Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
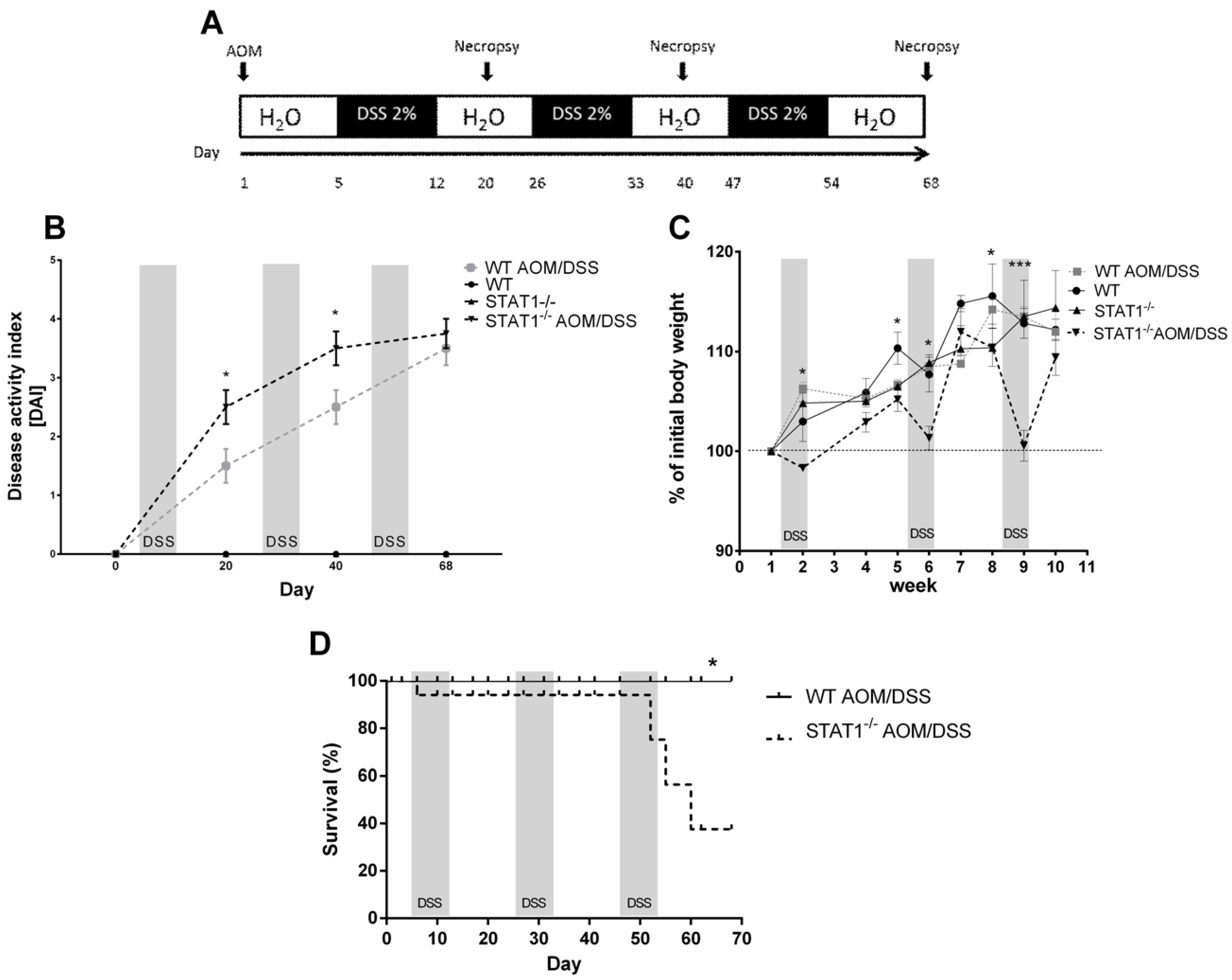
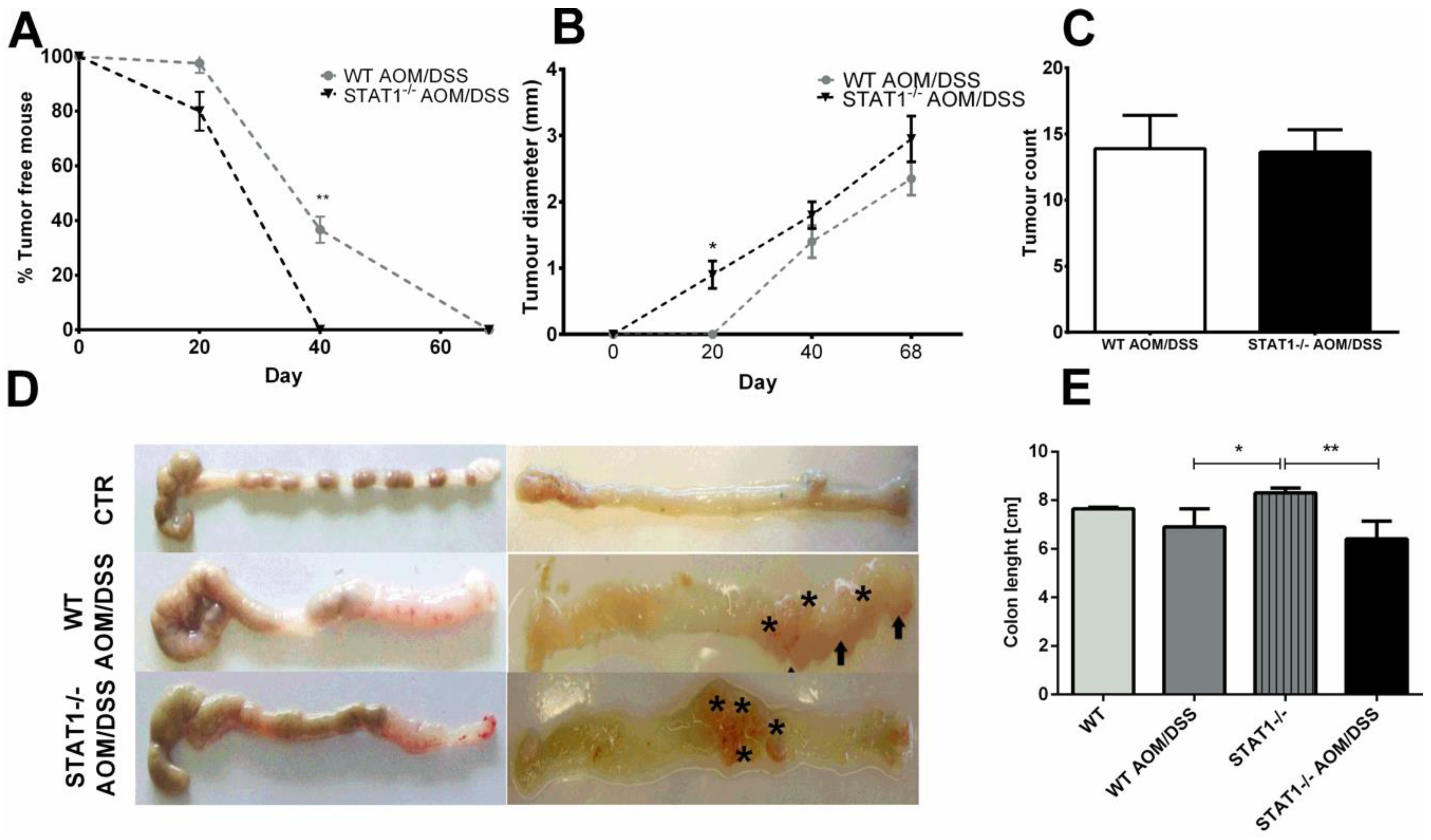
