circ-DNA still complexed with histones [27,77,78].
Circulating miRNAs can be released passively by dying tissue or can be actively exported by cells. These miRNAs are remarkably stable in blood and seem to be protected from RNAses that should destroy free RNA [79,80,81,82]. This stability is because most miRNAs (approximately 90%) are bound to proteins such as Argonaute2, while others are encapsulated in vesicles such as exosomes while in circulation [83]. Circulating lncRNAs also seem to be detectable and stable in bodily fluids; however the mechanism of secretion and stability in the bloodstream is poorly understood and some studies could not reliably detect circulating lncRNAs [84].
Circulating histones are typically isolated with their associated DNA as a nucleosome. In cancer, circulating levels of nucleosomes are generally elevated, but the difference between healthy controls and patient nucleosome levels is not usually significant [85]. Determining the origins of circulating histones can be a difficult process. Only recently have methods emerged that can identify a nucleosome’s tissue-of-origin using “nucleosome footprinting” [86].
3.3. Colorectal Cancer
It was initially thought that the liquid biopsy epigenome would be an accurate proxy for the tumor epigenome; however concordance of tumor vs. serum markers is not observed in many studies [87,88]. Colorectal cancer has the highest degree of concordance between tumor and serum markers which has allowed for the rapid development of liquid biopsies to detect this cancer type [89]. Additionally, colorectal cancer has several qualities that make it a good model to develop novel diagnostic markers: there is a predictable pre-cancerous phase, there is low-compliance with current screening/detection programs, and current non-invasive markers are relatively insensitive [90].
There are five commercially available DNA methylation-based diagnostic tests for colorectal cancer: 3/5 of the commercial tests (Epi proColon, ColoVantage, and RealTime mS9) are designed to detect hypermethylated SEPT9 in a blood sample. ColoSure detects hypermethylated vimentin in a stool sample, and Cologuard detects (among other markers) hypermethylated NDRG4 and BMP3 in stool [91,92,93,94,95,96,97,98]. While other biomarkers are being investigated in clinical trials, Epi proColon is the most clinically validated diagnostic epigenetic biomarker (Table 1). In a recent meta-analysis, serological SEPT9 methylation was the superior methylation-based marker for colorectal cancer [99]. Tests like Epi proColon show great clinical potential because patients already prefer these non-invasive tests over colonoscopy/sigmoidoscopy for screening, and SEPT9 methylation is superior at detecting early stage disease over other non-invasive tests like fecal occult blood testing [100,101,102]. Combinations of other hypermethylated genes as stool or blood biomarkers have not displayed the high sensitivity observed in the SEPT9 methylation assays [89,103].
Other epigenetic biomarkers that could complement the methylation-based tests are being investigated. For example, serum levels of miR-21 were higher in colorectal cancer patients vs. individuals with benign polyps; however, it is not clear if this would detect early stage disease [104]. Due to limitations in histone isolation techniques, evidence for histone biomarkers lags behind DNA/RNA-based markers. Initial studies determined that H3K9me3 and H4K20me3 levels were significantly reduced in the plasma of colorectal cancer patients as compared to healthy controls [105,106]; but subsequent studies failed to confirm that H3K9me3 levels were different between patients and controls. Alternative histone marks, H3K27me3 and H4K20me3, only distinguished 49.2% of colorectal cancer patients [107].
3.4. Circulating Gene-Specific Promoter Hypermethylation
Unlike colorectal cancer, most other tumor types have low concordance between the methylation profile seen in tumor tissue vs. that observed in blood samples—this has limited the development of sensitive DNA methylation biomarkers. For example, several attempts have been made to use genes that are commonly hypermethylated in breast tumors (GSTP1, RASSF1A, RARβ, APC, and DAPK) to generate a blood-based breast cancer methylation signature [108]. Though at least one of these genes is methylated in 57–100% of tumors, methylation is detected in only 20–76% of patient serum samples [109,110,111]. One study used a different hypermethylated gene panel (RASSF1A, ESR1, CDH1, TIMP3, SYK) in the hopes of finding more concordant data; however, only 17% of patients in that study had simultaneous methylation of a gene in the tumor and in the blood [112].
Regardless of concordance with tumor tissue, serum-based multigene panels are more robust than single gene DNA methylation biomarkers. For instance, an eight gene promoter hypermethylation panel (APC, BRCA1, BIN1, CST6, GSTP1, P16, P21, and TIMP3) had a reported sensitivity of 90% for identifying breast cancer patients [74,113]. A similar approach using 17 gene promoters achieved a sensitivity of 91.2% and 90.8% specificity in distinguishing between pancreatic cancer patients and those with chronic pancreatitis [114,115].
Other than blood, several other fluids are candidates for liquid biopsy including: nasopharyngeal brushings, oral rinses, urine, vaginal fluid, and sputum. Nasopharyngeal brushings as well as oral rinses are a source of tumor-associated promoter hypermethylation to detect oral, pharyngeal, and nasopharyngeal carcinomas. DNA methylation markers (RASSF1A, WIF1, DAPK1 and/or RARβ2 + 20 additional CpGs) in these samples were predictive of early stage carcinoma when compared to healthy controls [63,116]. Urine could be a source of tumor DNA for detecting prostate and bladder cancer, though hypermethylation of glutathione S-transferase P1 (GSTP1) or other gene panels (e.g., RASSF1A, APC, p14) do not have sufficient sensitivity in detecting early stage disease [117,118]. Vaginal fluid could be used to distinguish endometrial or cervical cancer patients from healthy controls using a DNA methylation signature composed of 500 CpGs, which seemed to reflect methylation patterns seen in tumor biopsies [119]. Finally, sputum or bronchial lavage fluid can be used to aid in the diagnosis of lung cancer. Commercially available Epi proLung detects promoter methylation of short stature homeobox 2 (SHOX2) in bronchial lavage of lung cancer patients with a sensitivity of 78% and specificity of 96% [120].
3.5. Circulating Non-Coding RNAs
Similar to DNA hypermethylation markers, circulating non-coding RNA diagnostic markers are more robust when multiple non-coding RNAs are used, though some studies still utilize single markers. For example, elevated levels of miR-378 in serum can distinguish gastric cancer patients from healthy controls, while increased miR-141 identified prostate cancer [48,121]. Pancreatic cancer has been the focus of many miRNA biomarker studies, with more than 30 candidate miRNA blood biomarkers. Elevated levels of a single miRNA (miR-223) in plasma could distinguish patients with malignant intraductal papillary mucinous neoplasm (IPMN) from those with benign IPMN [122]. However, most of these potential miRNA markers are used within a panel, such as the combination of eight miRNAs (miR-6075, miR-4294, miR-6880-5p, miR-6799-5p, miR-125a-3p, miR-4530, miR-6836-3p, and miR-4476) which could achieve 80% sensitivity and 97.6% specificity in detecting pancreatic cancer [123,124]. Though detection of circulating lncRNAs is fraught with difficulties, plasma levels of lncRNA fragments HOXA distal transcript antisense RNA (HOTTIP-005) and RP11-567G11.1 were stably and significantly elevated in pancreatic cancer patients compared to healthy controls [125]. In gastric cancer patients, plasma levels of urothelial cancer associated 1 (UCA1/CUDR), long stress-induced non-coding transcript 5 (LSINCT-5), phosphatase and tensin homolog pseudogene 1 (PTENP1), and cytoskeleton regulator RNA (CYTOR/LINC00152) were higher than in healthy individuals [126,127]. However, it is unclear if circulating lncRNAs will ever be suitable blood-based biomarkers.
3.6. Circulating Histone Modifications
Most studies have focused on identifying cancer-specific histone modifications using tumor tissue—due to the difficulty of isolating circulating histones. Unfortunately, this means that most data is going to be extrapolated from tumor biopsy to liquid biopsy without a thorough understanding of how concordant these samples are for histone modifications. For example, low H3K16Ac and H3K12Ac have been implicated as an early event in breast cancer progression, and proposed as circulating biomarkers [128,129]. Caution should be used when assuming tumor biopsy histone modifications are viable circulating biomarkers.
