Genomic Landscapes of EBV-Associated Nasopharyngeal Carcinoma vs. HPV-Associated Head and Neck Cancer


1
School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong 999077, China
2
Department of Anatomical and cellular Pathology, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong 999077, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2018, 10(7), 210; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10070210
Submission received: 17 May 2018
/
Revised: 9 June 2018
/
Accepted: 13 June 2018
/
Published: 21 June 2018
(This article belongs to the Special Issue Epstein–Barr Virus Associated Cancers)
Abstract
:Epstein-Barr virus-positive nasopharyngeal carcinoma (EBV(+) NPC), and human papillomavirus-positive head and neck squamous cell carcinoma (HPV(+) HNSCC) are two distinct types of aggressive head and neck cancers with early age onsets. Their recently identified genomic landscapes by whole-exome sequencing (WES) clearly reveal critical roles of: (1) inflammation via NF-kB activation, (2) survival via PI3K aberrations, and perhaps (3) immune evasion via MHC loss in these cancers as summarized in this review. Immediate outcomes of these WES studies include the identification of potential prognostic biomarkers, and druggable events for these cancers. The impact of these genomic findings on the development of precision medicine and immunotherapies will be discussed. For both of these cancers, the main lethality comes from metastases and disease recurrences which may represent therapy resistance. Thus, potential curing of these cancers still relies on future identification of key genomic drivers and likely druggable events in recurrent and metastatic forms of these intrinsically aggressive cancers of the head and neck.
1. Introduction
The global incidence of head and neck cancer has recently climbed to ~0.74 million new cases per year based on 2015 cancer statistics [1]. With the upper respiratory tract being the first site of contact with environmental carcinogens including certain chemicals (in cigarette smoke or alcohol), air pollutants [2], as well as oncogenic viruses [3,4], the incidence of head and neck cancer will probably continue to rise in the next decade. The collective term of head and neck cancer encompass epithelial malignancies of the oral cavity, tongue, floor of the mouth, pharynx, larynx, oropharynx, nasopharynx, paranasal sinuses, as well as the salivary glands [5,6]. Among which, cancers of the nasopharynx and oropharynx represent two major subtypes with unique and strong etiological association with two distinct oncogenic viruses, namely the Epstein-Barr virus (EBV) and the human papillomavirus (HPV), respectively. The Epstein-Barr Virus (EBV)-associated nasopharyngeal carcinoma (hereafter, abbreviated as EBV(+) NPC) is the most metastatic cancer of the head and neck with the highest prevalence in Southern China and Southeast Asia, while the HPV-associated head and neck squamous cell carcinoma (cancer of squamous epithelial cell origin, hereafter, abbreviated as HPV(+) HNSCC; mostly represented by HPV(+) oropharyngeal cancer) is a rising epidemic cancer of Western countries, including Europe and the US.
For a long time, our understanding on how these oncogenic viruses cause head and neck cancers has remained incomplete. Why are nasopharyngeal and oropharyngeal cancers, which are so close in their anatomical locations, affected by two different DNA viruses? Are there any commonalities in these viral-mediated processes that trigger transformation of the head and neck epithelium? How similar are these two cancers in terms of their underlying biology, which may translate into more effective treatments in the future? Yet, recent genomic characterizations of EBV(+) NPC and HPV(+) HNSCC have revealed multiple new dimensions and insights for the above questions. The big leap in the understanding of the genomic landscapes of these two virus-associated cancers may provide new therapeutic opportunities in the near future.
2. Background, Similarities and Differences between EBV(+) NPC and HPV(+) HNSCC
2.1. EBV(+) NPC
Nasopharyngeal carcinoma (NPC) is a squamous epithelial cancer arising from the mucosa of the nasopharynx. The nasopharynx is a structure that extends from the skull base to the soft palate’s upper surface, and it connects to the oropharynx [7]. In 2012, a total of 86,691 new cases of NPC were reported worldwide [8], with the highest incidence in endemic regions such as South China and Southeast Asia (especially among the Cantonese population with male ASR (age-standardization rate) of ~13–25/100,000 persons) and moderate rate in North Africa (male ASR of ~4–6/100,000 persons) [9]. In contrast, this unique type of head and neck cancer is rarely found in Western countries, with an incidence rate ~100-fold lower than that in the endemic regions. Interestingly, almost all NPC cases from the endemic regions (>95%) are EBV(+) non-keratinizing undifferentiated carcinoma, while those in non-endemic regions are EBV(−) keratinizing carcinomas with well-differentiated histologic features. The major cause of EBV(+) NPC is believed to be caused by latent infection of the oncogenic virus, EBV. Yet, as of today, it still remains perplexing as to why EBV causes EBV(+) NPC in the endemic regions, but mononucleosis in Caucasians. Unique dietary habits (salted fish consumption), as well as genetic susceptibility factors are likely etiological factors for NPC carcinogenesis. Studies have shown that descendants of emigrants from EBV(+) NPC endemic regions have a higher rate of EBV(+) NPC development even after their settlements in non-endemic regions [10,11,12,13]. This indicates a likely genetic factor behind EBV(+) NPC oncogenesis. The potential strong influence of environmental factors on NPC carcinogenesis was recently revealed by a French study, in which male NPC patients of French origin, but born or living in Maghreb (North Africa) appear to have a 5.7-times higher risk to develop NPC than those of French origin born in France [12]. This study highlights the important impact of environment factors on NPC development.
EBV(+) NPC is one of the most metastatic head and neck cancers which is often found to metastasize to lymph nodes as well as distant organs at the time of diagnosis. It often metastasizes to the liver, lung and brain for reasons yet to be identified. Pathologically, this intrinsically aggressive cancer is characterized by: (1) high intra-tumoral lymphocyte infiltrations; (2) the presence of EBV genome and expression of EBV latent genes including EBERs, EBNA1, LMP1, LMP2 and BARTs; (3) high levels of circulating EBV DNA in the plasma of NPC patients [14,15]. In fact, recurrences of EBV(+) NPC can be predicted by the increasing levels of plasma EBV DNA in patients [16].
Although at early stages (I or II), the curative rate of NPC can be ~90% with the most aggressive concurrent chemoradiotherapy. However, the majority of newly diagnosed cases are of advanced stages (III or IV), and these patients have a dismal 5-year survival of ~50–60%. The high mortality rate of NPC is due to the asymptomatic but aggressive nature of this cancer. Thus far, due to the lack of solid gene-drug knowledge and evidence, no NPC-specific targeted therapy has been approved for NPC treatment. Precision medicine for this cancer has not been developed [17,18,19].
2.2. HPV(+) HNSCC
HPV(+) HNSCC is largely represented by squamous cell carcinoma (SCC) of the oropharynx, and some other less frequently infected sites, such as the oral cavity, larynx and hypopharynx, etc. [20,21]. Oropharyngeal cancers are caused mainly by persistent infection of the oropharyngeal epithelium by the oncogenic HPV 16 via oral sex [22]. Note that cervical cancer is also caused by HPV 16, as well as HPV 18. The blooming epidemic of HPV(+) HNSCC first noted in the Western countries is starting to be seen in some Eastern countries as well [23]. As of today, as much as 80–100%, and 70% of oropharyngeal cancer from European countries and the US are HPV-positive, respectively [24]. In South Korea and Japan, HPV-associated oropharyngeal cancer is as high as 67% [25], while it remains as low as 15% in China and Hong Kong (where EBV(+) NPC is prevalent) [26].
Compared to non-HPV-associated head and neck cancer (named as HPV(−) HNSCC hereafter), HPV(+) HNSCC appears to be more aggressive with a higher tendency to metastasize to distant organs [27]. Standardized conventional treatment modalities are being currently applied for both HPV(+) HNSCC and HPV(−) HNSCC, which include surgery, radiation, chemotherapy (or combination), cetuximab (for all HNSCC), and two newly approved PD-L1 inhibitors (pembrolizumab and nivolumab). Interestingly, with the exception of smokers [28], it is recently noted that HPV(+) HNSCC patients, in general, have better clinical outcomes after treatments than HPV(−) HNSCC patients. This general observation leads to multiple clinical trials aiming at de-intensifying treatments for HPV(+) HNSCC. Although the HPV vaccine has been shown to be effective for the prevention of cervical cancer, and its impact on HPV(+) HNSCC is currently being validated with promising preventive results [29]. Notably, multiple genomic characterization studies of HNSCC, including those for HPV(+) HNSCC, have contributed to new molecular understanding of HNSCC, and opened up new opportunities for precision medicine for HPV(+) HNSCC, as well as HPV(−) HNSCC.
2.3. Similarities and Differences between these Two Virally Associated HNCs in General
Theoretically, the exposures to chemical or viral carcinogens are rather similar in these cancers as the nasopharynx and the oropharynx are anatomically connected [30]. Yet, EBV infects the nasopharynx, while HPV infects the oropharynx, giving rise to two distinct cancer types.
Not often noted, the two cancer types do share some degree of similarities and differences (Table 1): (1) both cancers are etiologically linked with double-stranded DNA viruses (i.e., with DNA genomes), with both EBV and HPV being Group I carcinogens to human (World Health Organization; WHO). Yet, the HPV genome (~8 kb; belonging to the human papillomavirus family) is much smaller than that of the EBV genome (~172 kb; belonging to the Herpesvirus family). The EBV genome found in EBV(+) NPC is episomal and clonal in nature [31], while the HPV genome is mostly found to be integrated into human chromosomes, but also observed to be episomal in some HPV(+) HNSCC cases [32]. (2) Clinically, both cancers are known to have young age onsets at ~40–45 years of age (as compared to 65 years old for HPV(−) HNSCC) [4], and both have a relatively higher male to female predominance of 3 or 4:1. (3) Interestingly, both tumors are small in size and potentially antigenic due to the presence of viral antigens. (4) Both cancers are responsive to some traditional treatments as EBV(+) NPC is sensitive to radiation and chemotherapy, while HPV(+) HNSCCs are known to be treatment sensitive in general. In fact, HPV-positivity has been recognized as a single independent favorable prognostic factor for HNSCC survival [33], even for patients with recurrences or metastases [34]. The treatment sensitivity natures of both cancers are unknown, but it has been postulated that the antigenic viral natures, and the p53 wildtype natures of both cancers may constitute to their treatment responsiveness in general [35]. (5) In terms of cancer diagnosis, both EBV and HPV nucleic acids can be detected in the patient’s body fluids, in addition to the patients’ tumors. EBV DNA can be readily detected in nasal discharge, saliva and plasma of the EBV(+) NPC patients [36,37,38], while HPV DNA can be detected in the saliva and plasma of HPV(+) HNSCC patients (though with limited sensitivity at the moment) [39,40]. (6) Theoretically, both cancers can be potentially prevented by vaccination. Currently, HPV vaccine is already available for HNSCC prevention (as well as cervical cancer prevention), while EBV vaccination has not been successfully developed yet.
