TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure

 , , , and
, , , and
Abstract
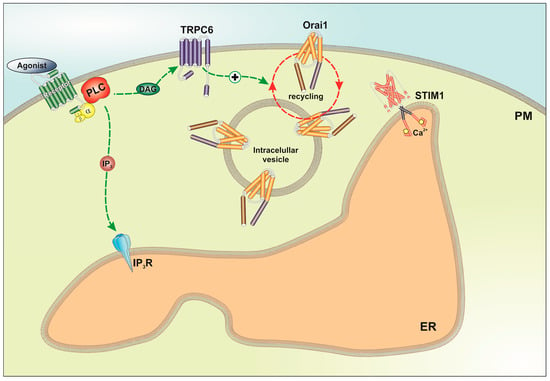
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
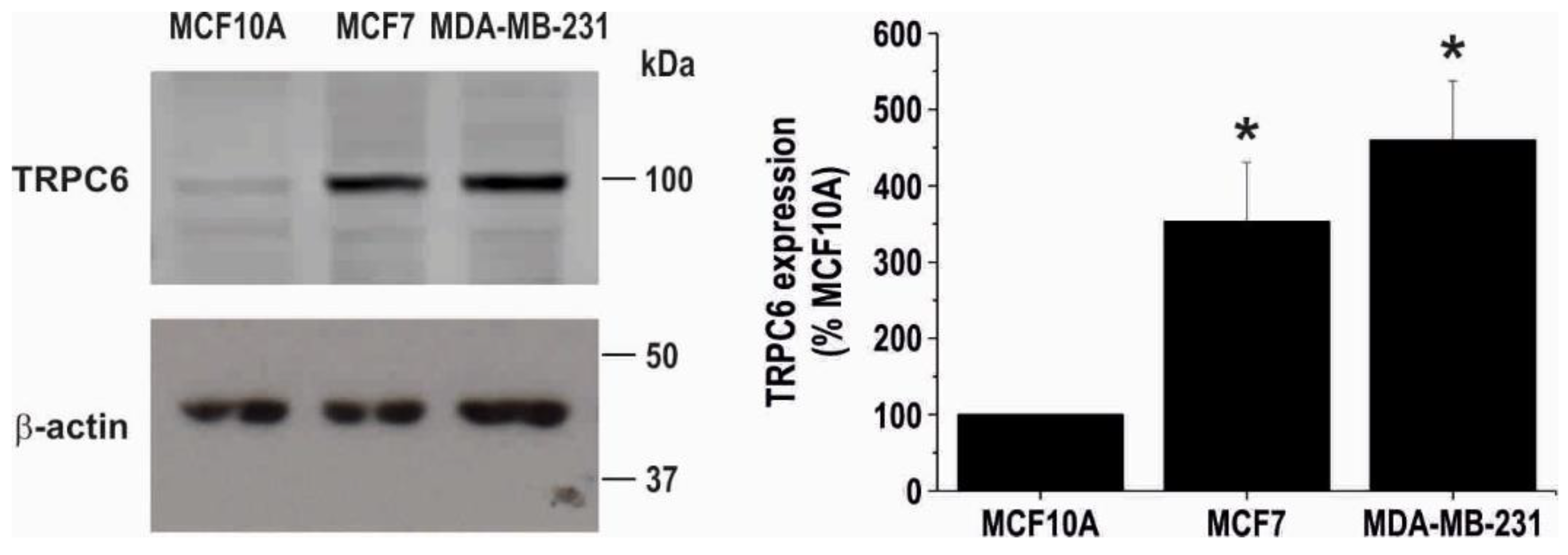
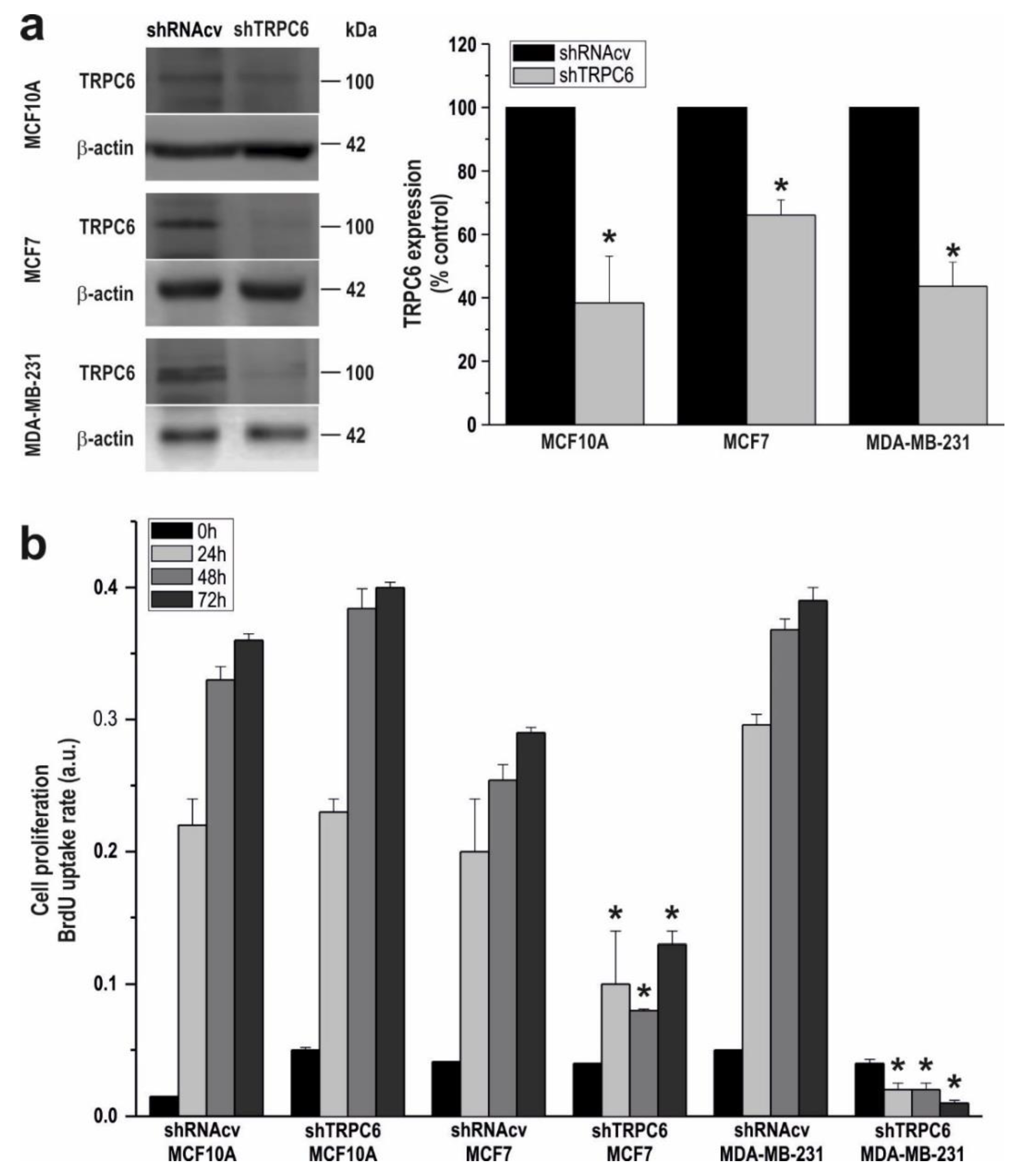
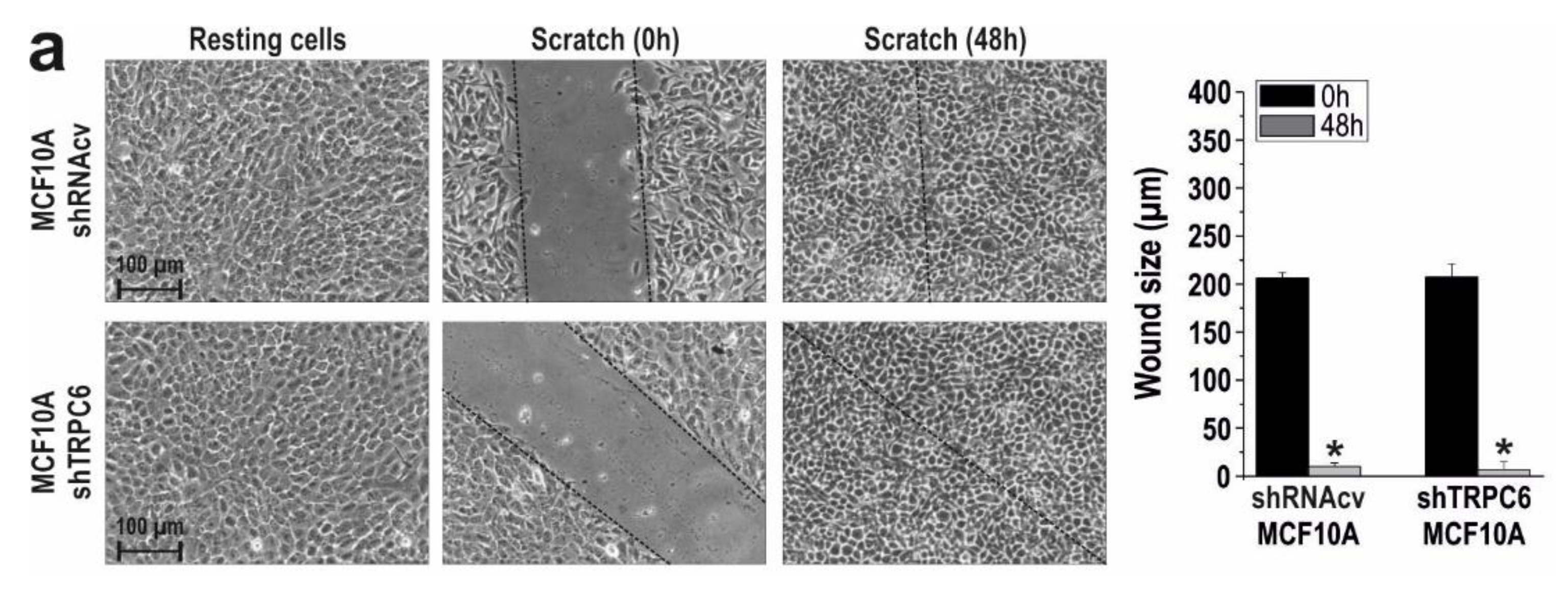
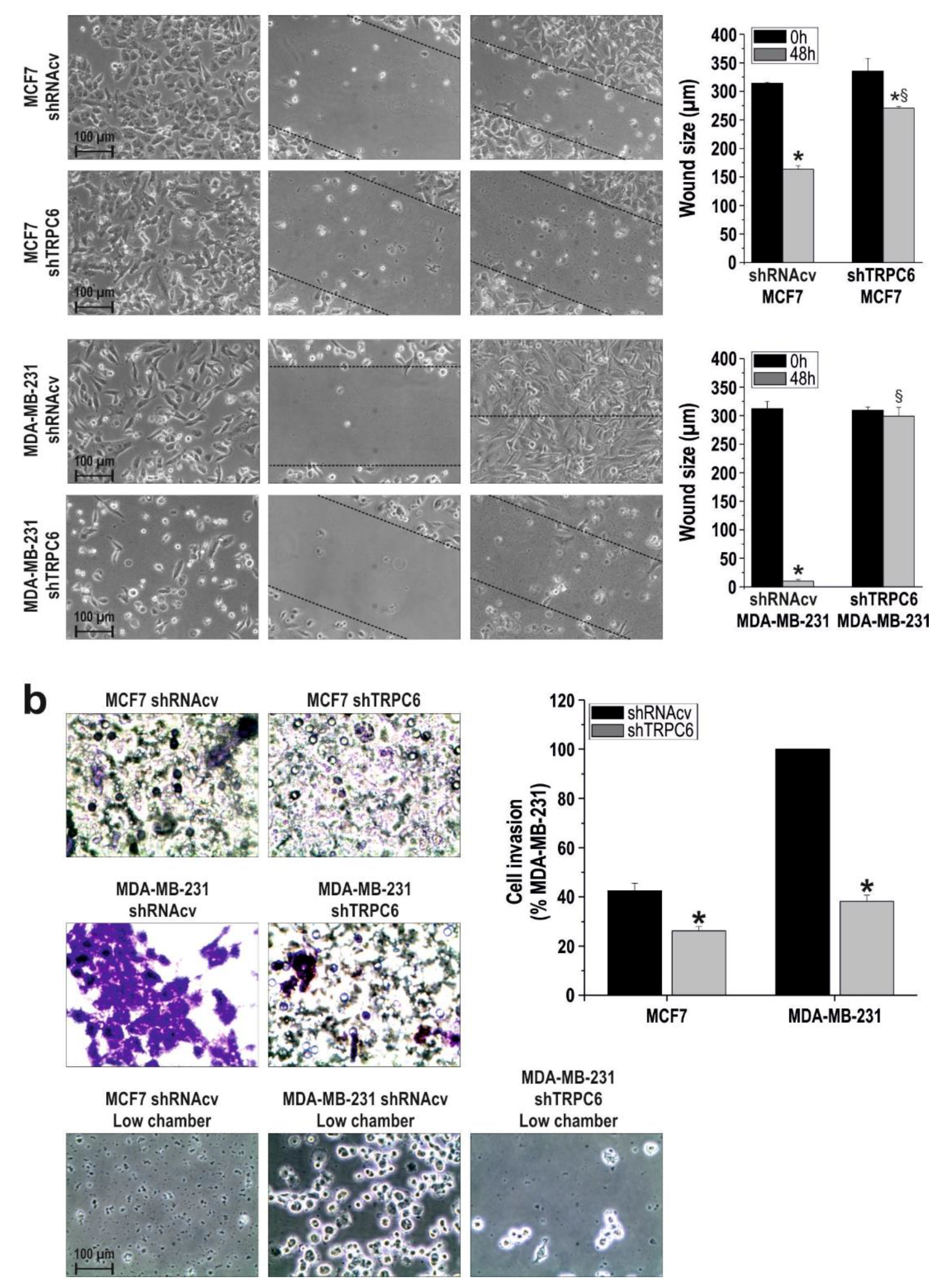
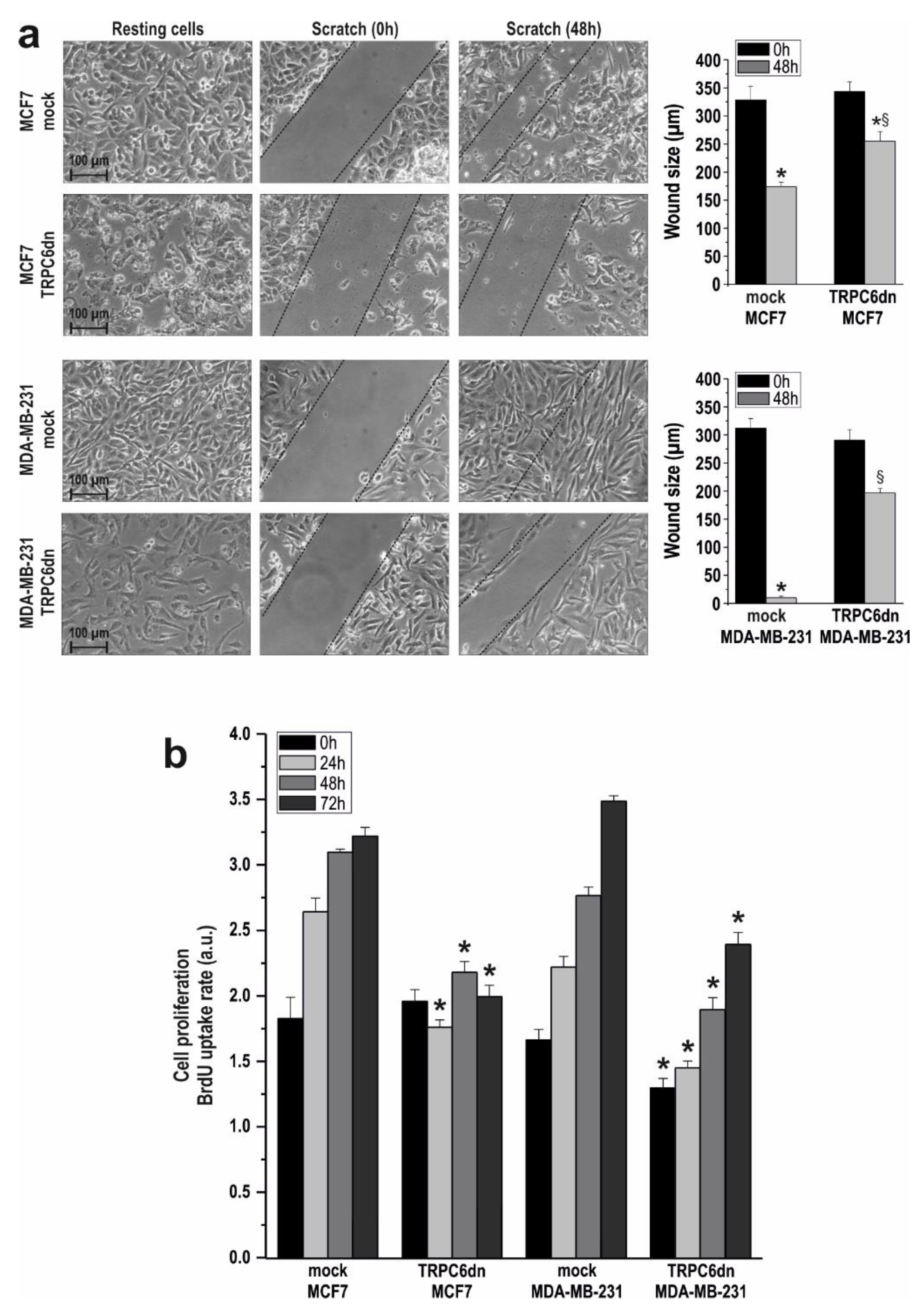
2.1. TRPC6 Is Overexpressed in MCF7 and MDA-MB-231 Breast Cancer Cell Lines and is Required for Breast Cancer Cell Proliferation, Migration and Invasion
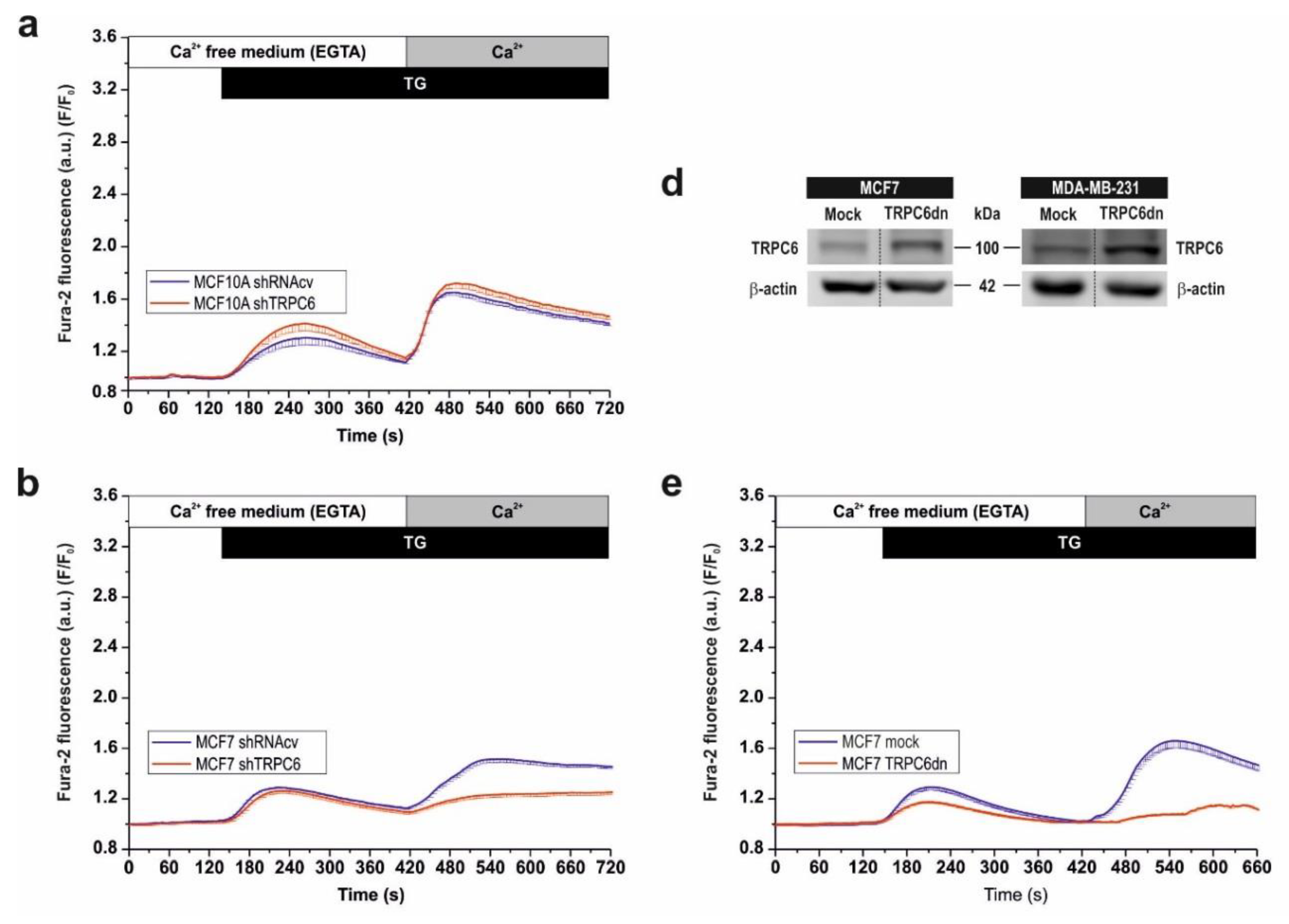
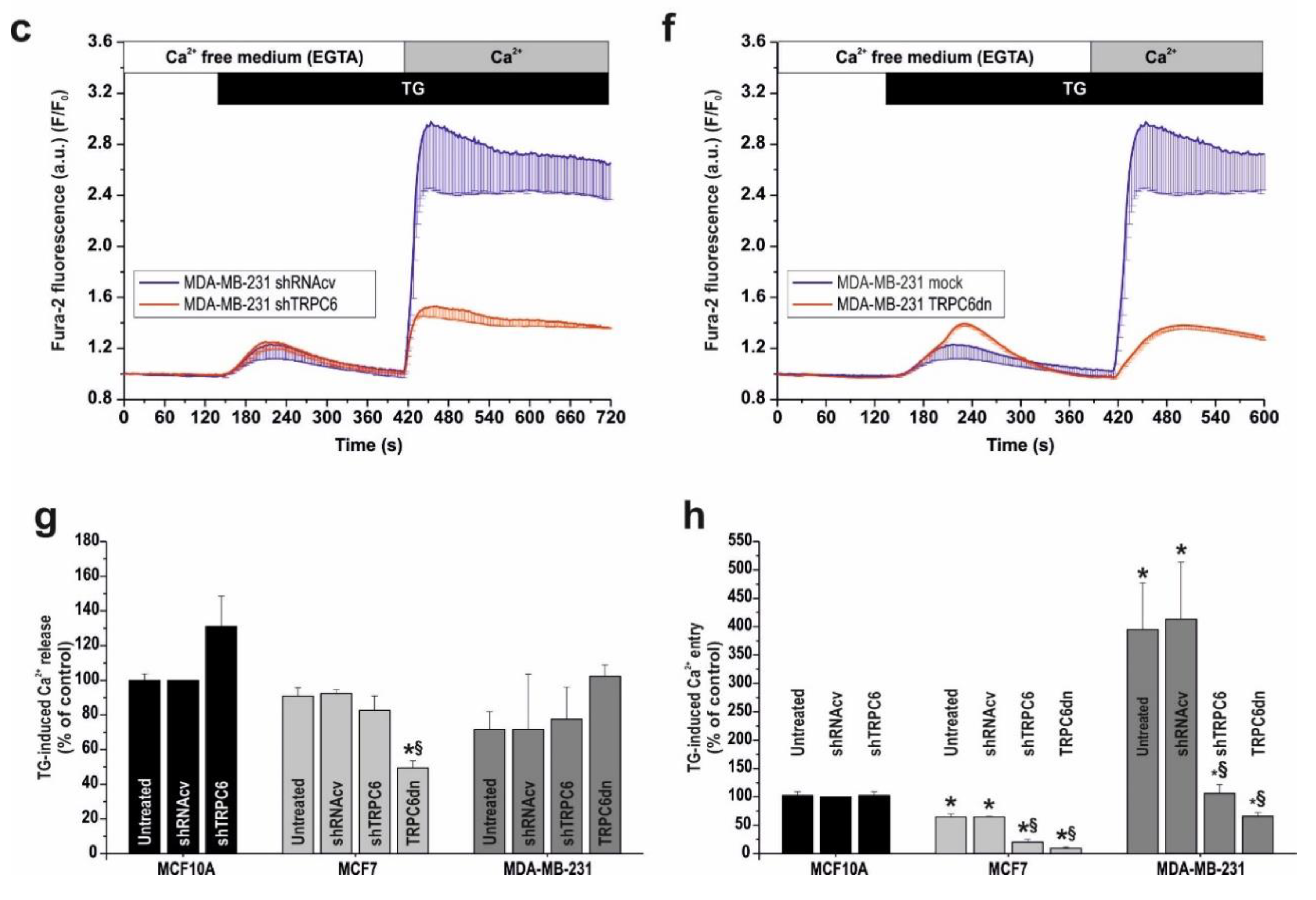
2.2. Functional Role of TRPC6 in SOCE in Breast Cancer Cell Lines
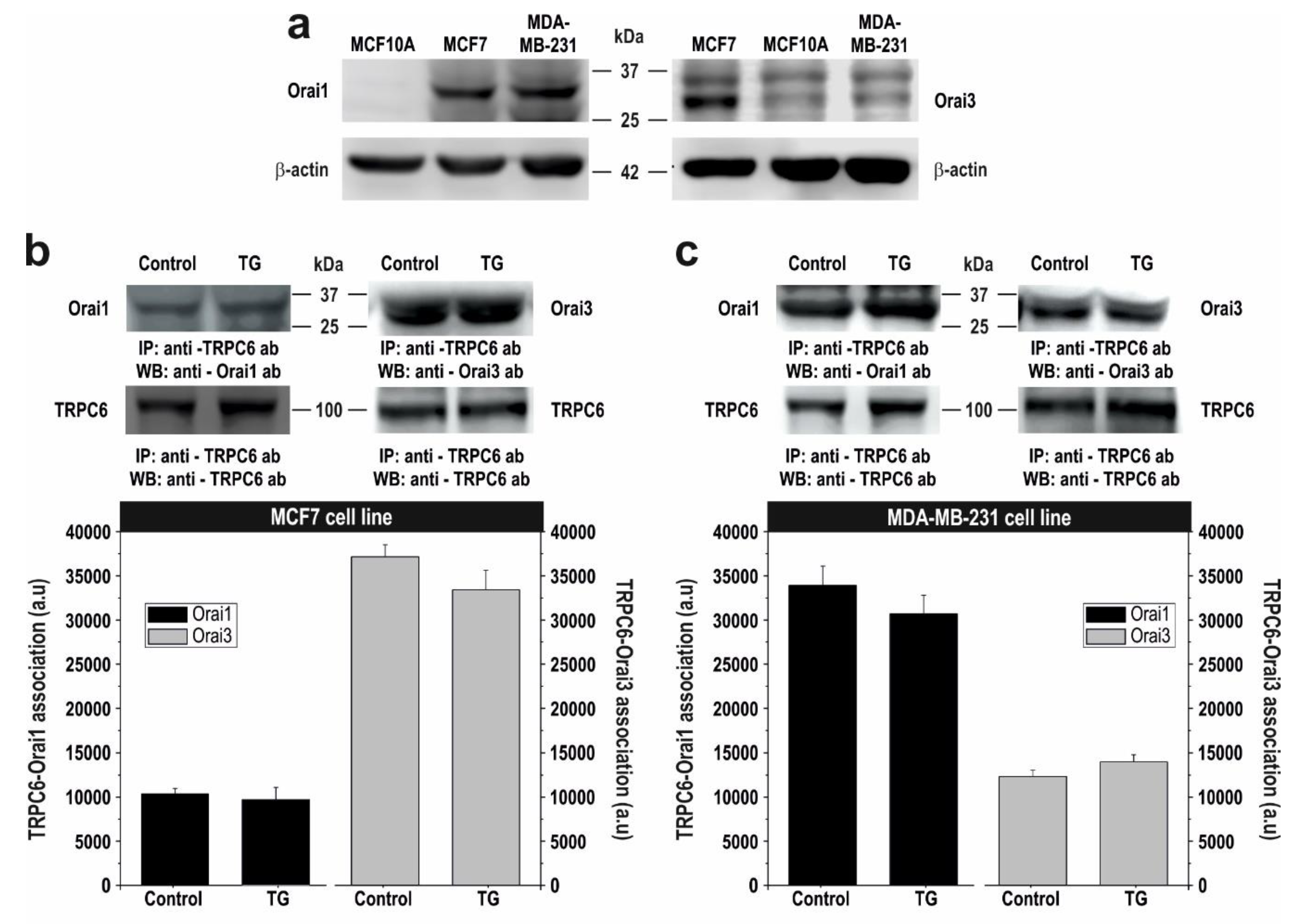
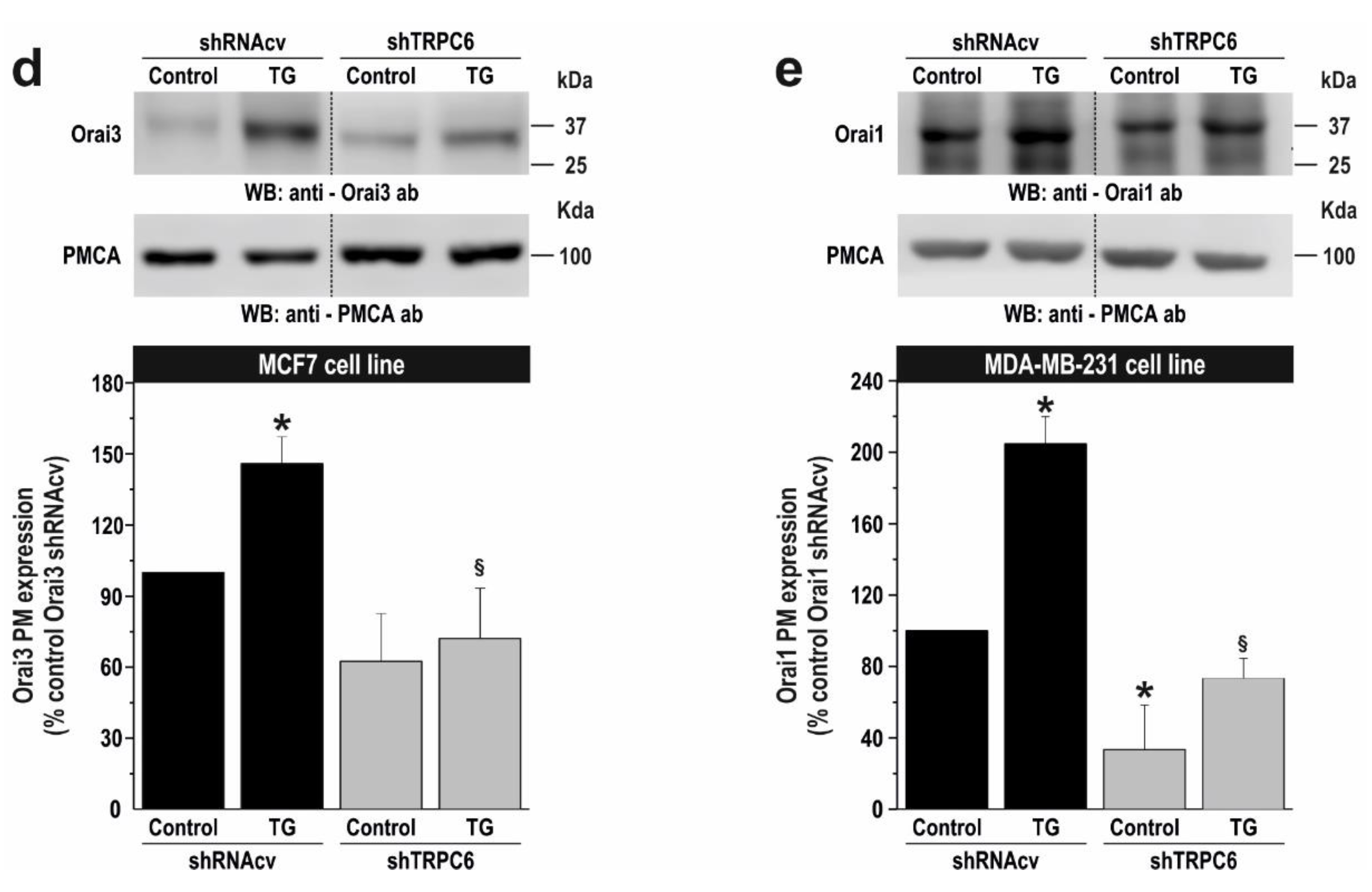
2.3. TRPC6 Expression Is Required for Plasma Membrane Localization of Orai1 and Orai3 in Breast Cancer Cells
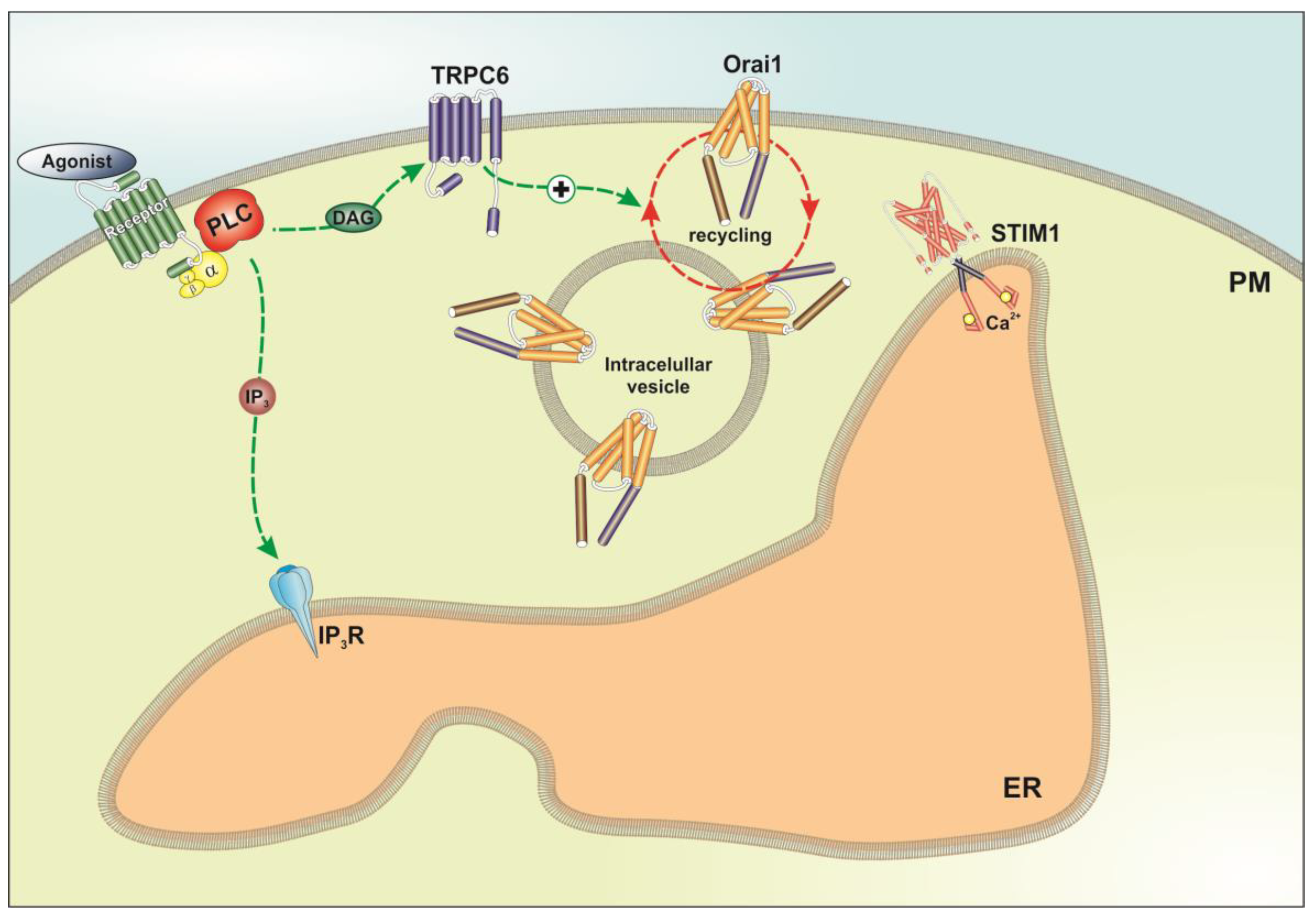
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Transfection
4.3. Measurement of Cytosolic Free-Calcium Concentration
4.4. Immunoprecipitation and Western Blotting
4.5. Transwell Migration Assay
4.6. Wound Healing Assay
4.7. Determination of Cell Proliferation
4.8. Biotinylation Protocol