Initial studies have focused on identifying histone modifications of circulating nucleosome isolated from cancer patients; though these modifications may not be effective at detecting early stage disease. For example, H3K9me3 and H4K20me3 have been detected on the circulating nucleosomes of multiple myeloma patients, but levels of these modifications were not compared the healthy controls and were not associated with disease stage [130]. In another study, the ratio of H3K9me3/nucleosome and H4K20me3/nucleosome was found to be higher in serum of breast and colorectal cancer patients compared to healthy individuals [131]. Similarly, an elevated ratio of H2AK119Ub/nucleosome H3K4me2/nucleosome was observed in pancreatic cancer patients [132].
Most histone variants have not been assessed as circulating biomarkers, so their utility is largely unknown; however, H2AZ has been assessed in a couple of studies. In the sera of colonoscopy patients, circulating levels of H2AZ did not distinguish between healthy controls and those diagnosed with colorectal cancer [107]. Circulating H2AZ levels (as part of an ELISA-based NuQ® 5 nucleosome marker signature) were elevated in early stage pancreatic cancer patients [132].
Circulating histones and their cancer-specific modifications may have potential as biomarkers; however an association with early stage disease has not been established and the assays are more difficult and expensive than those used in isolating circulating nucleic acids.
4. Epigenetic Markers Informing Therapeutic Decision-Making
Epigenetic biomarkers may impact therapeutic decision-making through (Section 4.1) stratifying high-risk patients for intense treatment (Section 4.2) or predicting response to a specific treatment (Section 4.3).
4.1. Epigenetic Prognostic Markers
Upon diagnosis, some cancer types are further classified based on immunohistochemical staining or other molecular markers. The resulting subtypes can be clinically relevant as in the case of breast cancer where the presence of estrogen receptor (ER), progesterone receptor (PR), and epidermal growth factor receptor (HER2) within the tumor directly impacts prognosis and treatment strategy [133]. Epigenetic profiling can complement these pre-existing prognostic biomarkers and further define the current subtypes; or there may be epigenetic markers that have prognostic relevance independent of our current subtyping strategies.
Several epigenetic markers correlate with known prognostic factors in breast cancer. For example, high expression of lysine-specific histone demethylase 1, low levels of H3K9ac, H3K18ac, H4K12ac, H3K4me2, H4K20me2 and H4K3me2, and elevated amounts of circulating miR-21 and miR-10b are associated with the aggressive ER-subtype [83,128,134]. Epigenome-wide profiling studies of DNA methylation, histone modifications, miRNA expression, and lncRNA expression have generally found that unsupervised clustering of patients based on differential epigenome markers recapitulates the existing breast cancer subtypes [135,136].
Perhaps the best characterized epigenetic subtype is the CpG island methylator phenotype (CIMP) which was first identified in colorectal cancer [137]. CIMP+ tumors are identified based on promoter CpG island methylation of CDKN2A and MLH1 genes, and these tumors tend to have a unique molecular and clinicopathological profile compared to CIMP lesions. While it is assumed that CIMP+ are generally more aggressive, some studies showed that once other important clinical factors were controlled for, CIMP+ status was not an independent predictor of poor prognosis [138].
There are plenty of other epigenetic markers that seem to be effective prognostic indicators such as high expression of lncRNAs HOTTIP-005 or RP11-567G11.1 and high serum miR196a predicting poor prognosis for pancreatic cancer [125,139]. While it is useful to define patients with poor prognoses and should encourage novel therapies for these underserved groups; here, the focus will be on describing epigenetic modifications that can more directly inform therapeutic strategies.
4.2. Anticipating the Benefit of Adjuvant Therapy
Epigenetic biomarkers could identify patients who would derive value from more intense or adjuvant therapies. In ER-/PR-/HER2- breast cancer patients, high miR-200b-3p and miR-190a along with low miR-512-5p levels in core biopsy was predictive of better response to neoadjuvant chemotherapy [140]. Similarly, in colorectal tumor samples, high expression of miR-16, miR-590-5p, and miR-153 predicted a complete response to neoadjuvant chemotherapy [141].
Retrospective analysis of patient cohorts which did not receive any adjuvant therapies can reveal patient subpopulations which would have benefitted from additional treatment. For example oral squamous cell carcinoma patients with a (AJAP1, SHANK2, FOXA2, MT1A, ZNF570, HOXC4, and HOXB4) methylation-based signature or stage I lung cancer patients with elevated H3K4me2 levels could have benefited from adjuvant chemotherapy [142,143]. Promoter methylation of TNFRSF25, EDNRB, RASSF1A, and APC could even be used to anticipate which bladder cancer patients require routine surveillance versus immediate radical treatment [144,145,146].
4.3. Epigenetic Biomarkers that Predict Response to Specific Therapies
Breast tumors that are immunohistochemically positive for ER expression are typically treated with the ER antagonist tamoxifen. It is unclear how methylation of the estrogen receptor 1 gene (ESR1) is connected with silencing of ER expression or response to tamoxifen. Unexpectedly, promoter hypermethylation of the ER gene is not generally predictive of decreased ER protein levels [147]; however, ESR1 methylation in circulating DNA actually does correlate with ER protein in the tumor [148]. It was hypothesized that ER silencing via ESR1 hypermethylation could indicate resistance to tamoxifen; but unexpectedly, ESR1 methylation was predictive of longer survival in tamoxifen-treated patients [149].
Cytotoxic chemotherapies like taxanes preferentially affect cancer cells based on their rapid proliferation and cell cycle checkpoint dysfunction. Checkpoint with forkhead and ring finger domains (CHFR) controls cell cycle progression at the G2/M checkpoint and can initiate mitotic arrest; thus downregulation of CHFR encourages uncontrolled cellular division [150,151]. Hypermethylation of CHFR is observed in breast, bladder, colorectal, gastric, nasopharyngeal, lung, esophageal, cervical, hepatocellular, oral squamous, head and neck, and endometrial cancer [152]; and is a potential marker of taxane sensitivity for many cancer types. In vitro evidence using nasopharyngeal carcinoma, colorectal cancer, esophageal, endometrial cancer, and gastric cancer cell lines suggests that CHFR hypermethylation contributes to docetaxel and paclitaxel sensitivity [153,154,155,156,157]. However, subsequent clinical studies have not shown a clear influence of CHFR methylation on taxane sensitivity. In advanced gastric cancer patients, CHFR methylation was not predictive of response to docetaxel or paclitaxel [158]. Clearly, randomized prospective clinical trials are necessary to confirm the clinical validity of CHFR methylation as a marker of taxane sensitivity [159].
Expression of the excision repair enzyme O-6-methylguanine–DNA methyltransferase (MGMT) has previously been associated with clinical resistance to alkylating agents [160]. This suggested that epigenetic silencing of MGMT may cause sensitivity to those same drugs [161]. This seems to generally be the case for glioblastoma patients as MGMT promoter hypermethylation is an independent favorable prognostic factor [162,163]. Patients with promoter methylation survived longer after temozolomide plus radiotherapy treatment compared to patients with unmethylated MGMT. Similarly, MGMT hypermethylation was observed in gliomas from patients who had survived longer after treatment with the alkylating agent carmustine [160]. However, not all trials have shown predictive value of MGMT promoter methylation [164].
5. Epigenetic Biomarkers to Monitor Patient Responses
Liquid biopsies could be used to monitor the real-time dynamics of a patient’s response to therapy or to monitor survivors for potential recurrence. Early response to therapy is often indicative of an overall better prognosis, so early response biomarkers would be a valuable prognostic tool and could also be used to make timely adjustments to treatment regimens [165]. For example, in patients with advanced lung cancer, a significant decrease in circulating nucleosome levels was indicative of a favorable response to the first chemotherapy cycle [166].