3. Genomic Characterizations of EBV(+) NPC and HPV(+) HNSCC Reveal Major Genetic Differences and Similarities
3.1. WES and WGS Studies on EBV(+) NPC and HPV(+) HNSCC
As of today, several whole-exome sequencing (WES) and whole-genome sequencing (WGS) studies have been conducted to specifically investigate the genetic aberrations of EBV(+) NPC tumors and HNSCC tumors, which include a small number of HPV(+) HNSCC tumors (Table 2). It has to be noted that the TCGA HNSCC cohort covers only a limited number of anatomic sites in the head and neck region, with only 7 major sites having more than 5% of cases in the cohort. These are cancers of the tongue, larynx, oral cavity, pharynx, lips, floor of the mouth, and the tonsils, etc. As expected from the ethnic and geographical prevalence of these cancers, sequencing and molecular information generated for EBV(+) NPC are largely Asian-relevant, while that for HPV(+) HNSCC are mainly Caucasian-relevant. These findings will be reviewed in details below.
3.2. EBV(+) NPC Has a Relatively Lower Mutational Burden than HPV(+) HNSCC
Three NPC WES studies to date showed that EBV(+) NPC has a relatively lower rate of somatic mutations than HPV(+) HNSCC as well as EBV(+) stomach adenocarcinoma [41,42,43]. This may imply that EBV is performing a reasonable big task on oncogenesis already. In fact, in contrast to the limited viral gene products expressed in HPV(+) HNSCC, EBV does express a number of viral proteins and noncoding RNAs to promote transformation via multiple cellular mechanisms and signaling pathways. With the majority of NPC bearing TP53 wildtype genes (~94% primary NPC tumors), and the low rate of somatic mutation detected in primary NPC tumors, it appears that early development of NPC does not require the accumulation of many somatic genetic events. During early oncogenesis, transformation of normal nasopharyngeal epithelium (e.g., by activation of telomerase) is believed to be a key process [14]. This is followed by losses of chromosomes 3p and 9p (including inactivation of RASSF1A and CDKN2A genes that were identified in precancerous lesions of NPC over a decade ago). Such a consistent loss of chr. 3p was recently confirmed by both WGS and WES in 100% cases of >111 microdissected EBV(+) NPC tumors of various stages (Stage I to IV) from Hong Kong, an endemic region in Southeast Asia [43]. It is plausible that both chr. 3p and 9p losses predispose the nasopharyngeal epithelial cells to perisistent EBV latent infection and allows clonal expansion of EBV-infected cells.
Interestingly, the prognostic value of mutational burden of EBV(+) NPC tumors has been recently examined. We showed that patients with EBV(+) NPC tumors harboring less somatic mutations were found to be associated with better overall survival and disease-free survival than those with higher mutational rates in their tumors [43]. Similarly, patients with HPV(+) HNSCC tumors (also known to have relatively lower mutational rates than HPV(−) HNSCC in general [43,50,52]) are also known to be associated with better clinical outcomes [23]. Though the biological mechanism(s) behind the observed favorable clinical outcomes of these two virally associated head and neck cancers remains largely unknown, plausible mechanisms may include the presence of wildtype TP53, and likely immunogenic viral antigens in these cancers.
The reported lower rates of somatic mutation in HPV(+) HNSCC vs. HPV(−) HNSCC are consistent with the fact that HPV has already contributed significantly to HNSCC oncogenesis, and only very few additional somatic mutations are likely needed for tumorigenesis to be full-bloomed. Similarly, a lower rate of somatic mutation was found in HPV(+) vulvar SCC vs. HPV(−) vulvar SCC [53]. However, in a recent Indian WES study on HNSCC patients with heavy carcinogen exposures (i.e., betel-nut or tobacco-chewing), the mutational burden of HPV(+) HNSCC (mainly OSCC) does not differ from that of HPV(−) HNSCCs [48]. This may imply an added mutagenic effect of betel-nut and tobacco-chewing on top of HPV-induced carcinogenesis among Indian HPV(+) HNSCC.
Thus far, the role of EBV in NPC oncogenesis is still not fully understood, while HPV is believed to contribute to HNSCC oncogenesis via alteration of the activities of two key tumor suppressors, p53 and RB proteins, by HPV E6 and E7 proteins, respectively [54]. In addition to p53 and RB alterations, recent WGS results of 35 HPV(+) HNSCC revealed for the first time that HPV genomic integration into certain important cellular genes, thus affecting their functions, may serve as additional mechanisms for HPV-medicated HNSCC oncogenesis. These include HPV integrations within the RAD51B and ETS2 genes causing their loss-of-function activities, or HPV integration resulting in altered expression of an alternative CD274 (PDL1) transcript [49]. Since viral integration can only be thoroughly studied by WGS, it is believed that larger cohorts of WGS-characterized HPV(+) HNSCC may reveal even more previously unknown oncogenic mechanisms by HPV integration events in HNSCC. As for EBV(+) NPC, it remains to be carefully examined in larger WGS cohorts if there is any EBV integration into the human genome or not.
3.3. EBV(+) NPC and HPV(+) HNSCC Have Different Mutational Signatures
Mutational signature analysis can inform us on the underlying mechanisms of DNA mutations in human tumors. In EBV(+) NPC, mutational analysis showed that spontaneous deamination of 5-methylcytosine (characterized by Signature 2, COSMIC database; implicating an endogenous mutational process) and defective DNA mismatch repair (characterized by co-occurring Signatures 6, 15, 20, and 26) are the possible underlying mechanisms for DNA mutagenesis in this tumor type [41,43]). The presence of Signature 6 in NPC (a signature implying defective DNA mismatch repair and microsatellite unstable tumors) does imply some potential involvement of microsatellite unstable genomic events in NPC mutagenesis. However, the APOBEC-related signature (Signatures 2 and 13) is only found in a small subset of NPC tumors (<20%), implying APOBEC-mediated mutagenesis is not the major event for NPC as compared to defective DNA mismatch repair. Whereas in HPV(+) HNSCC, it is predominated by the APOBEC signature, indicating a mutagenic mechanism due to viral infection, tissue inflammation or retrotransposon activity [55,56]. Chen et al. also reported the APOBEC signature as the major mutational signature for HPV(+) cervical cancer [57]. These findings implicate a clear etiologic contribution by HPV via APOBEC-mediated mutagenesis, which is different from the defective DNA mismatch repair signature contributed by EBV in NPC. These mutational signatures of EBV(+) NPC and HPV(+) HNSCC appear to reflect major differences in viral or environmental carcinogen-driven mutational forces for these cancers.
3.4. Overall Comparison of Genomic Profiles between EBV(+) NPC and HPV(+) HNSCC
Shortly after the completion of WES studies on these two virally associated head and neck cancers, it was immediately realized and genomically confirmed that previous scattered reports on low rates of TP53 and RB1 mutations are indeed key genomic features of these cancers [41,43,50,52]. More interestingly, independent conclusions from several studies suggest that distinct genomic drives are likely responsible for their tumorigenesis in general. Based on the WES data to date, major “NF-κB activating” somatic aberrations are the key driver events for EBV(+) NPC tumorigenesis, while HPV(+) HNSCC is driven by “PI3K pathway activating” genomic changes [43,50,52,58]. Furthermore, several other important signaling pathways are also genomically altered in subsets of patients with these two cancers.
3.4.1. An NF-kB Genomic Drive for EBV(+) NPC
Prior to the genomic characterization of EBV(+) NPC, various signaling pathways including the EGFR, STAT3, c-MET, NF-κB pathways were believed to be important for NPC tumorigenesis and progression. These previous views were supported by data generated from primary tumors and limited cell line models of EBV(+) NPC. Yet, our recent large scale whole-exome characterization of micro-dissected Asian EBV(+) NPC reveals that NF-κB signaling aberrations are the most critical genomic force driving NF-κB activation in EBV(+) NPC. Over 40% of microdissected EBV(+) NPC tumors were found to harbor genetic aberrations (including translocation, tandem duplication, homozygous deletion and mutation) of negative regulators of NF-κB pathways, which include: CYLD (18.6%), TRAF3 (17.5%), NFKBIA (6.7%) and NLRC5 (4.8%) [43]. CYLD is a tumor suppressor gene, when germline mutated, it can contribute to a rare form of inherited syndrome called the CYLD (cylindromatosis) cutaneous syndrome with clinical manifestation of multiple benign tumors in the head and neck region at early age [59]. CYLD is a deubiquitinase that drives the degradation of multiple regulators of the NF-κB pathway [43]. In our study, as high as 18.6% of EBV(+) NPC patient tumors were found to have either inactivating mutations, or gene rearrangements of CYLD with high allele frequencies. Somatic mutation of CYLD was also reported in another exome and target-sequencing study [42]. CYLD restoration in a CYLD-deficient NPC cell line, C666-1, confirmed its tumor-suppressive role in EBV(+) NPC cell context. On the other hand, ectopic expression of CYLD mutants was found to elevate NF-κB activity in NPC cells, thus demonstrating a mutational drive for NF-κB signaling by CYLD aberrations in NPC. In particular, activation of the atypical NF-κB pathway (as demonstrated by a marked increase in p50/p50/Bcl-3 nuclear complex) appears to be the main consequence driven by CYLD mutants in the EBV(+) NPC cell context [43]. It is note-worthy that CYLD serves multiple functions in normal and pathophysiology, it is plausible that CYLD mutations may affect other functions in EBV(+) NPC cells, such as cilia formation and innate immunity, which is consistent with the loss of ciliated structures during NPC development [60].
In addition to CYLD, another major negative regulator of NF-κB, namely TRAF3 (TNF receptor associated factor 3), was also found to be aberrant in as high as 17.5% of microdissected EBV(+) NPC tumors [43]. We also demonstrated that TRAF3 WT gene inhibited the non-canonical NF-κB signaling in NPC cells, while TRAF3 mutants lost the ability to suppress NF-κB activation in EBV(+) cell context. These genomic aberrations may cause constitutive activation of the NF-κB signaling pathway. These genome findings do suggest the importance of NF-κB targeting for EBV(+) NPC. Microdissection, which aims at enriching tumor cell content prior to sequencing, appears to be crucial for accurate identification of NPC somatic changes as heavy stromal content including lymphocyte infiltration can obscure genomic findings. Without microdissection, the detected frequencies for TRAF3, NFKBIA and NLRC5 mutations were much lower [41,42]. Nevertheless, similar to EBV(+) NPC, a subset of HNSCC with episomal HPV infection also harbors frequent somatic CYLD and TRAF3 mutations [61].
In addition to CYLD and TRAF3 aberrations, an EBV oncoprotein, the latent-membrane protein 1 (LMP1) is also known to be a potent activator for NF-κB signaling [62]. LMP1 has been shown to phosphorylate the IKK complex and induce IκBα degradation, thus resulting in constitutive NF-κB activation. Our exome study revealed for the first time that LMP1 overexpression and genomic aberrations of NF-κB pathway are mutually exclusive [43]. In sum, the LMP1-overexpressing NPC subset and the NF-κB altered NPC subset together account for >70% of all EBV(+) NPC cases. Thus, NF-κB activation is selected for, by both somatic and viral events during NPC pathogenesis in a large majority of patients. This is consistent with the finding that non-coding RNAs of EBV, called Epstein-Barr virus (EBV)-encoded small RNAs (EBERs) also function to promote NF-κB signaling via their interactions with TLR3 [63]. It has been proposed that LMP1 and EBERs form a regulatory loop feeding forward to activate NF-κB signaling for inflammatory responses in NPC.