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Velloso, F.J.; Bianco, A.F.; Farias, J.O.; Torres, N.E.; Ferruzo, P.Y.; Anschau, V.; Jesus-Ferreira, H.C.; Chang, T.H.; Sogayar, M.C.; Zerbini, L.F.; et al. The crossroads of breast cancer progression: Insights into the modulation of major signaling pathways. Onco. Targets Ther. 2017, 10, 5491–5524. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.; Santos, A.; Campbell, F.; Figueiredo, C.; Hammond, D.; Ellies, L.G.; Weyer-Czernilofsky, U.; Bogenrieder, T.; Schmid, M.; Mielgo, A. Blockade of insulin-like growth factors increases efficacy of paclitaxel in metastatic breast cancer. Oncogene 2018, 37, 2022–2036. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Paciga, J.E.; Feldman, R.I.; Yuan, Z.; Coppola, D.; Lu, Y.Y.; Shelley, S.A.; Nicosia, S.V.; Cheng, J.Q. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001, 61, 5985–5991. [Google Scholar] [PubMed]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.H.; Yue, W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 239–256. [Google Scholar] [CrossRef]
- Carboni, G.L.; Gao, B.; Nishizaki, M.; Xu, K.; Minna, J.D.; Roth, J.A.; Ji, L. CACNA2D2-mediated apoptosis in NSCLC cells is associated with alterations of the intracellular calcium signaling and disruption of mitochondria membrane integrity. Oncogene 2003, 22, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Peinelt, C.; Vig, M.; Koomoa, D.L.; Beck, A.; Nadler, M.J.; Koblan-Huberson, M.; Lis, A.; Fleig, A.; Penner, R.; Kinet, J.P. Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nat. Cell Biol. 2006, 8, 771–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.C.; Fang, Y.Y.; Chang, H.W.; Chuang, L.Y.; Lin, Y.D.; Hou, M.F.; Yang, C.H. Identifying association model for single-nucleotide polymorphisms of ORAI1 gene for breast cancer. Cancer Cell Int. 2014, 14, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanisz, H.; Saul, S.; Muller, C.S.; Kappl, R.; Niemeyer, B.A.; Vogt, T.; Hoth, M.; Roesch, A.; Bogeski, I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014, 27, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lkhagvadorj, S.; Lee, M.R.; Hwang, K.H.; Chung, H.C.; Jung, J.H.; Cha, S.K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.Y.; Zhong, L.X.; Feng, M.; Wang, J.F.; Liu, D.B.; Xiong, J.P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar] [PubMed]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Haglund, F.; Ma, R.; Huss, M.; Sulaiman, L.; Lu, M.; Nilsson, I.L.; Hoog, A.; Juhlin, C.C.; Hartman, J.; Larsson, C. Evidence of a functional estrogen receptor in parathyroid adenomas. J. Clin. Endocrinol. Metab. 2012, 97, 4631–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta 2017, 1864, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Fixemer, T.; Wissenbach, U.; Flockerzi, V.; Bonkhoff, H. Expression of the Ca2+-selective cation channel TRPV6 in human prostate cancer: A novel prognostic marker for tumor progression. Oncogene 2003, 22, 7858–7861. [Google Scholar] [CrossRef] [PubMed]