Epigenetic markers could be applied in two ways when monitoring for relapse: patients could be stratified based on risk of recurrence and routine monitoring could be intensified, or unique epigenetic markers could identify patients who are currently experiencing relapse. In urothelial carcinomas, hypermethylated insulin-like growth factor-binding protein 3 (IGFBP3) and apoptotic protease activating factor 1 (APAF-1) was indicative of a high risk of recurrence and identified patients who should be monitored closely; however, this was not a liquid biopsy and close monitoring could not be aided with epigenetic biomarkers [167]. Peritoneal fluid is relatively accessible and sentinel lymph nodes are occasionally dissected in gastrointestinal cancer patients, so several groups have used these samples to assess patients for recurrence. In the peritoneal fluid or sentinel lymph nodes of gastric cancer patients, hypermethylation of a 6-gene signature predicted an increased risk of recurrence or metastasis [168,169]. Blood samples are very accessible, and global hypermethylation was associated was increased relapse potential in acute lymphoblastic leukemia patients; however, global hypomethylation (LINE-1) was associated with relapse risk in oropharyngeal squamous cell carcinoma [170,171]. There are several markers that show promise for identifying patients who are experiencing relapse, such as three miRNAs that were upregulated and 8 miRNAs that were downregulated in the serum of recurrent breast cancer patients [172].
6. Future Directions
The detection and assessment of circulating nucleic acids is still in the developmental stage and will require standardization of methodologies before true clinical utility is achieved. Conclusive evidence for candidate biomarkers is often missing because meta-analyses are limited by the wide range of methodologies used between research groups. For example, there is not even a consensus on whether serum or plasma is a more appropriate sample type for DNA or lncRNA biomarkers; and there no clear evidence for the significance (if any) of contaminating leukocyte DNA [173]. So far, several recommendations have been made: miRNA biomarkers should be assayed from serum samples [48,83,174], and DNA methylation biomarker panels should not explicitly include leukocyte differentially methylated regions [175,176].
Finally, even though DNA methylation biomarkers have advanced into the clinic (e.g., Epi proColon); unbiased approaches using newer techniques should identify additional DNA methylation biomarkers. Most candidate genes in methylation biomarker panels were chose a priori based on the gene product’s function, and while these candidate gene approaches have led to the development of successful biomarkers, care should be taken in their interpretation. Due to the volume of differentially methylated sites and lack of established p-value correction protocols, one study showed that 30–50% of randomly selected methylation sites could be significantly associated with clinical factors [177]. We should not only re-assess our current biomarker panels, but also use unbiased approaches with stringent statistics and large patient cohorts. Excitingly, DNA methylation biomarkers can finally be directly functionally validated with the creation of a clustered regularly interspaced short palindromic repeats (CRISPR) method which uses site-specific targeting with DNA methyltransferase 3 (DNMT3A) or methylcytosine dioxygenase 1 (TET1) to add/subtract a methyl group [178,179]. These novel techniques should finally resolve which methylation sites play a vital role in cancer progression and could reveal novel biomarkers.
Acknowledgments
Support was provided by grant funding to P.M. from the Cancer Research Society in partnership with the Institute of Cancer Research of the Canadian Institutes of Health Research (CIHR). M.L.T. is supported by CGS-D award from the CIHR, a Nova Scotia Health Research Foundation studentship, an NS graduate scholarship, and a Killam Laureate scholarship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. Epigenetics: A historical overview. Epigenetics 2006, 1, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.B.; Coghill, R.D. Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the Tubercle bacillus. J. Am. Chem. Soc. 1925, 47, 2838–2844. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands—‘A rough guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA hypomethylation contributes to genomic instability and intestinal cancer initiation. Cancer Prev. Res. 2016, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Estécio, M.R.H.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.P.J. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Johansson, S.; Ekström, T.J. Using LUMA: A luminometric-based assay for global DNA-methylation. Epigenetics 2006, 1, 45–48. [Google Scholar] [PubMed]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Rodriguez-Antolin, C.; Vallespín, E.; Carpeño, J.D.; de Caceres, I.I. The impact of next-generation sequencing on the DNA methylation–based translational cancer research. Transl. Res. 2016, 169, 1–18.e1. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Bednar, J.; Horowitz, R.A.; Grigoryev, S.A.; Carruthers, L.M.; Hansen, J.C.; Koster, A.J.; Woodcock, C.L. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA 1998, 95, 14173–14178. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R.; Chadee, D.N. Regulation and regulatory parameters of histone modifications. J. Cell. Biochem. Suppl. 1998, 30–31, 203–213. [Google Scholar] [CrossRef]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Horikoshi, N.; Kumar, P.; Sharma, G.G.; Chen, M.; Hunt, C.R.; Westover, K.; Chowdhury, S.; Pandita, T.K. Genome-wide distribution of histone H4 Lysine 16 acetylation sites and their relationship to gene expression. Genome Integr. 2013, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.C.A.; Eskeland, R.; Hekimoglu-Balkan, B.; Pradeepa, M.M.; Bickmore, W.A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013, 23, 2053–2065. [Google Scholar] [CrossRef] [PubMed]
- Kurdistani, S.K. Histone modifications in cancer biology and prognosis. Prog. Drug Res. 2011, 67, 91–106. [Google Scholar] [PubMed]
- Fraga, M. F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- McAnema, P.; Brown, J.A.L.; Kerin, M.J. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers (Basel) 2017, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Doyen, C.-M.; Montel, F.; Gautier, T.; Menoni, H.; Claudet, C.; Delacour-Larose, M.; Angelov, D.; Hamiche, A.; Bednar, J.; Faivre-Moskalenko, C.; et al. Dissection of the unusual structural and functional properties of the variant H2A.Bbd nucleosome. EMBO J. 2006, 25, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Kallen, C.B.; Dhar, R.; Baquero, M.T.; Mason, C.E.; Russell, B.A.; Shah, P.K.; Liu, J.; Khramtsov, A.; Tretiakova, M.S. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008, 4, 188. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-H.; Miyai, K.; Sporn, J.C.; Luo, L.; Wang, J.Y.J.; Cosman, B.; Ramamoorthy, S. Loss of histone variant macroH2A2 expression associates with progression of anal neoplasm. J. Clin. Pathol. 2016, 69, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Gessi, M.; Gielen, G.H.; Hammes, J.; Dörner, E.; Mühlen, A.Z.; Waha, A.; Pietsch, T. H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: Possible diagnostic and therapeutic implications? J. Neurooncol. 2013, 112, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Giardina, C.; Rasmussen, T.P. Quantitation of nucleosome acetylation and other histone posttranslational modifications using microscale NU-ELISA. Methods Mol. Biol. 2013, 981, 167–176. [Google Scholar] [PubMed]
- Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mattick, J.S. The eukaryotic genome as an RNA machine. Science 2008, 319, 1787–1789. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Calin, G.A. Circulating free xeno-microRNAs—The new kids on the block. Mol. Oncol. 2016, 10, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Gutschner, T. Long non-coding RNAs in cancer and development: Where do we go from here? Int. J. Mol. Sci. 2015, 16, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.Y.-Y.; Lin, C.H.; Choi, S.C.; Yip, T.T.C.; Ngan, R.K.-C.; Tsao, G.S.-W.; Lung, M.L. Integrated mRNA and microRNA transcriptome sequencing characterizes sequence variants and mRNA-microRNA regulatory network in nasopharyngeal carcinoma model systems. FEBS Open Bio 2014, 4, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xue, S.; Liu, X.; Liu, H.; Hu, T.; Qiu, X.; Zhang, J.; Lei, M. Analyses of long non-coding RNA and mRNA profiling using RNA sequencing during the pre-implantation phases in pig endometrium. Sci. Rep. 2016, 6, 20238. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Calin, G.A.; Volinia, S.; Croce, C.M. MicroRNA expression profiling using microarrays. Nat. Protoc. 2008, 3, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Shang, J. Long noncoding RNA expression profiling using Arraystar LncRNA microarrays. Methods Mol. Biol. 2016, 1402, 43–61. [Google Scholar] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D. Cancer incidence and mortality in relation to body mass index in the million women study: Cohort study. BMJ Br. Med. J. 2007, 335, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Physical activity, obesity, and risk for colon cancer and adenoma in men. Ann. Intern. Med. 1995, 122, 327. [Google Scholar] [CrossRef] [PubMed]
- Smith-Warner, S.A.; Spiegelman, D.; Yaun, S.-S.; van den Brandt, P.A.; Folsom, A.R.; Goldbohm, R.A.; Graham, S.; Holmberg, L.; Howe, G.R.; Marshall, J.R.; et al. Alcohol and breast cancer in women. JAMA 1998, 279, 535. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Flanagan, J.M. Epigenome-wide association studies for breast cancer risk and risk factors. Trends Cancer Res. 2017, 12, 19–28. [Google Scholar] [PubMed]
- Kuhl, C.K.; Schrading, S.; Leutner, C.C.; Morakkabati-Spitz, N.; Wardelmann, E.; Fimmers, R.; Kuhn, W.; Schild, H.H. Mammography, breast ultrasound, and magnetic resonance imaging for surveillance of women at high familial risk for breast cancer. J. Clin. Oncol. 2005, 23, 8469–8676. [Google Scholar] [CrossRef] [PubMed]
- Richert-Boe, K.E.; Humphrey, L.L. Screening for cancers of the cervix and breast. Arch. Intern. Med. 1992, 152, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Locke, I.; Kote-Jarai, Z.; Fackler, M.J.; Bancroft, E.; Osin, P.; Nerurkar, A.; Izatt, L.; Pichert, G.; Gui, G.P.; Eeles, R.A. Gene promoter hypermethylation in ductal lavage fluid from healthy BRCA gene mutation carriers and mutation-negative controls. Breast Cancer Res. 2007, 9, R20. [Google Scholar] [CrossRef] [PubMed]
- Bean, G.R.; Bryson, A.D.; Pilie, P.G.; Goldenberg, V.; Baker, J.C.; Ibarra, C.; Brander, D.M.U.; Paisie, C.; Case, N.R.; Gauthier, M.; et al. Morphologically normal-appearing mammary epithelial cells obtained from high-risk women exhibit methylation silencing of INK4a/ARF. Clin. Cancer Res. 2007, 13, 6834–6841. [Google Scholar] [CrossRef] [PubMed]
- Kazarian, A.; Blyuss, O.; Metodieva, G.; Gentry-Maharaj, A.; Ryan, A.; Kiseleva, E.M.; Prytomanova, O.M.; Jacobs, I.J.; Widschwendter, M.; Menon, U.; et al. Testing breast cancer serum biomarkers for early detection and prognosis in pre-diagnosis samples. Br. J. Cancer 2017, 116, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Bolick, S.C.E.; DeRoo, L.A.; Weinberg, C.R.; Sandler, D.P.; Taylor, J.A. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. JNCI J. Natl. Cancer Inst. 2013, 105, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Anjum, S.; Fourkala, E.-O.; Zikan, M.; Wong, A.; Gentry-Maharaj, A.; Jones, A.; Hardy, R.; Cibula, D.; Kuh, D.; Jacobs, I.J.; et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2004, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Taslim, C.; Weng, D.Y.; Brasky, T.M.; Dumitrescu, R.G.; Huang, K.; Kallakury, B.V.S.; Krishnan, S.; Llanos, A.A.; Marian, C.; McElroy, J.; et al. Discovery and replication of microRNAs for breast cancer risk using genome-wide profiling. Oncotarget 2016, 7, 86457–86468. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.K.; Lim, M.Y.; Kanagalingam, J. Screening in nasopharyngeal carcinoma: Current strategies and future directions. Curr. Otorhinolaryngol. Rep. 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Yang, X.; Dai, W.; Kwong, D.L.; Szeto, C.Y.Y.; Wong, E.H.; Ng, W.T.; Lee, A.W.M.; Ngan, R.K.C.; Yau, C.C.; Tung, S.Y.; et al. Epigenetic markers for noninvasive early detection of nasopharyngeal carcinoma by methylation-sensitive high resolution melting. Int. J. Cancer 2015, 136, E127–E135. [Google Scholar] [CrossRef] [PubMed]
- Hutajulu, S.H.; Indrasari, S.R.; Indrawati, L.P.; Harijadi, A.; Duin, S.; Haryana, S.M.; Steenbergen, R.D.; Greijer, A.E.; Middeldorp, J.M. Epigenetic markers for early detection of nasopharyngeal carcinoma in a high risk population. Mol. Cancer 2011, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Thunnissen, F.B.J.M. Sputum examination for early detection of lung cancer. J. Clin. Pathol. 2003, 56, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; Liechty, K.C.; Gentry, F.D.; Wolf, H.J.; Rogers, J.; Vu, K.; Haney, J.; Kennedy, T.C.; Hirsch, F.R.; Miller, Y.; et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006, 6, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, I.W.Y.; Shin, V.Y.; Kwong, A. Detection of methylated circulating DNA as noninvasive biomarkers for breast cancer diagnosis. J. Breast Cancer 2017, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Witham, T.F.; Flickinger, J.C.; Kondziolka, D.; Lunsford, L.D. Comprehensive assessment of hemorrhage risks and outcomes after stereotactic brain biopsy. J. Neurosurg. 2001, 94, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Bokhorst, L.P.; Lepistö, I.; Kakehi, Y.; Bangma, C.H.; Pickles, T.; Valdagni, R.; Alberts, A.R.; Semjonow, A.; Strölin, P.; Montesino, M.F.; et al. Complications after prostate biopsies in men on active surveillance and its effects on receiving further biopsies in the Prostate cancer research international: Active surveillance (PRIAS) study. BJU Int. 2016, 118, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. In Lung Cancer and Personalized Medicine. Advances in Experimental Medicine and Biology; Ahmad, A., Gadgeel, S., Eds.; Springer: Cham, Switzerland, 2016; Volume 893, pp. 1–19. [Google Scholar]
- Islami, F.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Ward, E.M.; Jemal, A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA. Cancer J. Clin. 2017, 67, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Barekati, Z.; Kohler, C.; Lv, Q.; Bürki, N.; Diesch, C.; Bitzer, J.; Zheng, H.; Schmid, S.; Zhong, X.Y. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS ONE 2011, 6, e16080. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Chan, A.K.C.; Chiu, R.W.K.; Lo, Y.M.D. Clinical sciences reviews committee of the association of clinical biochemists. Cell-free nucleic acids in plasma, serum and urine: A new tool in molecular diagnosis. Ann. Clin. Biochem. 2003, 40, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.D.; Chan, K.C.A.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.F.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.R.; et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Bonnefoi, H.; Pelte, M.F.; Lyautey, J.; Lederrey, C.; Movarekhi, S.; Schaeffer, P.; Mulcahy, H.E.; Meyer, P.; Stroun, M.; et al. Telomerase RNA as a detection marker in the serum of breast cancer patients. Clin. Cancer Res. 2001, 6, 3823–3826. [Google Scholar]