In summary, multiple genomic and viral events aim to constitutively activate NF-κB signaling for cell proliferation, survival, and inflammation during NPC tumorigenesis. For the very first time, this unique NPC/NF-κB genomic signature unifies many previous scattered findings for this cancer. Whether such a unique NF-κB genomic landscape drives only the non-canonical NF-κB pathway or not, and whether it can help sustaining EBV infection and tumor growth will require further investigations. Yet, targeting of these NF-κB aberrations for NPC treatment development will require multiple representative EBV(+) NPC models, which are currently lacking.
3.4.2. A Prominent PI3K Genomic Drive for HPV(+) HNSCC
HPV is the causative agent of cervical cancer, genital warts, oral papillomas, HPV(+) HNSCC, and some benign tumors. The HPV family has more than 150 subtypes, and many of them have been classified as high-oncogenic risk subtypes, including HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59 for tumorigenesis [64,65]. In particular, HPV 16 and 18 have been the focus of study in cervical cancer. It has been well-recognized that the transforming activities of HPV 16 and HPV 18 are largely exercised through the E6 and E7 oncoproteins, which impair two most important tumor suppressors in mammalian cells. E6 targets p53 for proteasome degradation, resulting in cell cycle dysregulation and the loss of p53-mediated apoptosis in HPV infected cells. Whereas E7 binds to and inactivates RB1, and promotes RB1 degradation. This E7-mediated RB1 degradation results in the release of activated E2F transcription factor, which promotes S phase gene transcription, including that of the p16INK4a (or CDKN2A or p16). Thus, cell cycle progression is dysregulated by E6 and E7 proteins upon HPV infection. Furthermore, HPV integration into the human chromosomes can result in E6 and E7 amplification via viral-host concatemers in HPV(+) HNSCC tumor samples [32].
Prior to genomic characterization, HNSCC collectively, was believed to be governed by several major growth and survival pathways such as EGFR, JAK/STAT signaling, and p53 aberrations. Yet, the recently completed WES studies revealed a wide array of genomic aberrations affecting growth, differentiation, senescence, apoptosis, DNA repair, and immunity in HNSCC. These include genomic aberrations of TP53, CDKN2A, HRAS, NOTCH, PIK3CA, PTEN and CASP8, etc. [50]. Yet, with only 36 HPV(+) HNSCC being exome characterized by the TCGA [50] and reported by us [52], HPV(+) and HPV(−) HNSCCs immediately emerged as distinct types of HNSCC based on their differential genomic profiles. The distinct genomic profile of HPV(+) HNSCC has then been at least partially confirmed in several targeted sequencing studies [66,67,68] and is expected to be validated in even larger cohorts as there are many more incidences of HPV(+) HNSCC in Western countries. Yet, based on the published WES data of 36 HPV(+) HNSCC tumors from the TCGA, HPV(+) HNSCC were found to have almost no (or <1%) somatic TP53 mutation, while HPV(−) HNSCC were characterized by a very high rate of TP53 mutations (~85%; 206/243 cases). This particular low rate of TP53 mutation in HPV(+) HNSCC may simply reflect a “sufficient and efficient” p53-degradating activity of E6 protein on the infected epithelium during HPV-mediated HNSCC oncogenesis. Interestingly, Zevallos et al. reported a previously un-noticed effect of heavy smoking on HPV(+) HNSCC genomic profiles [69]. Zevallos et al. found that HPV(+) HNSCC tumors deriving from heavy smokers (>10 pack-years) have exclusive mutational events of TP53, CDKN2A, KRAS and NOTCH1, and have worse survival than those HPV-positive patients with <10 year-pack year of smoking history. This may suggest the added effects of smoking, on top of HPV, on HNSCC genomic landscapes, which may have future clinical implications, including biomarker development.
Another unexpected genomic finding is the presence of a relatively high rate of PIK3CA mutation and amplification in HPV(+) HNSCC (~40–50% of cases). As first reported by us [52], PIK3CA somatic aberrations were actually found to be 2 folds higher in HPV(+) HNSCC than that of the HPV(−) HNSCC. This is similar to the high rate of PIK3CA mutations in HPV(+) cervical cancer (39–41% cases; TCGA; www.cbioportal.org), implicating the biological importance of PI3K activation in these HPV-positive cancers. A recent study by Han et al. also demonstrated the presence of PIK3CA mutations in HPV(+) vulvar SCC [70].
Most of the identified HNSCC-associated PIK3CA mutations are hotspot mutations [71], and drivers for HNSCC cell growth [52]. By far, PIK3CA hotspot mutations rank the first among all recurrent mutations in HPV(+) HNSCC. These include: PIK3CA R88Q, E542K and E545K. It is possible that these gain-of-function PIK3CA activating genomic events in HPV(+) HNSCC may promote PI3K pathway activation, thus cell survival, growth, or perhaps even immune evasion [72].
It was recently speculated that PIK3CA-mutated HPV(+) HNSCC may confer sensitivity of these tumors for PI3K inhibitors. Yet, Brand et al. showed that some HPV(+) HNSCC cell models, and patient-derived xenografts (PDXs) could be resistant to the PI3K pathway inhibitor, BYL-719 [73]. Subsequent mechanistic investigation showed that PI3K targeting in HPV(+) HNSCC cells could result in a feedback upregulation of ERBB3 signaling via E6 and E7 induction, thus conferring PI3K resistance. Importantly, it was then demonstrated that co-targeting of PI3K and ERBB3 could be an effective approach circumventing this ERBB3 feedback, thus resulting in good antitumor efficacy for HPV(+) HNSCC [74,75].
3.4.3. Systematic Pathway Comparisons between EBV(+) NPC and HPV(+) HNSCC Reveal Commonalities in NF-κB and PI3K Pathway Activation in Both Cancers
The original messages from the WES studies are rather clear that EBV(+) NPC is a NF-κB-driven cancer, while HPV(+) HNSCC is a PI3K-driven cancer. Yet, in this review, based on the pathways that were found to be important for both cancers, we re-analyzed the WES data and systematically compared all key pathways in both cancers to better comprehend how similar or how different they are genomically. (Note that we only include the genomic data of HPV(−) oropharyngeal cancer for reference purposes as HPV(+) HNSCC is mostly represented by cancer arising from the oropharynx).
As shown in Figure 1, apart from NF-κB aberrations (34.2% cases) that was published to be the most frequently altered pathway, PI3K pathway mutations rank the second most commonly mutated signaling pathway in EBV(+) NPC (20.7% cases), followed by the MAPK pathway (11.7% cases), and JAK/STAT, NOTCH, WNT (all with 10.8% cases mutated). Interestingly, in HPV(+) HNSCC, aberrations of the PI3K pathway (52.8% cases; previously published) was seconded by aberrations of the NF-κB pathway genes (38.9% cases) [50] (Figure 1), followed by aberrations of the NOTCH (27.8% cases), JAK/STAT (19.4% cases), WNT (19.4% cases), and the MAPK (13.9% cases) pathways. As for HPV(−) oropharyngeal cancer, which does appear to be mutationally different from the HPV(+) HNSCC, the most common aberrations are that of the PI3K pathway, WNT pathway, as well as the MHC Class I genes (all are noted in 27.3% cases respectively). Therefore, NF-κB and PI3K pathways are, in fact, the top two most commonly mutated signaling pathways in these two virally associated head and neck cancers (Figure 1a and b). Furthermore, as far as MHC Class I/II genes are concerned, both of these virally associated cancer types, as well as HPV(−) oropharyngeal cancer, all showed relatively higher rates of mutations of MHC Class I gene than that of MHC Class II genes (Figure 1). This suggests that MHC Class I gene defects can be more important and relevant for both viral and non-viral mediated tumorigenesis or disease progression. Yet, particularly for EBV(+) NPC and HPV(+) HNSCC, it remains to be determined if defective viral antigen presentation via MHC Class I-dependent pathway is involved in their carcinogenesis. (Figure 1).
As far as the PI3K pathway is concerned, mutations of the well-known oncogene, PIK3CA, occur far more frequently in HPV(+) HNSCC than in EBV(+) NPC (Table 3; 36% cases vs. 4% cases, respectively). Interestingly, in microdissected NPC, PTEN mutation is the most common event of the PI3K aberrations (5/28 PI3K mutated tumors), followed by mutations of a diverse group of PI3K players, such as PIK3CA (4/28), PIK3C2G (4/28), MTOR (2/28), PIK3R4 (2/28), PIK3AP1 (2/28)., PIK3CB/G (2/28 each), TSC1 (2/28) and RICTOR (2/28). In fact, some of these recurrent mutations have also been identified in another NPC exome study in Asia [PIK3CA (1/56), MTOR (1/56), PIK3C2G (1/56) and PIK3CG (1/56)] [41]. Note that PIK3CA mutations only occur in a small subset of HPV(−) oropharyngeal cancer (9% cases) as compared to HPV(+) HNSCC (36% cases).
Whilst for the NF-κB pathway, CYLD and TRAF3 mutations appear to occur at a similar high rate in both cancers (8–11% rates), followed by the same rate of mutations of NLRC5 (6% rates) (Table 3). This may suggest that CYLD, TRAF3 and NLRC5 aberrations are critical for oncogenesis for a major subset of these cancers. Interestingly, mutations of NFKBIA, an inhibitor of the NF-κB /Rel A complex is only observed in EBV(+) NPC (7/111, 6%), but not in HPV(+) HNSCC. Note that CYLD, TRAF3 and NLRC5 are not found in HPV(–) oropharyngeal cancer (Table 3). In sum, a “favorable NF-κB niche” is perhaps permissive or advantageous for both EBV and HPV-associated cancer cells to grow, survive, and evade the immune surveillance.
Enrichment of PIK3CA mutations in HPV(+) HNSCC was first noted in WES studies [50,52], which was later confirmed by another targeted sequencing study of 51 cases of mostly advanced HPV(+) HNSCC (617 cancer-associated genes [76]). This study also revealed enrichments of MLL3, DDX3X, FGFR2, FGFR3, NOTCH1, NF1, KRAS and FBXW7 mutations in this cancer. Further signaling network analysis indicated a connection with aberrations in DNA damage, FGF/FGFR, JAK/STAT and immune genes (HLA-A, HLA-B) [76]. Interestingly, some of these genes are also found to be altered in EBV(+) NPC, especially those related to antigen presentation. In particular, subsets of EBV(+) NPC tumors do harbor loss-of-function of MHC Class I genes (such as HLA-A, HLA-B, HLA-C, B2M, NLRC5) [43]. It is worth-noting that NPC patients with somatic MHC Class I gene defects do have poorer overall survival and disease-free survival [43]. Further, germline MHC haplotypes, HLA-A2 and HLA-B46, have also been previously shown to be associated with EBV(+) NPC susceptibility in familial and population studies of NPC [77]. These cumulative findings indicate a key role of MHC genes in immune evasion for EBV(+) NPC. Similarly, such an immune evasion mechanism, predominantly offered by MHC class I gene aberrations, is likely to be active in HPV(+) HNSCC as well.z
Although the intricate interactions between the viral genomes and somatic events initiating/ facilitating oncogenesis of these two head and neck cancers are incompletely understood, the central message is becoming much clearer than before. In both cancers, two major signaling pathways are likely critical for, or in support of an inflammatory (NF-κB) and survival-promoting (PI3K) environment for full-bloom oncogenesis.