2.1. STAT1 Deficiency Leads to Rapidly Increase Development of CAC
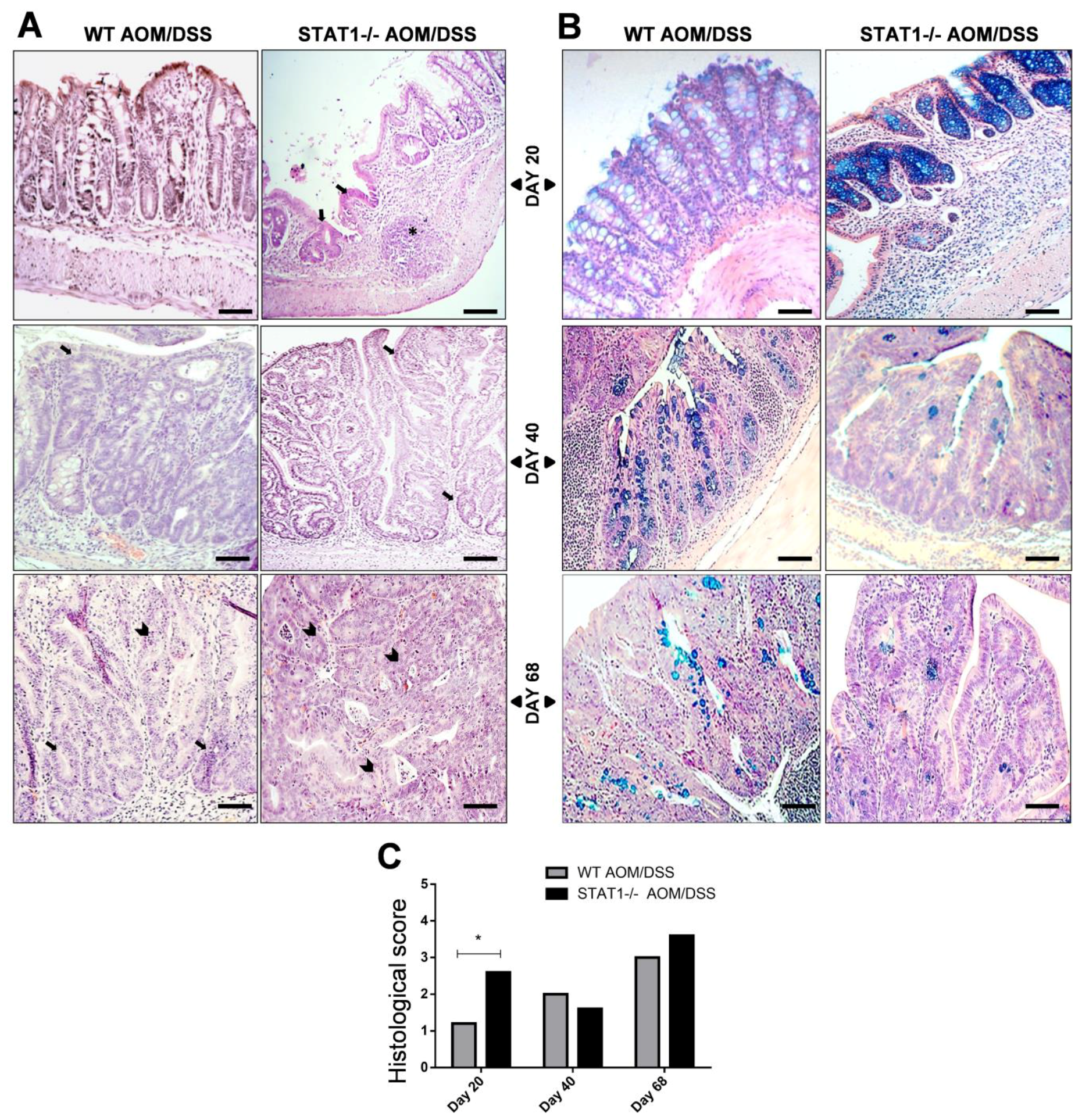