- Hasselmann, D.O.; Rappl, G.; Rössler, M.; Ugurel, S.; Tilgen, W.; Reinhold, U. Detection of tumor-associated circulating mRNA in serum, plasma and blood cells from patients with disseminated malignant melanoma. Oncol. Rep. 2001, 8, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Kopreski, M.S.; Benko, F.A.; Gocke, C.D. Circulating RNA as a tumor marker: Detection of 5T4 mRNA in breast and lung cancer patient serum. Ann. N. Y. Acad. Sci. 2001, 945, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kopreski, M.S.; Benko, F.A.; Kwak, L.W.; Gocke, C.D. Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clin. Cancer Res. 1999, 5, 1961–1965. [Google Scholar] [PubMed]
- Casey, M.C.; Sweeney, K.J.; Brown, J.A.L.; Kerin, M.J. Exploring circulating micro-RNA in the neoadjuvant treatment of breast cancer. Int. J. Cancer 2016, 139, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Schlosser, K.; Hanson, J.; Villeneuve, P.J.; Dimitroulakos, J.; McIntyre, L.; Pilote, L.; Stewart, D.J. Assessment of circulating LncRNAs under physiologic and pathologic conditions in humans reveals potential limitations as biomarkers. Sci. Rep. 2016, 6, 36596. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Khade, B.; Pandya, R.; Gupta, S. A novel method for isolation of histones from serum and its implications in therapeutics and prognosis of solid tumours. Clin. Epigenetics 2017, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.K.; Shim, Y.R.; Choi, J.H.; Kim, M.J.; Gabrielson, E.; Lee, S.J.; Hwang, T.Y.; Shin, S.O. Gene promoter hypermethylation in tumors and plasma of breast cancer patients. Cancer Res. Treat. 2005, 37, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-L.; Leung, W.K.; Chan, M.W.Y.; Ng, E.K.W.; Tong, J.H.M.; Lo, K.-W.; Chung, S.C.S.; Sung, J.J.Y.; To, K.-F. Detection of gene promoter hypermethylation in the tumor and serum of patients with gastric carcinoma. Clin. Cancer Res. 2002, 8, 1761–1766. [Google Scholar] [PubMed]
- Salehi, R.; Salehi, A.; Emami, M.; Mohammadi, M. Methylation pattern of SFRP1 promoter in stool sample is a potential marker for early detection of colorectal cancer. Adv. Biomed. Res. 2012, 1, 87. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.; Park, S.; Jeon, H.; Lee, W.; Kim, J.K.; Cho, M.; Kim, M.; Lim, J.; Kang, C.S.; et al. Gastrointestinal tract cancer screening using fecal carcinoembryonic antigen. Ann. Clin. Lab. Sci. 2003, 33, 32–38. [Google Scholar] [PubMed]
- Ned, R.M.; Melillo, S.; Marrone, M. Fecal DNA testing for colorectal cancer screening: The ColoSureTM test. PLoS Curr. 2011, 3, RRN1220. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Lee, Y.M.; Kim, J.S.; Kim, H.S.; Lee, K.H.; Juhng, S.W.; Lee, J.H. Aberrant promoter methylation of the vimentin gene in colorectal cancer associated with the adenoma-carcinoma sequence. Korean J. Pathol. 2010, 44, 179–186. [Google Scholar] [CrossRef]
- Shirahata, A.; Sakata, M.; Sakuraba, K.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y.; et al. Vimentin methylation as a marker for advanced colorectal carcinoma. Anticancer Res. 2009, 29, 279–281. [Google Scholar] [PubMed]
- Zou, H.; Harrington, J.J.; Shire, A.M.; Rego, R.L.; Wang, L.; Campbell, M.E.; Oberg, A.L.; Ahlquist, D.A. Highly methylated genes in colorectal neoplasia: Implications for screening. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-D.; Han, Z.J.; Skoletsky, J.; Olson, J.; Sah, J.; Myeroff, L.; Platzer, P.; Lu, S.; Dawson, D.; Willis, J.; et al. Detection in fecal DNA of colon cancer—Specific methylation of the nonexpressed vimentin gene. JNCI J. Natl. Cancer Inst. 2005, 97, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- YLamb, N.; Dhillon, S. Epi proColon® 2.0 CE: A blood-based screening test for colorectal cancer. Mol. Diagn. Ther. 2017, 21, 225–232. [Google Scholar]
- Journal of the American Medical Association (JAMA). A stool DNA test (Cologuard) for colorectal cancer screening. JAMA 2014, 312, 2566. [Google Scholar]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Levin, T.R.; Lavin, P.; Lidgard, G.P.; Ahlquist, D.A.; Berger, B.M. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 370, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gan, A.; Chen, X.; Wang, X.; He, W.; Zhang, X.; Huang, R.; Zhou, S.; Song, X.; Xu, A. Diagnostic performance of DNA hypermethylation markers in peripheral blood for the detection of colorectal cancer: A meta-analysis and systematic review. PLoS ONE 2016, 11, e0155095. [Google Scholar] [CrossRef] [PubMed]
- Schroy, P.C.; Heeren, T.C. Patient perceptions of stool-based DNA testing for colorectal cancer screening. Am. J. Prev. Med. 2005, 28, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Turnbull, B.A.; Ross, M.E.; Colorectal cancer study group. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population. N. Engl. J. Med. 2004, 351, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Molnár, B.; Tóth, K.; Barták, B.K.; Tulassay, Z. Plasma methylated septin 9: A colorectal cancer screening marker. Expert Rev. Mol. Diagn. 2015, 15, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Salehi, R.; Atapour, N.; Vatandoust, N.; Farahani, N.; Ahangari, F.; Salehi, A. Methylation pattern of ALX4 gene promoter as a potential biomarker for blood-based early detection of colorectal cancer. Adv. Biomed. Res. 2015, 4, 252. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Takahashi, M.; Hur, K.; Nagasaka, T.; Tanaka, K.; Inoue, Y.; Kusunoki, M.; Boland, C.R.; Goel, A. Serum miR-21 as a diagnostic and prognostic biomarker in colorectal cancer. JNCI J. Natl. Cancer Inst. 2013, 105, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Gezer, U.; Üstek, D.; Yörüker, E.E.; Cakiris, A.; Abaci, N.; Leszinski, G.; Dalay, N.; Holdenrieder, S. Characterization of H3K9me3- and H4K20me3-associated circulating nucleosomal DNA by high-throughput sequencing in colorectal cancer. Tumor Biol. 2013, 34, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.-F.; Druez, A.; Faugeras, L.; Martinet, J.; Géhénot, M.; Josseaux, E.; Herzog, M.; Micallef, J.; George, F.; Delos, M.; et al. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin. Epigenetics 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Hoon, D.S.B.; Giuliano, A.E.; Hansen, N.M.; Wang, H.-J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Nakayama, T.; Kajita, M.; Miyake, T.; Iwamoto, T.; Kim, S.J.; Sakai, A.; Ishihara, H.; Tamaki, Y.; Noguchi, S. Detection of aberrant promoter methylation of GSTP1, RASSF1A, and RARβ2 in serum DNA of patients with breast cancer by a newly established one-step methylation-specific PCR assay. Breast Cancer Res. Treat. 2012, 132, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dulaimi, E.; Hillinck, J.; de Caceres, I.I.; Al-Saleem, T.; Cairns, P. Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin. Cancer Res. 2004, 10, 6189–6193. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.O.; Feng, Q.; Toure, P.; Dem, A.; Critchlow, C.W.; Hawes, S.E.; Wood, T.; Jeronimo, C.; Rosenbaum, E.; Stern, J.; et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J. Clin. Oncol. 2006, 24, 4262–4269. [Google Scholar] [CrossRef] [PubMed]
- Kajabova, V.; Smolkova, B.; Zmetakova, I.; Sebova, K.; Krivulcik, T.; Bella, V.; Kajo, K.; Machalekova, K.; Fridrichova, I. RASSF1A promoter methylation levels positively correlate with estrogen receptor expression in breast cancer patients. Transl. Oncol. 2013, 6, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Barekati, Z.; Kohler, C.; Schumacher, M.M.; Grussenmeyer, T.; Jenoe, P.; Hartmann, N.; Moes, S.; Letzkus, M.; Bitzer, J.; et al. Integrated epigenetics of human breast cancer: Synoptic investigation of targeted genes, microRNAs and proteins upon demethylation treatment. PLoS ONE 2011, 6, e27355. [Google Scholar] [CrossRef] [PubMed]