Another similarity between these two virally associated cancers is that they seem to employ both specific somatic (genetic) and viral events for the achievement of a common goal, i.e. to constitutively activate a critical signaling pathway that is essential for their oncogenesis. This is apparent in EBV(+) NPC, as both high rates of NF-κB pathway mutations and LPM1 overexpression (which also drives NF-κB activation) are found to be existing in mutually exclusive subsets of NPC tumors, totally more than 70% of all NPC cases. These findings do strongly imply that both somatic and viral events are exerting common efforts driving constitutive NF-κB activation during NPC carcinogenesis [43]. Similarly, a very high rate of PI3K pathway mutations (>50–60% cases) in HPV(+) HNSCC clearly implicates the importance of PI3K activation in HPV-mediated HNSCC oncogenesis. In fact, this is supported by previous findings that HPV-16 E7 oncoprotein is a driver for PI3K pathway activation [78], highlighting the biological importance of PI3K activation during HPV-mediated carcinogenesis of the head and neck. Yet, whether PI3K mutations and the expression levels of E7 oncoprotein in HPV(+) HNSCC patient tumors also follow a mutually exclusive relationship (as NF-κB aberrations and LMP1 overexpression in NPC) remains to be examined.
The questions remaining are: Would these pathway activations serve to facilitate viral-mediated oncogenesis that are critical for both EBV(+) NPC and HPV(+) HNSCC? How do the NF-κB defects (e.g. CYLD, TRAF3 aberrations) and the immune system defects (including MHC loss) lead to tumor formation, or immune evasion (e.g., via dysregulation of innate and adaptive immune responses) [79,80]?
4. Limited Genomic Profiles of EBV(+) NPC and HPV(+) HNSCC Recurrences and Metastases
With the understanding of somatic alterations in a reasonable number of primary NPC tumors, there are limited genomic information for recurrent or metastatic NPC. Thus far, there are reported somatic changes of 11 local recurrences and 22 metastatic EBV(+) NPC tumors only [43]. Interestingly, similar predominant mutation signatures were found in both primary and recurrent tumors, suggesting the absence of any new or drastically different etiological factors during NPC recurrences or metastasis. As mutation rate is concerned, TP53 mutations were identified to be >2-fold enriched in recurrent or metastatic tumors (15.2% cases with TP53 mutation) vs. primary tumors (6.4% mutation rate of TP53). This appears to support the notion that NPC clones with genomic instability are selected for during progression to more advanced diseases.
In addition to TP53 mutations, NPC patients bearing PI3K/MAPK pathway-mutated recurrent and metastatic tumors were found to have significantly poorer overall survival than those with WT tumors [43]. These PI3K/MAPK pathway mutations include NRAS, KRAS, PIK3CA, PTEN, NF1, TSC1 and FGFR3. In two of the three pairs of primary NPC and local recurrence samples, RAS activating mutations were shown to be progression drivers. Activations of the MAPK and PI3K-AKT-mTOR signaling are known to control multiple cellular processes and promote resistance to anticancer therapies in human cancers. Therefore, it is plausible that tumors with acquired activating RAS mutations can cause recurrences via resistant mechanisms to chemotherapy and radiotherapy through MAPK pathway activation. The WES data thus far revealed a noticeable frequency of 5% tumors with inactivating PTEN mutations, and 4% cases with PIK3CA hotspot mutations and 20% cases with PIK3CA amplification. Whether these PI3K aberrations can contribute to therapy resistance and subsequent disease recurrences remain unknown. Lastly, recurrent mutations of ABL1, BUB1B, NCOR1, CARS, HSP90AB1, and NCOA1 were uniquely found in recurrent or metastatic NPC tumors, but not in primary NPC. Yet, it remains to be further validated if these unique mutations are determinant of NPC progression or not. Future genomic investigations on larger recurrent and metastatic NPC cohorts will allow more accurate delineation of critical drivers for NPC progression and recurrences for both prognosis and treatment advancement.
When compared to NPC, the genomic landscape of recurrent or metastatic HPV(+) HNSCC is even more obscure. The most comprehensive TCGA cohorts [50] as well as the recently completed larger cohort only represent the landscape of primary tumors, but not that of recurrences or metastasis. Currently, there is no reported comprehensive exome data on recurrent and metastatic HPV(+) HNSCC. This is largely due to the rarity of recurrent HPV(+) HNSCC, as HPV-positivity is associated with favorable outcomes. In short, the recurrent and metastatic genome of these two virally associated head and neck cancers are largely unknown. Yet, it is believed that the ultimate answers for curative treatments do lie partially on these important genomic information, as recurrent and metastatic forms of these tumors are mostly fatal.
5. Implications for Precision Medicine Development
Based on the survival data of a medium size cohort of 111 EBV(+) NPC cases with primary tumors, several major druggable events and genetic biomarkers have emerged [43]. In contrast, the small HPV(+) HNSCC cohort of 36 cases appears to be limited for biomarkers discovery, though some druggable events can be identified.
The genomic data for both cancer types reveal a promising druggable target, the fibroblast growth factor receptor 3 (FGFR3). FGFR3 is found to be mutated or rearranged in 11–14% of HPV(+) HNSCC (4/36 cases) [50,76] vs. 0% of HPV(−) HNSCC (0/243 cases; TCGA), and in 1.8% of EBV(+) NPC (2/111 cases) [81]. In particular, the FGFR3-TACC3 gene rearrangement has been identified in many other tumor types, including cervical cancer (1.3–1.9%; [82]), lung cancer (1.9%; [83]), esophageal cancer (2.1%) [84], lung adenocarcinoma [85], bladder cancer [86], endometrial adenocarcinoma, gall bladder carcinoma, non-small cell lung cancer (NSCLC), pancreatic exocrine carcinoma, renal cell carcinoma [87], etc. Importantly, the druggability of FGFR3 aberrations has been demonstrated in FGFR3-TACC3 rearranged glioma, bladder and cervical cancer cells, which were found to have high sensitivity to FGFR3 inhibitors [88,89,90]. Interesting, the sensitivity of FGFR3-TACC3 rearranged cervical cancer cells towards FGFR3 inhibition (BGJ398) was found to be dependent on the wildtype PI3K status of the cells [82].
In cervical epithelial cell models, expression of the FGFR3-TACC3 fusion results in transformation to squamous cell carcinoma. Similarly, such a fusion gene has oncogenic or transforming activities in glioblastoma [91], and nasopharyngeal cancer [81], as well as in the Ba/F3 cell model [85]. Recent findings in head and neck cancer show that FGFR3-TACC3 fusion is capable of substituting for EGFR signaling in HNSCC models, and confers resistance to EGFR targeting in HNSCC and possibly in other cancers [92]. In fact, subsequent studies showed that such a fusion gene can confer resistance to all three generations of EGFR tyrosine kinase inhibitors in EGFR-mutated NSCLC [93]. It is believed that this fusion protein can substitute EGFR signaling and may act to promote inflammation in the cancer site as well. The clinical efficacy of targeting such as a fusion is currently under investigation.
In addition to FGFR3, other prominent drug targets include: BRCA1/2, SRC, LYN, YES1, ALK, VEGFA, EGFR, FGFR1/2/3/4, ERBB2/3/4, PDGFRA/B, JAK1/2/3, NTRK1/2/3, MET, PIK3CA, TSC1/2, MTOR, H/NRAS, BRAF, MAPK1/3, MAP2K1/2, NF1, NR2C2, PTPRD/T, PDCD1 and CD274. (EBV(+)NPC 36/111, 32%; HPV(+)HNSCC 21/36, 58%). Although these potentially druggable targets are mutated in EBV(+) NPC and HPV(+) HNSCC at notable but different frequencies, their druggabilities in patients remain to be investigated. Preclinical PDXs results are supportive of PIK3CA-mutated HNSCC for PI3K pathway inhibitors [52]. Clinical trials are ongoing to determine the clinical efficacy of PI3K targeting in HNSCC in general, including HPV(+) HNSCC. It still remains to be examined if mutations of other major players of the PI3K pathway (e.g., MTOR, TSC1/2) can also confer sensitivity to PI3K/mTOR inhibitors in HPV(+) HNSCC or not. Overall, there is a sizeable subset of HNSCC patients with receptor-tyrosine kinase (RTK) mutations (e.g., NTRK1/2/3, ERBB1/2/3/4, PDGFRs, MET, etc.) and it remains to be carefully defined if these RTK mutations will cause any drug sensitivity in preclinical models or in patients. Yet, ERBB1 (EGFR) mutations are not likely to contribute to responses in most human cancers besides demonstrated efficacies in NSCLC.
It is important to realize that an integrative genomic-proteomic approach for drug target discovery is gaining practical importance in precision medicine development. Several such studies discovered new links between PTPRT and PTPRD mutations and STAT3 activation in HNSCC patient tumors and preclinical models. These findings strongly imply a potential druggability of PTPRD- and PTPRT-mutant tumors with STAT3 inhibitors [94,95]. Lastly, results are anticipated to see if HRAS mutations can confer sensitivity to the new RAS inhibitor, tipifarinib in clinical trial settings or not.
6. Implications for Immunotherapy
The genomic profiles of both EBV(+) NPC and HPV(+) HNSCC alert us that there are subsets of patients bearing tumors with antigen-presentation defects. In EBV(+) NPC, as high as 28.8% of patients have MHC Class I gene aberrations (Figure 1), and this subgroup of patients was found to have a poorer overall survival with conventional chemotherapies and/or radiation therapies, when compared with the MHC Class I wildtype patients [43]. This finding suggests that an immune component, such as defective antigen presentation, may be one underlying reason for poor survival in NPC. In fact, a recent study by Ma et al. showed that an EBV(+) NPC patient with aggressive recurrence had a complete response to a PD-L1 inhibitor, Nivolumab [96], highlighting the potential impact of an active immune recognition in NPC treatment response and outcome. Similar to findings in most other cancers, the predictive biomarkers behind such a dramatic response to checkpoint inhibitors in EBV(+) NPC is still unknown. Whether MHC defects in NPC affect the efficacies of immunotherapies remain to be examined carefully. Similarly, as high as 22.2% cases of HPV(+) HNSCC harbor MHC Class I gene aberrations (Figure 2). It is likely that HPV(+) HNSCC tumors may adopt this potential MHC defect for immune evasion. Again, the potential impact of these MHC losses on PD-L1 targeting in HPV(+) HNSCC are unknown. As mentioned earlier, both EBV(+) NPC and HPV(+) HNSCC can express viral antigens in addition to tumor neoantigens arising from somatic mutations. Whether these “extra” viral antigens may act like neo-antigens (as in MSI-high tumors), and be able to confer better clinical outcome in patients towards checkpoint inhibitors may worth future investigations.