- Raphael, M.; Lehen’kyi, V.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Vanden Abeele, F.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A.; et al. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef] [PubMed]
- Smani, T.; Shapovalov, G.; Skryma, R.; Prevarskaya, N.; Rosado, J.A. Functional and physiopathological implications of TRP channels. Biochim. Biophys. Acta 2015, 1853, 1772–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkika, D.; Lemonnier, L.; Shapovalov, G.; Gordienko, D.; Poux, C.; Bernardini, M.; Bokhobza, A.; Bidaux, G.; Degerny, C.; Verreman, K.; et al. TRP channel-associated factors are a novel protein family that regulates TRPM8 trafficking and activity. J. Cell Biol. 2015, 208, 89–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobos, C.; Sobradillo, D.; Hernandez-Morales, M.; Nunez, L. Calcium remodeling in colorectal cancer. Biochim. Biophys. Acta 2017, 1864, 843–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, R.; Ding, X.; Zhou, K.; Shi, Y.; Ge, R.; Ren, G.; Jin, Y.; Wang, Y. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int. J. Cancer 2009, 125, 2281–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thebault, S.; Flourakis, M.; Vanoverberghe, K.; Vandermoere, F.; Roudbaraki, M.; Lehen’kyi, V.; Slomianny, C.; Beck, B.; Mariot, P.; Bonnal, J.L.; et al. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006, 66, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; He, Z.; Zhou, K.; Cheng, J.; Yao, H.; Lu, D.; Cai, R.; Jin, Y.; Dong, B.; Xu, Y.; et al. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010, 102, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yang, Y.; Xie, R.; Liu, J.; Nie, X.; An, J.; Wen, G.; Liu, X.; Jin, H.; Tuo, B. The NCX1/TRPC6 complex mediates TGFbeta-driven migration and invasion of human hepatocellular carcinoma cells. Cancer Res. 2018, 78, 2564–2576. [Google Scholar] [CrossRef] [PubMed]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Dhennin-Duthille, I.; Gautier, M.; Sevestre, H.; Ahidouch, A. TRP calcium channel and breast cancer: Expression, role and correlation with clinical parameters. Bull. Cancer 2012, 99, 655–664. [Google Scholar] [PubMed]
- Aydar, E.; Yeo, S.; Djamgoz, M.; Palmer, C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: A potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Cellular Physiology Research Group, Department of Physiology, Institute of Molecular Pathology Biomarkers, University of Extremadura, Caceres, Spain; Department of Medical Physiology and Biophysic, Institute of Biomedicine of Sevilla, Sevilla, Spain. Unpublished work. 2018. [Google Scholar]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether a-gogo K(+) channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Montell, C.; Birnbaumer, L.; Flockerzi, V. The TRP channels, a remarkably functional family. Cell 2002, 108, 595–598. [Google Scholar] [CrossRef]
- Fliniaux, I.; Germain, E.; Farfariello, V.; Prevarskaya, N. TRPs and Ca(2+) in cell death and survival. Cell Calcium. 2018, 69, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Choong, L.Y.; Jin, T.H.; Mon, N.N.; Chong, S.; Liew, C.S.; Putti, T.; Lu, S.Y.; Harteneck, C.; Lim, Y.P. TRPV4 plays a role in breast cancer cell migration via Ca(2+)-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncogenesis 2017, 6, e338. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.; Singh, H.; Tan, T.Z.; et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci. Rep. 2016, 6, 27903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Cai, C.; Wu, J.; Cai, S.; Ye, C.; Chen, H.; Yang, Z.; Zeng, H.; Shen, Q.; Zou, F. TRPM7 mediates breast cancer cell migration and invasion through the MAPK pathway. Cancer Lett. 2013, 333, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell Physiol. 2009, 297, C493–C502. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gou, H.; Zhu, J.; Tian, S.; Yu, L. Lidocaine inhibits the invasion and migration of TRPV6-expressing cancer cells by TRPV6 downregulation. Oncol. Lett. 2016, 12, 1164–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilbert, A.; Dhennin-Duthille, I.; Hiani, Y.E.; Haren, N.; Khorsi, H.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Expression of TRPC6 channels in human epithelial breast cancer cells. BMC Cancer 2008, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Dionisio, N.; Smani, T.; Salido, G.M.; Rosado, J.A. Transient receptor potential ankyrin-1 (TRPA1) modulates store-operated Ca(2+) entry by regulation of STIM1-Orai1 association. Biochim. Biophys. Acta 2013, 1833, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 2012, 287, 35612–35620. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. FKBP52 is involved in the regulation of SOCE channels in the human platelets and MEG 01 cells. Biochim. Biophys. Acta 2013, 1833, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, N.; Smani, T.; Woodard, G.E.; Castellano, A.; Salido, G.M.; Rosado, J.A. Homer proteins mediate the interaction between STIM1 and Cav1.2 channels. Biochim. Biophys. Acta 2015, 1853, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Woodard, G.E.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry-associated regulatory factor (SARAF) plays an important role in the regulation of arachidonate-regulated Ca2+ (ARC) channels. J. Biol. Chem. 2016, 291, 6982–6988. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Lopez, J.J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem. J. 2016, 473, 3581–3595. [Google Scholar] [CrossRef] [PubMed]
- Zbidi, H.; Jardin, I.; Woodard, G.E.; Lopez, J.J.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A. STIM1 and STIM2 Are Located in the Acidic Ca2+ Stores and Associates with Orai1 upon Depletion of the Acidic Stores in Human Platelets. J. Biol. Chem. 2011, 286, 12257–12270. [Google Scholar] [CrossRef] [PubMed]
- Albarran, L.; Berna-Erro, A.; Dionisio, N.; Redondo, P.C.; Lopez, E.; Lopez, J.J.; Salido, G.M.; Brull Sabate, J.M.; Rosado, J.A. TRPC6 participates in the regulation of cytosolic basal calcium concentration in murine resting platelets. Biochim. Biophys. Acta 2014, 1843, 789–796. [Google Scholar] [CrossRef] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10090331
Jardin I, Diez-Bello R, Lopez JJ, Redondo PC, Salido GM, Smani T, Rosado JA. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers. 2018; 10(9):331. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10090331
Chicago/Turabian StyleJardin, Isaac, Raquel Diez-Bello, Jose J. Lopez, Pedro C. Redondo, Ginés M. Salido, Tarik Smani, and Juan A. Rosado. 2018. "TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure" Cancers 10, no. 9: 331. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10090331