- Liggett, T.; Melnikov, A.; Yi, Q.; Replogle, C.; Brand, R.; Kaul, K.; Talamonti, M.; Abrams, R.A.; Levenson, V. Differential methylation of cell-free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer 2010, 116, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.A.; Scholtens, D.; Talamonti, M.S.; Bentrem, D.J.; Levenson, V.V. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J. Surg. Oncol. 2009, 99, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Eliot, M.; Butler, R.A.; Cheong, A.; Zhang, X.; McClean, M.D.; Koestler, D.C.; Kelsey, K.T. CpG island methylation profile in non-invasive oral rinse samples is predictive of oral and pharyngeal carcinoma. Clin. Epigenetics 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Gonzalgo, M.L.; Yegnasubramanian, S.; Lin, X.; de Marzo, A.M.; Nelson, W.G. GSTP1 CpG island hypermethylation as a molecular biomarker for prostate cancer. J. Cell. Biochem. 2004, 91, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Woodson, K.; O’Reilly, K.J.; Hanson, J.C.; Nelson, D.; Walk, E.L.; Tangrea, J.A. The usefulness of the detection of GSTP1 methylation in urine as a biomarker in the diagnosis of prostate cancer. J. Urol. 2008, 179, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Doufekas, K.; Zheng, S.C.; Ghazali, S.; Wong, M.; Mohamed, Y.; Jones, A.; Reisel, D.; Mould, T.; Olaitan, A.; Macdonald, N.; et al. DNA methylation signatures in vaginal fluid samples for detection of cervical and endometrial cancer. Int. J. Gynecol. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Kneip, C.; Raji, O.; Liloglou, T.; Seegebarth, A.; Schlegel, T.; Flemming, N.; Rausch, S.; Distler, J.; Fleischhacker, M.; et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int. J. Oncol. 2011, 40, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, L.; Liu, B.; Yang, L.; Meng, X.; Zhang, W.; Ma, Y.; Xiao, H. Genome-wide microRNA profiles identify miR-378 as a serum biomarker for early detection of gastric cancer. Cancer Lett. 2012, 316, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Ichikawa, D.; Miyamae, M.; Kawaguchi, T.; Morimura, R.; Hirajima, S.; Okajima, W.; Ohashi, T.; Imamura, T.; Konishi, H.; et al. Malignant potential in pancreatic neoplasm; new insights provided by circulating miR-223 in plasma. Expert Opin. Biol. Ther. 2015, 15, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.A.; Dehlendorff, C.; Jensen, B.V.; Bjerregaard, J.K.; Nielsen, K.R.; Bojesen, S.E.; Calatayud, D.; Nielsen, S.E.; Yilmaz, M.; Holländer, N.H.; et al. MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA 2014, 311, 392. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Sudo, H.; Kawauchi, J.; Takizawa, S.; Kondou, S.; Nobumasa, H.; Ochiai, A. MicroRNA markers for the diagnosis of pancreatic and biliary-tract cancers. PLoS ONE 2015, 10, e0118220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Z.; Zheng, S.; Zhou, Y.; Zhao, L.; Ye, H.; Zhao, X.; Gao, W.; Fu, Z.; Zhou, Q.; et al. Expression profile of long non-coding RNAs in pancreatic cancer and their clinical significance as biomarkers. Oncotarget 2015, 6, 35684–35698. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yin, C.; Dang, Y.; Ye, F.; Zhang, G. Identification of the long non-coding RNA H19 in plasma as a novel biomarker for diagnosis of gastric cancer. Sci. Rep. 2015, 5, 11516. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Shao, Y.; Zhang, X.; Zheng, T.; Miao, M.; Qin, L.; Wang, B.; Ye, G.; Xiao, B.; Guo, J. Plasma long noncoding RNA protected by exosomes as a potential stable biomarker for gastric cancer. Tumor Biol. 2015, 36, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Chen, Y.-Y.; Scott, G.K.; DeVries, S.; Chin, K.; Benz, C.C.; Waldman, F.M.; Hwang, E.S. Protein acetylation and histone deacetylase expression associated with malignant breast cancer progression. Clin. Cancer Res. 2009, 15, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Deligezer, U.; Akisik, E.E.; Erten, N.; Dalay, N. Sequence-specific histone methylation is detectable on circulating nucleosomes in plasma. Clin. Chem. 2008, 54, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Leszinski, G.; Gezer, U.; Siegele, B.; Stoetzer, O.; Holdenrieder, S. Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Res. 2012, 32, 2199–2205. [Google Scholar] [PubMed]
- Bauden, M.; Pamart, D.; Ansari, D.; Herzog, M.; Eccleston, M.; Micallef, J.; Andersson, B.; Andersson, R. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin. Epigenetics 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Van Grembergen, O.; Bizet, M.; de Bony, E.J.; Calonne, E.; Putmans, P.; Brohée, S.; Olsen, C.; Guo, M.; Bontempi, G.; Sotiriou, C.; et al. Portraying breast cancers with long noncoding RNAs. Sci. Adv. 2016, 2, e1600220. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, Z.; Yang, Y.; Xiang, T.; Song, W.; Liu, S. Microarray expression profiling of dysregulated long non-coding RNAs in triple-negative breast cancer. Cancer Biol. Ther. 2015, 16, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Jansen, L.; Walter, V.; Tagscherer, K.; Roth, W.; Herpel, E.; Kloor, M.; Bläker, H.; Chang-Claude, J.; Brenner, H.; et al. No association of CpG island methylator phenotype and colorectal cancer survival: Population-based study. Br. J. Cancer 2016, 115, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Du, Y.; Wang, G.; Gao, J.; Gong, Y.; Li, L.; Zhang, Z.; Zhu, J.; Jing, Q.; Qin, Y.; et al. Detection of differentially expressed microRNAs in serum of pancreatic ductal adenocarcinoma patients: miR-196a could be a potential marker for poor prognosis. Dig. Dis. Sci. 2011, 56, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Kolacinska, A.; Morawiec, J.; Fendler, W.; Malachowska, B.; Morawiec, Z.; Szemraj, J.; Pawlowska, Z.; Chowdhury, D.; Choi, Y.E.; Kubiak, R.; et al. Association of microRNAs and pathologic response to preoperative chemotherapy in triple negative breast cancer: Preliminary report. Mol. Biol. Rep. 2014, 41, 2851–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheirelseid, E.A.H.; Miller, N.; Chang, K.H.; Curran, C.; Hennessey, E.; Sheehan, M.; Newell, J.; Lemetre, C.; Balls, G.; Kerin, M.J. miRNA expressions in rectal cancer as predictors of response to neoadjuvant chemoradiation therapy. Int. J. Colorectal Dis. 2013, 28, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, G.; Shi, Q.; Zhang, R.; Zhao, Y.; Wei, Y.; Chen, F.; Christiani, D.C. Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma. Clin. Epigenetics 2017, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Barlési, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.E.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non-small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef] [PubMed]
- Yates, D.R.; Rehman, I.; Abbod, M.F.; Meuth, M.; Cross, S.S.; Linkens, D.A.; Hamdy, F.C.; Catto, J.W.F. Promoter hypermethylation identifies progression risk in bladder cancer. Clin. Cancer Res. 2007, 13, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Catto, J.W.F.; Hartmann, A.; Stoehr, R.; Bolderson, E.; Rehman, I.; Rosario, D.J.; Hamdy, F.C.; Meuth, M. Multifocal urothelial cancers with the mutator phenotype are of monoclonal origin and require panurothelial treatment for tumor clearance. J. Urol. 2006, 175, 2323–2330. [Google Scholar] [CrossRef]