The role of PDL1 gene rearrangement on head and neck cancer response to PD-L1 targeting remains undefined. Recently, Bellone et al. reported an exceptional pembrolizumab responder with PDL1-rearranged metastatic ovarian cancer [95]. Although a PD-L1-rearranged HPV(+) HNSCC case has recently been reported [97], the ability of such an aberration in conferring pembrolizumab sensitivity to HNSCC remains unclear. A better understanding of responder genetics for immune checkpoint inhibitors in both clinical and preclinical models will facilitate the use of immune checkpoint inhibitors for all head and neck cancers, including both EBV(+) NPC and HPV(+) HNSCC.
7. Conclusions
In conclusion, the completion of WES characterization of a reasonable number of EBV(+) NPC and a small number of HPV(+) HNSCC tumors has already contributed to a big leap to the understanding of these virally associated head and neck cancers. Our next step is taking these genomic data on to identify key driver events for treatment exploration. Functional annotations of these potential drivers and druggable genetic events will greatly facilitate rapid translation of these genomic findings into clinics. Lastly, the cure of these often aggressive cancers may actually lie in future collaborative endeavors to comprehensively characterize the genomics of large and representative cohorts of recurrent and metastatic EBV(+) NPC and HPV(+) HNSCC.
Author Contributions
H.-L.N., L.W., K.-W.L., and V.W.Y.L. contributed to the drafting and editing of this review.
Funding
This research was funded by the Research Grant Council, Hong Kong, grant number 17114814, 17121616, 14168517, 470312, 1404415, 14138016, 14117316, 14104415, and 1413016, Theme-based Research Scheme T12-401/13-R; by Faculty of Medicine, the Chinese University of Hong Kong, direct grant for research number 2016.095, Focused Innovations Scheme and Faculty Strategic Research grant number 4620513; and The Chinese University of Hong Kong, Start-up Fund from the School of Biomedical Sciences, and Core Utilities of Cancer Genome and Pathobiology, and VC’s One-off Discretionary Fund grant number VCF2014017 and VCF2014015; and Hong Kong government (Innovation and Technology Fund) and Lee’s Pharmaceutical (HK) Limited for University Industry Collaboration Program grant number UIM/329.
Acknowledgments
We acknowledge the generation donation of the Hong Kong Cancer Fund to support our research (to V.W.Y.L.).
Conflicts of Interest
V.W.Y.L. receives a research funding (UIM/329) through University Industry Collaboration Program by the Innovation Technology Fund, Hong Kong government and Lee’s Pharmaceutical (HK) Limited. The other authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| EBV | Epstein-Barr virus |
| EBV(+) | Epstein-Barr virus-positive |
| NPC | Nasopharyngeal carcinoma |
| HPV | Human papillomavirus |
| HNC | Head and Neck Cancer |
| HNSCC | Head and neck squamous carcinoma |
| HPV(+) | Human papillomavirus-positive |
| HPV(−) | Human papillomavirus-negative |
| WES | Whole-exome sequencing |
| SCC | squamous cell carcinoma |
| WGS | Whole-genome sequencing |
| CYLD | Cylindromatosis |
| EBERs | Epstein-Barr virus (EBV)-encoded small RNAs |
| LMP1 | Latent-membrane protein 1 |
| PDXs | Patient-derived xenografts |
| NSCLC | Non-small cell lung cancer |
| TRAF3 | TNF receptor associated factor 3 |
| RTK | receptor-tyrosine kinase |
References
- Psyrri, E. Epidemiology, Risk Factors and Pathogenesis of Squamous Cell Tumours. Available online: http://oncologypro.esmo.org/content/download/113133/1971849/file/2017-ESMO-Essentials-for-Clinicians-Head-Neck-Cancers-Chapter-1.pdf (accessed on 14 April 2018).
- Wong, I.C.K.; Ng, Y.K.; Lui, V.W.Y. Cancers of the lung, head and neck on the rise: Perspectives on the genotoxicity of air pollution. Chin. J. Cancer 2014, 33, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, T.R. HPV-associated head and neck cancers: An update. J. Pathol. 2017, 243, S8. [Google Scholar]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the united states. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). Head and Neck Cancers. Available online: https://www.cancer.gov/types/head-and-neck/head-neck-fact-sheet (accessed on 14 April 2018).
- Word Health Organization. World Cancer Report; Word Health Organization: Geneva, Switzerland, 2014; Chapter 5.8. [Google Scholar]
- Maryland, B.; Uniformed Services University of the Health Sciences School of Medicine. Clinical Head and Neck and Functional Neuroscience Course Notes; Uniformed Services University of the Health Sciences School of Medicine: Bethesda, MD, USA, 2008–2009. [Google Scholar]
- Ferlay, J.S.I.; Erivk, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Globocan 212: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. Iarc Cancerbase No. 11; International Agency for Research on Cancer: Lyon, France, 2012. [Google Scholar]
- Bray, F.C.M.; Mery, L.; Piñeros, M.; Znaor, A.; Zanetti, R.; Ferlay, J. Cancer Incidence in Five Continents, Volume XI (Electronic Version); International Agency for Research: Lyon, France, 2017. [Google Scholar]
- Buell, P. The effect of migration on the risk of nasopharyngeal cancer among Chinese. Cancer Res. 1974, 34, 1189–1191. [Google Scholar] [PubMed]
- Yu, M.C.; Yuan, J.M. Epidemiology of nasopharyngeal carcinoma. Semin. Cancer Biol. 2002, 12, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Jeannel, D.; Ghnassia, M.; Hubert, A.; Sanchogarnier, H.; Eschwege, F.; Crognier, E.; Dethe, G. Increased risk of nasopharyngeal carcinoma among males of french origin born in maghreb (North-Africa). Int. J. Cancer 1993, 54, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, A.; Wang, C.P. Genetic predisposition factors and nasopharyngeal carcinoma risk: A review of epidemiological association studies, 2000–2011 rosetta stone for NPC: Genetics, viral infection, and other environmental factors. Semin. Cancer Biol. 2012, 22, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A consensus statement from the international diabetes federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.D.; Chan, L.Y.S.; Chan, A.T.C.; Leung, S.F.; Lo, K.W.; Zhang, J.; Lee, J.C.K.; Hjelm, N.M.; Johnson, P.J.; Huang, D.P. Quantitative and temporal correlation between circulating cell-free Epstein-Barr virus DNA and tumor recurrence in nasopharyngeal carcinoma. Cancer Res. 1999, 59, 5452–5455. [Google Scholar] [PubMed]
- Ma, B.B.Y.; Hui, E.P.; Chan, A.T.C. Investigational drugs for nasopharyngeal carcinoma. Expert Opin. Investig. Drugs 2017, 26, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.Y.; Lim, W.T.; Goh, B.C.; Hui, E.P.; Lo, K.W.; Pettinger, A.; Foster, N.R.; Riess, J.W.; Agulnik, M.; Chang, A.Y.C.; et al. Antitumor activity of nivolumab in recurrent and metastatic nasopharyngeal carcinoma: An international, multicenter study of the mayo clinic phase 2 consortium (NCI-9742). J. Clin. Oncol. 2018, 36, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Lee, S.H.; Ejadi, S.; Even, C.; Cohen, R.B.; Le Tourneau, C.; Mehnert, J.M.; Algazi, A.; van Brummelen, E.M.J.; Saraf, S.; et al. Safety and antitumor activity of pembrolizumab in patients with programmed death-ligand 1-positive nasopharyngeal carcinoma: Results of the keynote-028 study. J. Clin. Oncol. 2017, 35, 4050–4056. [Google Scholar] [CrossRef] [PubMed]
- Paz, I.B.; Cook, N.; Odom-Maryon, T.; Xie, Y.; Wilczynski, S.P. Human papillomavirus (HPV) in head and neck cancer. An association of hpv 16 with squamous cell carcinoma of waldeyer’s tonsillar ring. Cancer 1997, 79, 595–604. [Google Scholar] [CrossRef]
- Hobbs, C.G.; Sterne, J.A.; Bailey, M.; Heyderman, R.S.; Birchall, M.A.; Thomas, S.J. Human papillomavirus and head and neck cancer: A systematic review and meta-analysis. Clin. Otolaryngol. 2006, 31, 259–266. [Google Scholar] [CrossRef] [PubMed]
- CDC. HPV and Oropharyngeal Cancer. Available online: https://www.cdc.gov/cancer/hpv/basic_info/hpv_oropharyngeal.htm (accessed on 14 April 2018).
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, H.; Beech, T.; Nicholson, T.; El-Hariry, I.; McConkey, C.; Paleri, V.; Roberts, S. The prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer: Systematic review and meta-analysis of trends by time and region. Head Neck 2013, 35, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, J.Y.; Lim, S.H.; Park, K.; Sun, J.M.; Ko, Y.H.; Baek, C.H.; Son, Y.I.; Jeong, H.S.; Ahn, Y.C.; et al. Association between pd-l1 and hpv status and the prognostic value of pd-l1 in oropharyngeal squamous cell carcinoma. Cancer Res. Treat 2016, 48, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.; Chan, J.Y.; Chan, A.B.; Ng, C.S.; Lo, S.T.; Lam, V.S.; Chan, M.M.; Ngai, C.M.; Vlantis, A.C.; Ma, R.K.; et al. Prevalence, clinicopathological characteristics, and outcome of human papillomavirus-associated oropharyngeal cancer in southern chinese patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Ruzevick, J.; Olivi, A.; Westra, W.H. Metastatic squamous cell carcinoma to the brain: An unrecognized pattern of distant spread in patients with hpv-related head and neck cancer. J. Neurooncol. 2013, 112, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Nuyts, S. HPV positive head and neck cancers: Molecular pathogenesis and evolving treatment strategies. Cancers (Basel) 2016, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Rusan, M.L.Y.; Hammerman, P.S. Genomic landscape of human papillomavirus-associated cancers. Clin. Cancer Res. 2015, 21, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Swenson, R. The Pharynx and Larynx. Available online: https://www.dartmouth.edu/~humananatomy/part_8/chapter_53.html (accessed on 14 April 2018).
- Pathmanathan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of hpv integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Tahtali, A.; Hey, C.; Geissler, C.; Filman, N.; Diensthuber, M.; Leinung, M.; Stover, T.; Wagenblast, J. HPV status and overall survival of patients with oropharyngeal squamous cell carcinoma—A retrospective study of a german head and neck cancer center. Anticancer Res. 2013, 33, 3481–3485. [Google Scholar] [PubMed]
- Trosman, S.J.; Koyfman, S.A.; Ward, M.C.; Al-Khudari, S.; Nwizu, T.; Greskovich, J.F.; Lamarre, E.D.; Scharpf, J.; Khan, M.J.; Lorenz, R.R.; et al. Effect of human papillomavirus on patterns of distant metastatic failure in oropharyngeal squamous cell carcinoma treated with chemoradiotherapy. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Liu, Z.Y.; Myers, J.N. TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N. Epstein-Barr virus and nasopharyngeal carcinoma. Semin. Cancer Biol. 1992, 3, 297–307. [Google Scholar] [PubMed]
- Lin, J.C.; Wang, W.Y.; Chen, K.Y.; Wei, Y.H.; Liang, W.M.; Jan, J.S.; Jiang, R.S. Quantification of plasma Epstein-Barr virus DNA in patients with advanced nasopharyngeal carcinoma. N. Engl. J. Med. 2004, 350, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.C.A.; Woo, J.K.S.; King, A.; Zee, B.C.Y.; Lam, W.K.J.; Chan, S.L.; Chu, S.W.I.; Mak, C.; Tse, I.O.L.; Leung, S.Y.M.; et al. Analysis of plasma Epstein-Barr virus DNA to screen for nasopharyngeal cancer. N. Engl. J. Med. 2017, 377, 513–522. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Dempsey, A. The role of HPV in head and neck cancer and review of the HPV vaccine. Prev. Med. 2011, 53, S5–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capone, R.B.; Pai, S.I.; Koch, W.M.; Gillison, M.L.; Danish, H.N.; Westra, W.H.; Daniel, R.; Shah, K.V.; Sidransky, D. Detection and quantitation of human papillomavirus (HPV) DNA in the sera of patients with HPV-associated head and neck squamous cell carcinoma. Clin. Cancer Res. 2000, 6, 4171–4175. [Google Scholar] [PubMed]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zheng, H.; Cheung, A.K.; Tang, C.S.; Ko, J.M.; Wong, B.W.; Leong, M.M.; Sham, P.C.; Cheung, F.; Kwong, D.L.; et al. Whole-exome sequencing identifies mst1r as a genetic susceptibility gene in nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 3317–3322. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; Woo, J.K.; Yip, K.Y.; Seo, J.; Hui, E.P.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-κB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; MacIsaac, K.D.; Zhou, T.; Huang, P.Y.; Xin, C.L.; Dobson, J.R.; Yu, K.; Chiang, D.Y.; Fan, Y.; Pelletier, M.; et al. Genomic analysis of nasopharyngeal carcinoma reveals TME-based subtypes. Mol. Cancer Res. 2017, 15, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Chow, Y.P.; Tan, L.P.; Chai, S.J.; Aziz, N.A.; Choo, S.W.; Lim, P.V.H.; Pathmanathan, R.; Kornain, N.K.M.; Lum, C.L.; Pua, K.C.; et al. Exome sequencing identifies potentially druggable mutations in nasopharyngeal carcinoma. Sci. Rep. UK 2017, 7, 42980. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- India Project Team of the International Cancer Genome Cell. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently mutated genes and molecular subgroups. Nat. Commun. 2013, 4, 2873. [Google Scholar]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of hpv and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [Green Version]
- Hedberg, M.L.; Goh, G.; Chiosea, S.I.; Bauman, J.E.; Freilino, M.L.; Zeng, Y.; Wang, L.; Diergaarde, B.B.; Gooding, W.E.; Lui, V.W.; et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Investig. 2016, 126, 1606. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.Y.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.L.; Zhang, Q.H.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Barzi, A.; Brezden-Masley, C.; Puccini, A.; Berger, M.D.; Tokunaga, R.; Battaglin, F.; Soni, S.; McSkane, M.; Zhang, W.; et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat. Rev. 2018, 66, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Scheffner, M.; Huibregtse, J.M.; Howley, P.M. Interactions of HPV E6 and E7 oncoproteins with tumor suppressor gene-products. Cancer Surv. 1992, 12, 197–217. [Google Scholar] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. Apobec-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- COSMIC. Signatures of Mutational Processes in Human Cancer. Available online: https://cancer.sanger.ac.uk/cosmic/signatures (accessed on 14 April 2018).
- Chen, L.; Qiu, X.; Zhang, N.; Wang, Y.; Wang, M.; Li, D.; Wang, L.; Du, Y. Apobec-mediated genomic alterations link immunity and viral infection during human papillomavirus-driven cervical carcinogenesis. Biosci. Trends 2017, 11, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Amal, M.; Marwa, A.; Hussein, H.; Samar, K. Retraction note: Human papillomavirus-16 (HPV-16) infection association with CIAP-2 expression in head and neck cancer. Med. Oncol. 2012, 29, 2459–2465. [Google Scholar]
- Verhoeft, K.R.; Ngan, H.L.; Lui, V.W. The cylindromatosis (CYLD) gene and head and neck tumorigenesis. Cancers Head Neck 2016, 1, 10. [Google Scholar] [CrossRef]
- Eguether, T.; Ermolaeva, M.A.; Zhao, Y.; Bonnet, M.C.; Jain, A.; Pasparakis, M.; Courtois, G.; Tassin, A.M. The deubiquitinating enzyme CYLD controls apical docking of basal bodies in ciliated epithelial cells. Nat. Commun. 2014, 5, 4585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajek, M.; Sewell, A.; Kaech, S.; Burtness, B.; Yarbrough, W.G.; Issaeva, N. TRAF3/CYLD mutations identify a distinct subset of human papillomavirus-associated head and neck squamous cell carcinoma. Cancer 2017, 123, 1778–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shair, K.H.Y.; Reddy, A.; Cooper, V.S. New insights from elucidating the role of LMP1 in nasopharyngeal carcinoma. Cancers (Basel) 2018, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Duan, Y.M.; Cheng, S.Y.; Chen, Y.; Hu, Y.X.; Zhang, L.; He, J.; Liao, Q.; Yang, L.F.; Sun, L.Q. EBV-encoded RNA via TLR3 induces inflammation in nasopharyngeal carcinoma. Oncotarget 2015, 6, 24291–24303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low and High-Oncogenic Risk Human Papillomaviruses: Every Rule has its Exception. Available online: http://www.dst.uff.br/revista23-4-2011/3.EDITORIAL-INGLES.pdf (accessed on 14 April 2018).
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.J.L.M.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Lechner, M.; Frampton, G.M.; Fenton, T.; Feber, A.; Palmer, G.; Jay, A.; Pillay, N.; Forster, M.; Cronin, M.T.; Lipson, D.; et al. Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV plus and HPV-tumors. Genome Med. 2013, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, J.H.; de Leng, W.W.J.; de Bree, R.; van Es, R.J.J.; Willems, S.M. Targeted sequencing reveals tp53 as a potential diagnostic biomarker in the post-treatment surveillance of head and neck cancer. Oncotarget 2016, 7, 61575–61586. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Guthrie, V.B.; Masica, D.L.; Tokheim, C.; Kang, H.; Richmon, J.; Agrawal, N.; Fakhry, C.; Quon, H.; Subramaniam, R.M.; et al. Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Ann. Oncol. 2015, 26, 1216–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zevallos, J.P.; Yim, E.; Brennan, P.; Liu, A.Y.; Taylor, J.M.; Weissler, M.; Anantharaman, D.; Abedi-Ardekani, B.; Olshan, A.F.; Hayes, N.N. Molecular profile of human papillomavirus-positive oropharyngeal squamous cell carcinoma stratified by smoking status. Int. J. Radiat. Oncol. 2016, 94, 864. [Google Scholar] [CrossRef]
- Bellone, S.; Buza, N.; Choi, J.; Zammataro, L.; Gay, L.; Elvin, J.A.; Rimm, D.L.; Liu, Y.; Ratner, E.; Schwartz, P.E.; et al. Exceptional response to pembrolizumab in a metastatic, chemotherapy/radiation resistant ovarian cancer patient harboring a CD274/PD-L1-genetic rearrangement. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.K.S.; Li, J.; Jeong, K.J.; Shao, S.; Chen, H.; Tsang, Y.H.; Sengupta, S.; Wang, Z.X.; Bhavana, V.H.; Tran, R.; et al. Systematic functional annotation of somatic mutations in cancer. Cancer Cell 2018, 33, 450. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.H.; Kang, T.H.; Kim, J.H.; Pai, S.I.; Lin, K.Y.; Hung, C.F.; Wu, T.C.; Kim, T.W. Activation of akt as a mechanism for tumor immune evasion. Mol. Ther. 2009, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Hartmann, S.; Bhola, N.E.; Li, H.; Zeng, Y.; O’Keefe, R.A.; Ranall, M.V.; Bandyopadhyay, S.; Soucheray, M.; Krogan, N.J.; et al. Crosstalk signaling between HER3 and HPV16 E6 and E7 mediates resistance to PI3K inhibitors in head and neck cancer. Cancer Res. 2018, 78, 2383–2395. [Google Scholar] [CrossRef] [PubMed]