- Dulaimi, E.; Uzzo, R.G.; Greenberg, R.E.; Al-Saleem, T.; Cairns, P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin. Cancer Res. 2004, 10, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, M.M.; Campan, M.; Figueroa, J.D.; Yang, X.R.; Lissowska, J.; Peplonska, B.; Brinton, L.A.; Rimm, D.L.; Laird, P.W.; Garcia-Closas, M.; et al. DNA hypermethylation of ESR1 and PGR in breast cancer: pathologic and epidemiologic associations. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Galán, J.; Torres-Torres, B.; Núñez, M.I.; López-Peñalver, J.; del Moral, R.; de Almodóvar, J.M.R.; Menjón, S.; Concha, Á.; Chamorro, C.; Ríos, S.; et al. ESR1 gene promoter region methylation in free circulating DNA and its correlation with estrogen receptor protein expression in tumor tissue in breast cancer patients. BMC Cancer 2014, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Siegmund, K.D.; Müller, H.M.; Fiegl, H.; Marth, C.; Müller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Liu, J.; Jing, H.; Dong, S.X.; Wu, J. The diagnostic and prognostic value of CHFR hypermethylation in colorectal cancer, a meta-analysis and literature review. Oncotarget 2017, 8, 89142–89148. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, F.; Zhang, H.; Sun, J.; Ma, Y. CHFR hypermethylation, a frequent event in acute myeloid leukemia, is independently associated with an adverse outcome. Genes Chromosom. Cancer 2016, 55, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Derks, S.; Cleven, A.H.G.; Melotte, V.; Smits, K.M.; Brandes, J.C.; Azad, N.; van Criekinge, W.; de Bruïne, A.P.; Herman, J.G.; van Engeland, M. Emerging evidence for CHFR as a cancer biomarker: From tumor biology to precision medicine. Cancer Metastasis Rev. 2013, 33, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, W.; Li, H.; Ma, Y.; He, G.; Tan, G. Genomic methylation profiling combined with gene expression microarray reveals the aberrant methylation mechanism involved in nasopharyngeal carcinoma taxol resistance. Anticancer Drugs 2012, 23, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Pelosof, L.; Yerram, S.R.; Ahuja, N.; Delmas, A.; Danilova, L.; Herman, J.G.; Azad, N.S. CHFR silencing or microsatellite instability is associated with increased antitumor activity of docetaxel or gemcitabine in colorectal cancer. Int. J. Cancer 2014, 134, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.; Liu, Y.; Gao, D.; Linghu, E.; Brock, M.V.; Yin, D.; Zhan, Q.; Herman, J.G.; Guo, M. Methylation of CHFR sensitizes esophageal squamous cell cancer to docetaxel and paclitaxel. Genes Cancer 2015, 6, 38–48. [Google Scholar] [PubMed]
- Wang, X.; Yang, Y.; Xu, C.; Xiao, L.; Shen, H.; Zhang, X.; Li, T.; Li, X. CHFR suppression by hypermethylation sensitizes endometrial cancer cells to paclitaxel. Int. J. Gynecol. Cancer 2011, 21, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Toyota, M.; Itoh, F.; Sasaki, Y.; Suzuki, H.; Ogi, K.; Kikuchi, T.; Mita, H.; Yamashita, T.; Kojima, T. Epigenetic inactivation of CHFR and sensitivity to microtubule inhibitors in gastric cancer. Cancer Res. 2003, 63, 8606–8613. [Google Scholar] [PubMed]
- Yoshida, K.; Hamai, Y.; Suzuki, T.; Sanada, Y.; Oue, N.; Yasui, W. DNA methylation of CHFR is not a predictor of the response to docetaxel and paclitaxel in advanced and recurrent gastric cancer. Anticancer Res. 2006, 26, 49–54. [Google Scholar] [PubMed]
- Kelly, R.J.; Wrangle, J.; Hales, R.K.; Molena, D.; Yang, S.C.; Rodgers, K.; Lang, M.; Reynolds, J.; Beckman, T.; Choflet, A.; et al. An interim analysis of a phase II study using an epigenetic biomarker (CHFR methylation status) to personalize chemotherapy in patients with operable esophageal cancer. J. Clin. Oncol. 2014, 32, 76. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst). 2007, 6, 1079–1099. [Google Scholar]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Godard, S.; Dietrich, P.-Y.; Regli, L.; Ostermann, S.; Otten, P.; van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT promoter methylation in malignant gliomas: Ready for personalized medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Minckwitz, G.; Kummel, S.; Vogel, P.; Hanusch, C.; Eidtmann, H.; Hilfrich, J.; Gerber, B.; Huober, J.; Costa, S.D.; Jackisch, C.; et al. Intensified neoadjuvant chemotherapy in early-responding breast cancer: phase III randomized GeparTrio study. JNCI J. Natl. Cancer Inst. 2008, 100, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Holdenrieder, S.; Stieber, P.; von Pawel, J.; Raith, H.; Nagel, D.; Feldmann, K.; Seidel, D. Circulating nucleosomes predict the response to chemotherapy in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 5981–5987. [Google Scholar] [CrossRef] [PubMed]
- Phé, V.; Cussenot, O.; Rouprêt, M. Interest of methylated genes as biomarkers in urothelial cell carcinomas of the urinary tract. BJU Int. 2009, 104, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Koga, Y.; Tanaka, T.; Nakamura, J.; Hashiguchi, K.; Noshiro, H.; Miyazaki, K. Aberrant gene methylation is a biomarker for the detection of cancer cells in peritoneal wash samples from advanced gastric cancer patients. Ann. Surg. Oncol. 2011, 18, 3013–3019. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Kitajima, Y.; Sato, S.; Mitsuno, M.; Koga, Y.; Nakamura, J.; Hashiguchi, K.; Noshiro, H.; Miyazaki, K. Aberrant gene methylation in the lymph nodes provides a possible marker for diagnosing micrometastasis in gastric cancer. Ann. Surg. Oncol. 2010, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Van Otterdijk, S.D.; Norden, J.; Dickinson, A.M.; Pearce, M.S.; Relton, C.L.; Mathers, J.C.; Strathdee, G. Aberrations in DNA methylation are detectable during remission of acute lymphoblastic leukemia and predict patient outcome. Epigenomics 2015, 7, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Furlan, C.; Polesel, J.; Barzan, L.; Franchin, G.; Sulfaro, S.; Romeo, S.; Colizzi, F.; Rizzo, A.; Baggio, V.; Giacomarra, V.; et al. Prognostic significance of LINE-1 hypomethylation in oropharyngeal squamous cell carcinoma. Clin. Epigenetics 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Sueta, A.; Yamamoto, Y.; Tomiguchi, M.; Takeshita, T.; Yamamoto-Ibusuki, M.; Iwase, H. Differential expression of exosomal miRNAs between breast cancer patients with and without recurrence. Oncotarget 2017, 8, 69934–69944. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Komatsu, S.; Ichikawa, D.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Arita, T.; Konishi, H.; Shiozaki, A.; et al. Liquid biopsy in patients with pancreatic cancer: Circulating tumor cells and cell-free nucleic acids. World J. Gastroenterol. 2016, 22, 5627. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, Y.; Cho, J.H.; McClarty, S.; Baxter, D.; Galas, D.J. Comparing the MicroRNA Spectrum between serum and plasma. PLoS ONE 2012, 7, e41561. [Google Scholar] [CrossRef] [PubMed]
- Langevin, S.M.; Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Nelson, H.H.; Karagas, M.R.; Marsit, C.J.; Wiencke, J.K.; Kelsey, K.T. Leukocyte-adjusted epigenome-wide association studies of blood from solid tumor patients. Epigenetics 2014, 9, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Koestler, D.C.; Marsit, C.J.; Christensen, B.C.; Accomando, W.; Langevin, S.M.; Houseman, E.A.; Nelson, H.H.; Karagas, M.R.; Wiencke, J.K.; Kelsey, K.T. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Shabalin, A.A.; Aberg, K.A.; van den Oord, E.J. Candidate gene methylation studies are at high risk of erroneous conclusions. Epigenomics 2015, 7, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247.e17. [Google Scholar] [CrossRef] [PubMed]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Types of epigenetic biomarkers that can be detected by liquid biopsy. Compared to normal cells, cancer cells shed a disproportionate amount of circulating free nucleic acids, microvesicle/exosome encapsulated nucleic acids, and nucleosomes; as well as shedding whole tumor cells into circulation. Epigenetic biomarkers can be detected in sputum (lung), urine (bladder and prostate), stool (colorectal), and blood (many cancer types). miRNA: microRNAs; lncRNA: long non-coding RNA.
Figure 1.