- Pollock, N.I.; Grandis, J.R. HER2 as a therapeutic target in head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Hartmann, S.F.; Bhola, N.E.; Peyser, N.D.; Li, H.; Zeng, Y.; Wechsler, E.I.; Ranall, M.V.; Bandyopadhyay, S.; Duvvuri, U.; et al. Human papillomavirus regulates HER3 expression in head and neck cancer: Implications for targeted HER3 therapy in HPV+ patients. Clin. Cancer Res. 2017, 23, 3072–3083. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Zuo, Z.X.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fasano, R.; Wang, E.; Yao, K.T.; Marincola, F.M. HLA associations with nasopharyngeal carcinoma. Curr. Mol. Med. 2009, 9, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase b pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1247. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, Y.Y.; Lim, J.Y.; Seo, G.J.; Kim, J.; Park, S.I.; Park, B.J. Opposite role of ras in tumor necrosis factor-α-induced cell cycle regulation: Competition for RAF kinase. Biochem. Biophys. Res. Commun. 2001, 287, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Liu, Z.H.; Lin, Z.R.; Xu, L.H.; Zhong, Q.; Zeng, M.S. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol. Ther. 2014, 15, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, R.; Yoshihara, K.; Saito, T.; Ishimura, R.; Martinez-Ledesma, J.E.; Xin, H.; Ishiguro, T.; Mori, Y.; Yamawaki, K.; Suda, K.; et al. Novel therapeutic strategy for cervical cancer harboring FGFR3-TACC3 fusions. Oncogenesis 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewski, I.J.; Mittempergher, L.; Davidson, N.M.; Bosma, A.; Willems, S.M.; Horlings, H.M.; de Rink, I.; Greger, L.; Hooijer, G.K.J.; Peters, D.; et al. Identification of recurrent FGFR3 fusion genes in lung cancer through kinome-centred rna sequencing. J. Pathol. 2013, 230, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.X.; Zhong, Q.; Liu, Y.; Yan, S.M.; Chen, Z.H.; Jin, S.Z.; Xia, T.L.; Li, R.Y.; Zhou, A.J.; Su, Z.; et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multi-region whole-exome sequencing. Nat. Commun. 2017, 8, 524. [Google Scholar] [CrossRef] [PubMed]
- Capelletti, M.; Dodge, M.E.; Ercan, D.; Hammerman, P.S.; Park, S.I.; Kim, J.; Sasaki, H.; Jablons, D.M.; Lipson, D.; Young, L.; et al. Identification of recurrent FGFR3-TACC3 fusion oncogenes from lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 6551–6558. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Carneiro, B.A.; Taxter, T.; Tavora, F.A.; Kalyan, A.; Pai, S.A.; Chae, Y.K.; Giles, F.J. FGFR3-TACC3 fusion in solid tumors: Mini review. Oncotarget 2016, 7, 55924–55938. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Annala, M.J.; Cogdell, D.E.; Granberg, K.J.; Sun, Y.; Ji, P.; Li, X.; Gumin, J.; Zheng, H.; Hu, L.; et al. The tumorigenic FGFR3-TACC3 gene fusion escapes MIR-99A regulation in glioblastoma. J. Clin. Investig. 2013, 123, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Lamont, F.R.; Tomlinson, D.C.; Cooper, P.A.; Shnyder, S.D.; Chester, J.D.; Knowles, M.A. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br. J. Cancer 2011, 104, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Castanaro, C.; Zhang, W.; Zhang, Q.; Wei, Y.; Ni, M.; Young, T.M.; Zhang, L.; Burova, E.; Thurston, G. FGFR3-TACC3 fusion proteins ACT as naturally occurring drivers of tumor resistance by functionally substituting for EGFR/ERK signaling. Oncogene 2017, 36, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.I.; Horn, L.; Cruz, M.; Vafai, D.; Lovly, C.M.; Spradlin, A.; Williamson, M.J.; Dagogo-Jack, I.; Johnson, A.; Miller, V.A.; et al. Emergence of FGFR3-TACC3 fusions as a potential by-pass resistance mechanism to egfr tyrosine kinase inhibitors in EGFR mutated NSCLC patients. Lung Cancer 2017, 111, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.Y.; Peyser, N.D.; Ng, P.K.S.; Hritz, J.; Zeng, Y.; Lu, Y.L.; Li, H.; Wang, L.; Gilbert, B.R.; General, I.J.; et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyser, N.D.; Du, Y.; Li, H.; Lui, V.; Xiao, X.; Chan, T.A.; Grandis, J.R. Loss-of-function PTPRD mutations lead to increased STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. PLoS ONE 2015, 10, e0135750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.D.; Xu, X.Q.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS. Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.C.; Xu, C.; Mitchell, R.M.; Zhang, B.; Zhao, D.; Li, Y.; Huang, X.; Fan, W.H.; Wang, H.W.; Lerma, L.A.; et al. Tumor evolution and intratumor heterogeneity of an oropharyngeal squamous cell carcinoma revealed by whole-genome sequencing. Neoplasia 2013, 15, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Bar graphs showing percentage of tumors harboring somatic mutations of 6 major signaling pathways (NF-κB, PI3K, JAK/STAT, MAPK, NOTCH, WNT), and of the MHC Class I, and MHC Class II genes in (a) EBV(+) NPC cohort (n = 111), (b) HPV(+) HNSCC cohort (n = 36; TCGA) and (c) HPV(−) oropharyngeal cancer cohort (n = 11; TCGA). The NF-κB pathway is defined as TAB1/2/3, MAP3K7/14, CHUK, IKBKB, IKBKG, NFKBIA, NFKBIE, REL, RELA/B, NFKB1/2, LTBR, TNF, TNFAIP3, TNFSF11/13B, TNFRSF1A/8/11A/13C, BTRC, CYLD, NLRC5, TRADD, CD40, CD40LG, LTA, TRAF2/3/5/6, IL1B and IL1R1. The PI3K pathway is defined as AKT1/2/3, PIK3CA/B/D/G/2A/2B/2G, PIK3AP1, PIK3IP1, PDK1, MTOR, TSC1, TSC2, PTEN, RICTOR, RPTOR, RHEB and PIK3R1/2/3/4/5/6. The JAK/STAT pathway is defined as JAK1/2/3, STAT1/2/3/4/5A/5B/6, PTPN11, IL6, IL6R, IL6ST and SOCS3. The MAPK pathway is defined as SHC1/2/3, GRB2, H/N/KRAS, A/BRAF, RAF1, MAP2K1/2, MAPK1/3, RPS6KA1 and DUSP1/2/3/4/5/6/7/9. The NOTCH pathway is defined as DLL1/3/4, JAG1/2, NOTCH1/2/3/4, NUMB, DTX1/3L, NEDD4, MAML1, RBPJ, POFUT1, HES1/5 and HEY1/2/L. The WNT pathway is defined as WNT1/3A/5A/5B/7A, CTNNB1, HNF1A, FZD1/2/3/7/8/9/10, AXIN1, LEF1, LOXL2, DVL2/3, NKD1/2, TAB1/2, GSK3B, CSNK1A1, NLK and LRP5/6. The MHC Class I genes are defined as HLA-A/B/C/E/F/G/H/K/L/J, B2M and NLRC5. The MHC Class II genes are defined as HLA-DMA/B, HLA-DOA/B, HLA-DPA1/A2/B1/B2, HLA-DQA1/A2/B1/B2/B3 and HLA-DRA/B1/B2/B3/B4/B5/B9.
Figure 1.
Bar graphs showing percentage of tumors harboring somatic mutations of 6 major signaling pathways (NF-κB, PI3K, JAK/STAT, MAPK, NOTCH, WNT), and of the MHC Class I, and MHC Class II genes in (a) EBV(+) NPC cohort (n = 111), (b) HPV(+) HNSCC cohort (n = 36; TCGA) and (c) HPV(−) oropharyngeal cancer cohort (n = 11; TCGA). The NF-κB pathway is defined as TAB1/2/3, MAP3K7/14, CHUK, IKBKB, IKBKG, NFKBIA, NFKBIE, REL, RELA/B, NFKB1/2, LTBR, TNF, TNFAIP3, TNFSF11/13B, TNFRSF1A/8/11A/13C, BTRC, CYLD, NLRC5, TRADD, CD40, CD40LG, LTA, TRAF2/3/5/6, IL1B and IL1R1. The PI3K pathway is defined as AKT1/2/3, PIK3CA/B/D/G/2A/2B/2G, PIK3AP1, PIK3IP1, PDK1, MTOR, TSC1, TSC2, PTEN, RICTOR, RPTOR, RHEB and PIK3R1/2/3/4/5/6. The JAK/STAT pathway is defined as JAK1/2/3, STAT1/2/3/4/5A/5B/6, PTPN11, IL6, IL6R, IL6ST and SOCS3. The MAPK pathway is defined as SHC1/2/3, GRB2, H/N/KRAS, A/BRAF, RAF1, MAP2K1/2, MAPK1/3, RPS6KA1 and DUSP1/2/3/4/5/6/7/9. The NOTCH pathway is defined as DLL1/3/4, JAG1/2, NOTCH1/2/3/4, NUMB, DTX1/3L, NEDD4, MAML1, RBPJ, POFUT1, HES1/5 and HEY1/2/L. The WNT pathway is defined as WNT1/3A/5A/5B/7A, CTNNB1, HNF1A, FZD1/2/3/7/8/9/10, AXIN1, LEF1, LOXL2, DVL2/3, NKD1/2, TAB1/2, GSK3B, CSNK1A1, NLK and LRP5/6. The MHC Class I genes are defined as HLA-A/B/C/E/F/G/H/K/L/J, B2M and NLRC5. The MHC Class II genes are defined as HLA-DMA/B, HLA-DOA/B, HLA-DPA1/A2/B1/B2, HLA-DQA1/A2/B1/B2/B3 and HLA-DRA/B1/B2/B3/B4/B5/B9.

Figure 2.
Figure showing percentage of tumors harboring at least one targetable event in (a) EBV(+) NPC cohort (n = 111) and (b) HPV(+) HNSCC cohorts (n = 36; TCGA). The targetable genes include: BRCA1/2, SRC, LYN, YES1, ALK, VEGFA, EGFR, FGFR1/2/3/4, ERBB2/3/4, PDGFRA/B, JAK1/2/3, NTRK1/2/3, MET, PIK3CA, TSC1/2, MTOR, H/NRAS, BRAF, MAPK1/3, MAP2K1/2, NF1, NR2C2, PTPRD/T, PDCD1 and CD274 (PDCD1LG1). The percent cases with targetable genes mutation in each cohort are shown below each pie chart.
Figure 2.
Figure showing percentage of tumors harboring at least one targetable event in (a) EBV(+) NPC cohort (n = 111) and (b) HPV(+) HNSCC cohorts (n = 36; TCGA). The targetable genes include: BRCA1/2, SRC, LYN, YES1, ALK, VEGFA, EGFR, FGFR1/2/3/4, ERBB2/3/4, PDGFRA/B, JAK1/2/3, NTRK1/2/3, MET, PIK3CA, TSC1/2, MTOR, H/NRAS, BRAF, MAPK1/3, MAP2K1/2, NF1, NR2C2, PTPRD/T, PDCD1 and CD274 (PDCD1LG1). The percent cases with targetable genes mutation in each cohort are shown below each pie chart.