Types of epigenetic biomarkers that can be detected by liquid biopsy. Compared to normal cells, cancer cells shed a disproportionate amount of circulating free nucleic acids, microvesicle/exosome encapsulated nucleic acids, and nucleosomes; as well as shedding whole tumor cells into circulation. Epigenetic biomarkers can be detected in sputum (lung), urine (bladder and prostate), stool (colorectal), and blood (many cancer types). miRNA: microRNAs; lncRNA: long non-coding RNA.


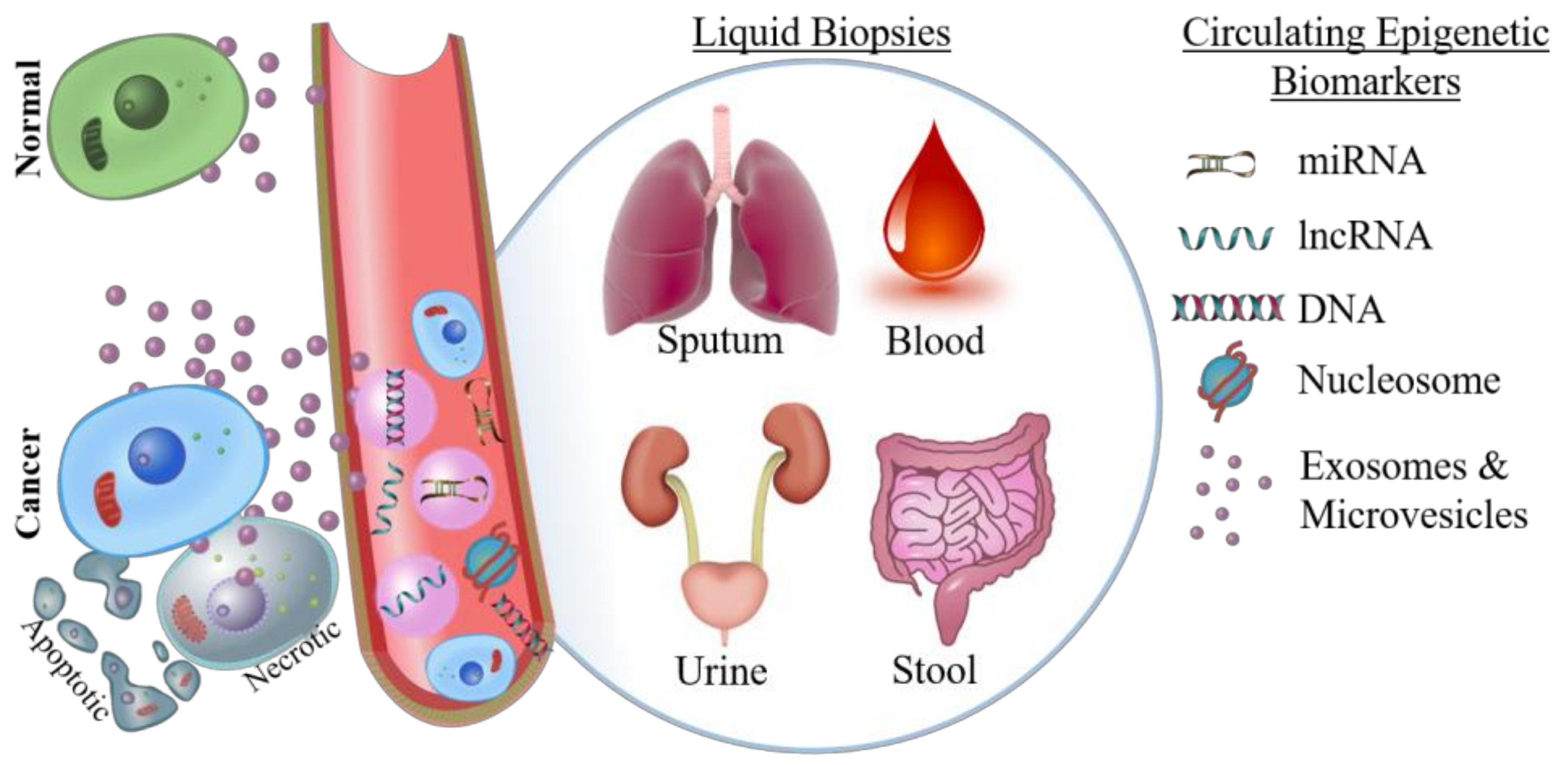{kind=link}
Table 1.
Clinical trials using diagnostic, prognostic, or predictive biomarkers based on DNA methylation.
Table 1.
Clinical trials using diagnostic, prognostic, or predictive biomarkers based on DNA methylation.
| Clinical Trial ID | Marker | Test | Sample | Neoplasm | Biomarker Type | Result/Status |
|---|---|---|---|---|---|---|
| NCT01329718 | Hypermethylated Septin-9 (SEPT9) | Epi proColon | Blood | Colorectal cancer | Diagnostic | Completed |
| NCT03218423 | Colorectal cancer | Longitudinal (Diagnostic) | Recruiting | |||
| NCT00696345 | Colorectal cancer | Diagnostic | Completed | |||
| NCT02198092 | Hereditary colorectal cancer | Diagnostic | Recruiting | |||
| NCT03311152 | Hepatocellular carcinoma | Diagnostic | Recruiting | |||
| NCT02540850 | Hypermethylated SEPT9 | Blood | Colorectal cancer | Diagnostic | Completed | |
| NCT00855348 | Colorectal cancer | Diagnostic | Completed | |||
| NCT02419716 | Hypermethylated NDRG family member 4 (NDRG4), bone morphogenetic protein 3 (BMP3) | ColoGuard | Stool | Colorectal cancer | Diagnostic | Active/Not Recruiting |
| NCT02715141 | Recruiting | |||||
| NCT01793207 | 7 Cpgs | Blood | Colorectal cancer | Diagnostic | Completed | |
| NCT03146520 | Hypermethylated syndecan 3 (SDC2) | EarlyTect | Stool | Colorectal cancer | Diagnostic | Enrolling by invitation |
| NCT02159339 | Hypermethylated cyclin dependent kinase inhibitor 2A (p16) | Tumor | Gastric cancer | Prognostic- Metastasis | Completed | |
| NCT00835341 | Biopsy | Oral cancer | Diagnostic | Completed | ||
| NCT01695018 | Mucosal Biopsy | Oral cancer | Diagnostic | Completed | ||
| NCT01774266 | Hypermethylated TP53-dependent G2 arrest mediator homolog (REPRIMO) | Serum | Gastric cancer | Diagnostic | Recruiting | |
| NCT01715233 | Hypermethylated checkpoint with forkhead and ring finger domains (CHFR) | Biopsy | Esophageal, gastroesophageal, gastric cancer | Predictive-Taxane Response | Recruiting | |
| NCT01372202 | Biopsy | Esophageal cancer | Active/Not Recruiting | |||
| NCT03217097 | Hypermethylated O-6-methylguanine-DNA methyltransferase (MGMT) | Biopsy | Neuroendocrine tumors | Predictive-Oxaliplatin/Alkylating Agent Response | Not Yet Recruiting | |
| NCT02700464 | 15 CpGs | EpiCheck | Urine | Bladder eurothelial cell carcinoma | Recurrence | Recruiting |
| NCT02647112 | Recruiting | |||||
| NCT02688491 | 5 CpGs | Surgical Specimen | Clear cell renal carcinoma (Stage III) | Predictive-Sunitinib Response | Not Yet Recruiting |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thomas, M.L.; Marcato, P. Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum. Cancers 2018, 10, 101. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040101
AMA Style
Thomas ML, Marcato P. Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum. Cancers. 2018; 10(4):101. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040101
Chicago/Turabian StyleThomas, Margaret L., and Paola Marcato. 2018. "Epigenetic Modifications as Biomarkers of Tumor Development, Therapy Response, and Recurrence across the Cancer Care Continuum" Cancers 10, no. 4: 101. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040101
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.