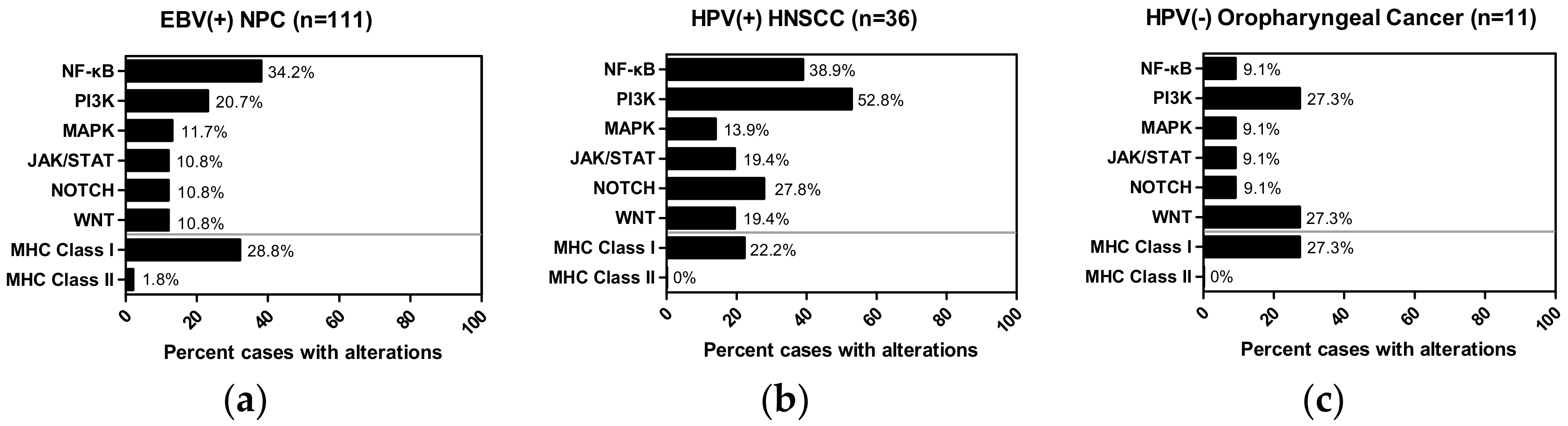{kind=link}
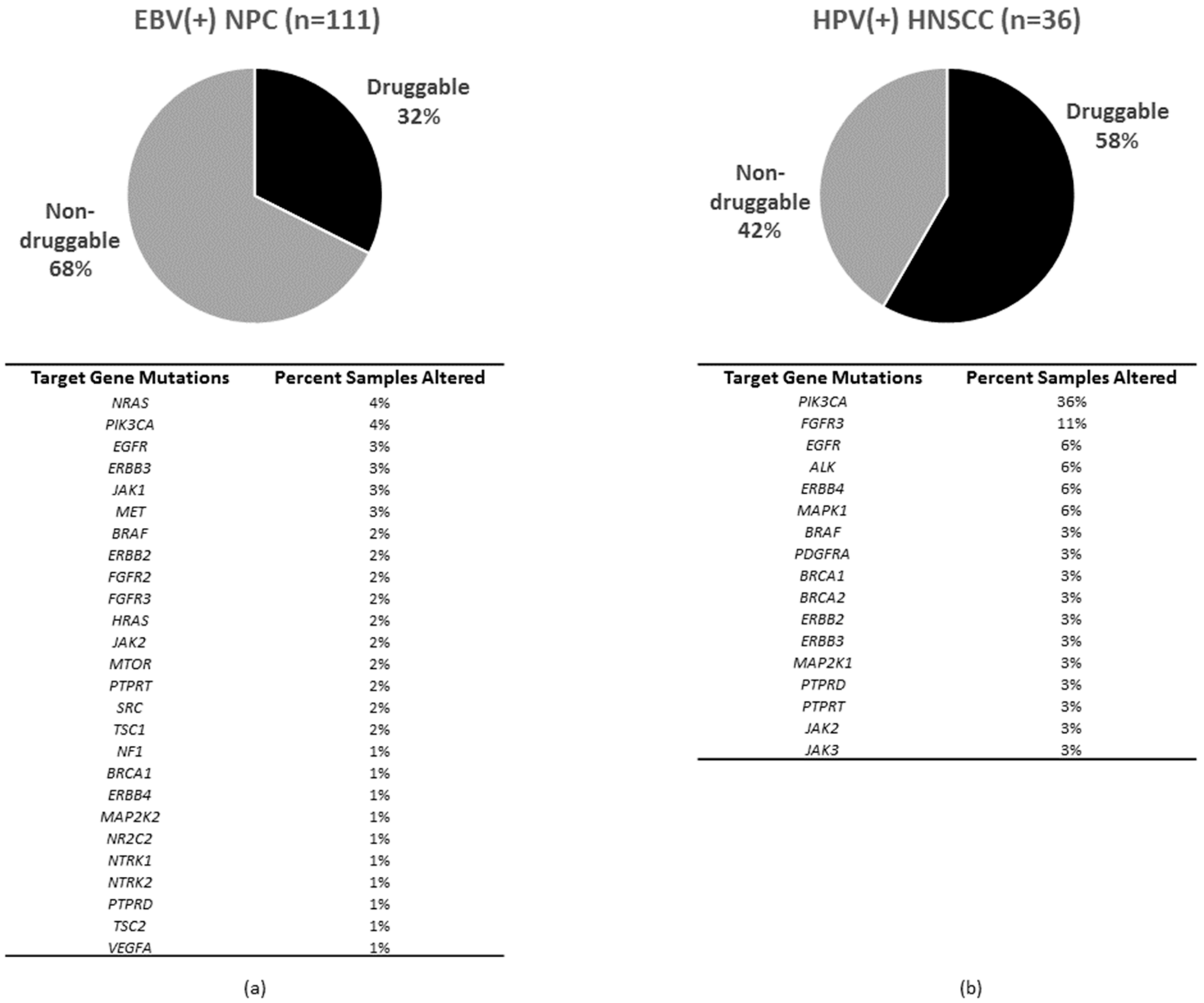{kind=link}
Table 1.
Similarities and differences between EBV(+) NPC and HPV(+) HNSCC.
| Features | EBV(+) NPC | HPV(+) HNSCC |
|---|---|---|
| Site | Nasopharynx | Mainly orapharynx |
| Onset age | Early young (~45) | Early young (~40–45) |
| Oncovirus infection | Herpesvirus, EBV (~172 kb), clonal, episomal | Human papillomavirus, HPV16(~7–8 kb), 50–70% intergraded, 30–50% episomal |
| Tumor size | Small | Small |
| Major tumor cell type | Most non-keratinizing squamous cell carcinoma | Squamous cell carcinoma (SCC) |
| Clinical outcome | Poor at advanced stages | Favorable |
| Treatment sensitivity | Sensitive | Sensitive |
EBV(+) NPC, Epstein-Barr virus-positive nasopharyngeal carcinoma; HPV(+) HNSCC, Human papillomavirus-positive head and neck squamous cell carcinoma.
Table 2.
WES or WGS studies for NPC and HNSCC.
| Publications | Ethnicity | WES or WGS | |
|---|---|---|---|
| NPC | Lin, et al. [41] | Asian (n = 61) | 56 tumors and 5 cell lines (WES) |
| Zheng H, et al. [42] | Asian (n = 59) | 51 tumors and 8 recurrent tumors (WES) | |
| Li, et al. [43] | Asian (n = 97) Caucasian (n = 8) | 111 micro-dissected EBV(+) NPCs from 105 unique subjects (WES, and 15 NPCs with WGS) | |
| Li Zhang, et al. [44] | Asian (n = 111) | 111 EBV(+) NPCs (WES) | |
| Yock Ping Chow, et al. [45] | Asian (n = 10) | 10 tumors | |
| HNSCC | Nicolas Stransky, et al. [46] | Caucasian (n = 92) | 92 tumors (7X WES with 14 HPV(+) HNSCC, 2 tumors with WGS) |
| Nishant Agrawal, et al. [47] | Caucasian (n = 32) | 32 tumors (WES; 4 HPV(+) HNSCC) | |
| India Project Team of the International Cancer Genome Consortium [48] | Asian (n = 50) | 50 tumors (WES; 13 HPV(+) HNSCC) | |
| Micheal Parfenov, et al. [49] | Caucasian (n = 150) | 150 tumors (WES; WGS, 35 HPV(+) HNSCC) | |
| TCGA [50] | Caucasian (n = 279) | 279 tumors (WES; 36 HPV(+) HNSCC) | |
| Matthew L. Hedberg, et al. [51] | Caucasian (n = 23) | 23 tumors (WES; 1 HPV(+) HNSCC) | |
| TCGA (provisional) | Caucasian (n = 530) | 530 tumors (WES) |
Whole-exome sequencing (WES) or whole-genome sequencing (WGS).
Table 3.
Tables showing the percentage of tumor with somatic mutations in the most affected NF-κB pathway and PI3K pathway in EBV(+) NPC, HPV(+) HNSCC and HPV(−) oropharyngeal cancer, respectively.
Table 3.
Tables showing the percentage of tumor with somatic mutations in the most affected NF-κB pathway and PI3K pathway in EBV(+) NPC, HPV(+) HNSCC and HPV(−) oropharyngeal cancer, respectively.
| NF-κB Pathway Aberrations | PI3K Pathway Aberrations | ||||||
|---|---|---|---|---|---|---|---|
| Gene | EBV(+) NPC (n = 111) | HPV(+) HNSCC (n = 36) | HPV(−) Oropharyngeal Cancer (n = 11) | Gene | EBV(+) NPC (n = 111) | HPV(+) HNSCC (n = 36) | HPV(−) Oropharyngeal Cancer (n = 11) |
| CYLD | 10% | 11% | 0% | PTEN | 5% | 3% | 0% |
| TRAF3 | 8% | 11% | 0% | PIK3C2G | 4% | 3% | 0% |
| NLRC5 | 6% | 6% | 0% | PIK3CA | 4% | 36% | 9% |
| NFKBIA | 6% | 0% | 0% | MTOR | 2% | 0% | 0% |
| CHUK | 2% | 0% | 0% | PIK3AP1 | 2% | 0% | 9% |
| DTL | 2% | 0% | 0% | PIK3CB | 2% | 3% | 0% |
| IL1B | 2% | 0% | 0% | PIK3CG | 2% | 3% | 0% |
| BTRC | 1% | 0% | 0% | PIK3R4 | 2% | 0% | 0% |
| CD40LG | 1% | 0% | 0% | RICTOR | 2% | 3% | 0% |
| IKBKG | 1% | 0% | 0% | TSC1 | 2% | 0% | 0% |
| IL1R1 | 1% | 3% | 0% | AKT2 | 1% | 0% | 0% |
| LTBR | 1% | 3% | 0% | PIK3C2A | 1% | 0% | 0% |
| NFKB2 | 1% | 0% | 0% | PIK3CD | 1% | 3% | 0% |
| REL | 1% | 0% | 0% | PIK3R1 | 1% | 3% | 0% |
| RELA | 1% | 0% | 0% | PIK3R5 | 1% | 3% | 0% |
| TAB1 | 1% | 6% | 0% | RPTOR | 1% | 3% | 9% |
| TAB3 | 1% | 3% | 0% | TSC2 | 1% | 0% | 0% |
| TNF | 1% | 0% | 0% | AKT1 | 0% | 3% | 0% |
| TNFAIP3 | 1% | 3% | 0% | PDK1 | 0% | 3% | 0% |
| TNFRSF1A | 1% | 0% | 0% | PIK3R3 | 0% | 3% | 9% |
| TNFRSF13C | 0% | 3% | 0% | PIK3R6 | 0% | 3% | 0% |
| TRAF5 | 0% | 3% | 0% | ||||
| MAP3K7 | 0% | 3% | 0% | ||||
| TNFRSF13B | 0% | 0% | 9% | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ngan, H.-L.; Wang, L.; Lo, K.-W.; Lui, V.W.Y. Genomic Landscapes of EBV-Associated Nasopharyngeal Carcinoma vs. HPV-Associated Head and Neck Cancer. Cancers 2018, 10, 210. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10070210
AMA Style
Ngan H-L, Wang L, Lo K-W, Lui VWY. Genomic Landscapes of EBV-Associated Nasopharyngeal Carcinoma vs. HPV-Associated Head and Neck Cancer. Cancers. 2018; 10(7):210. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10070210
Chicago/Turabian StyleNgan, Hoi-Lam, Lan Wang, Kwok-Wai Lo, and Vivian Wai Yan Lui. 2018. "Genomic Landscapes of EBV-Associated Nasopharyngeal Carcinoma vs. HPV-Associated Head and Neck Cancer" Cancers 10, no. 7: 210. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10070210
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.