2.2. STAT1−/− AOM/DSS Animals Have More Histological Damage Compared to WT AOM/DSS Mice
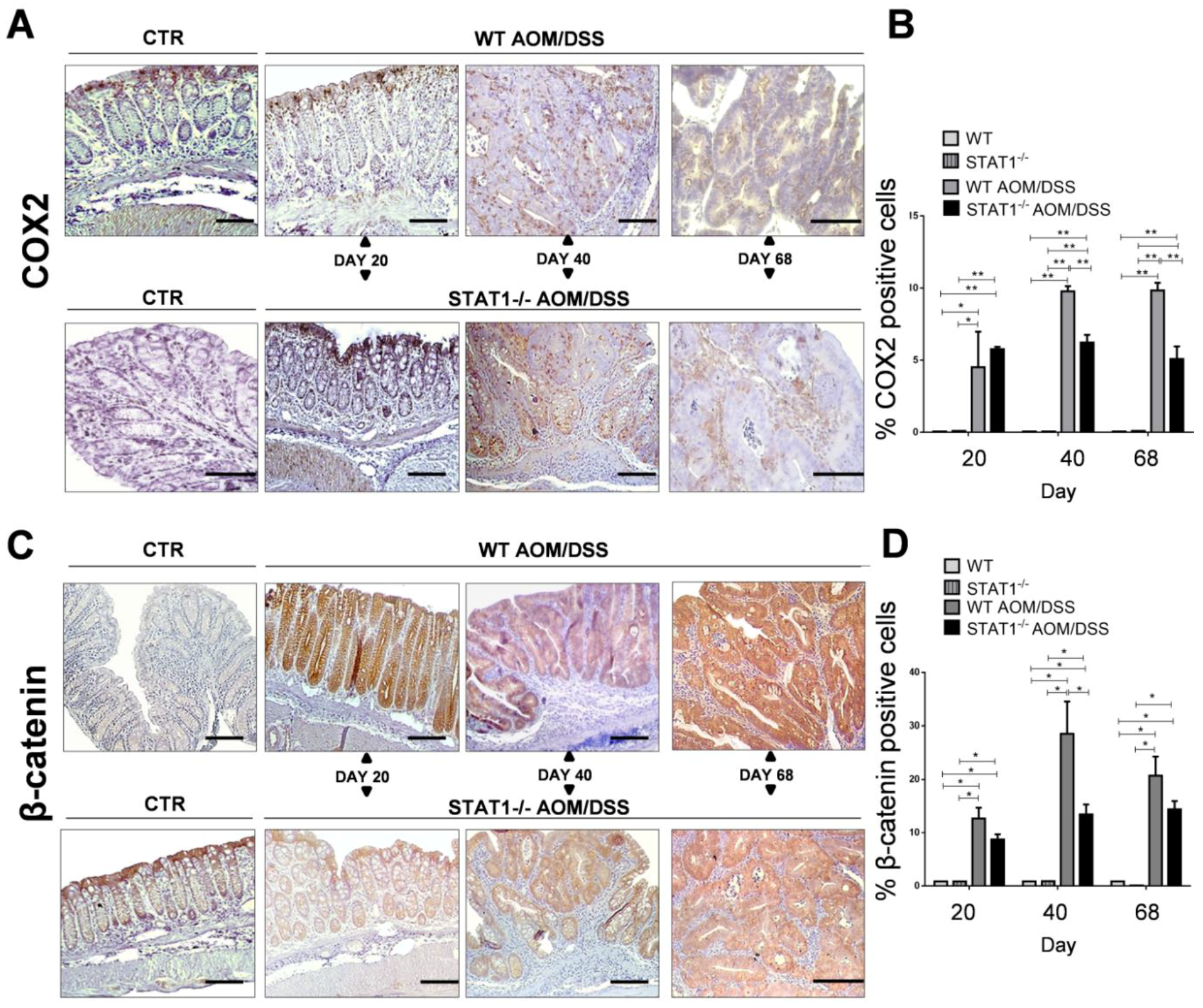
2.3. The Absence of STAT1 Does Not Alter the Early Expression of Markers for Tumorigenesis
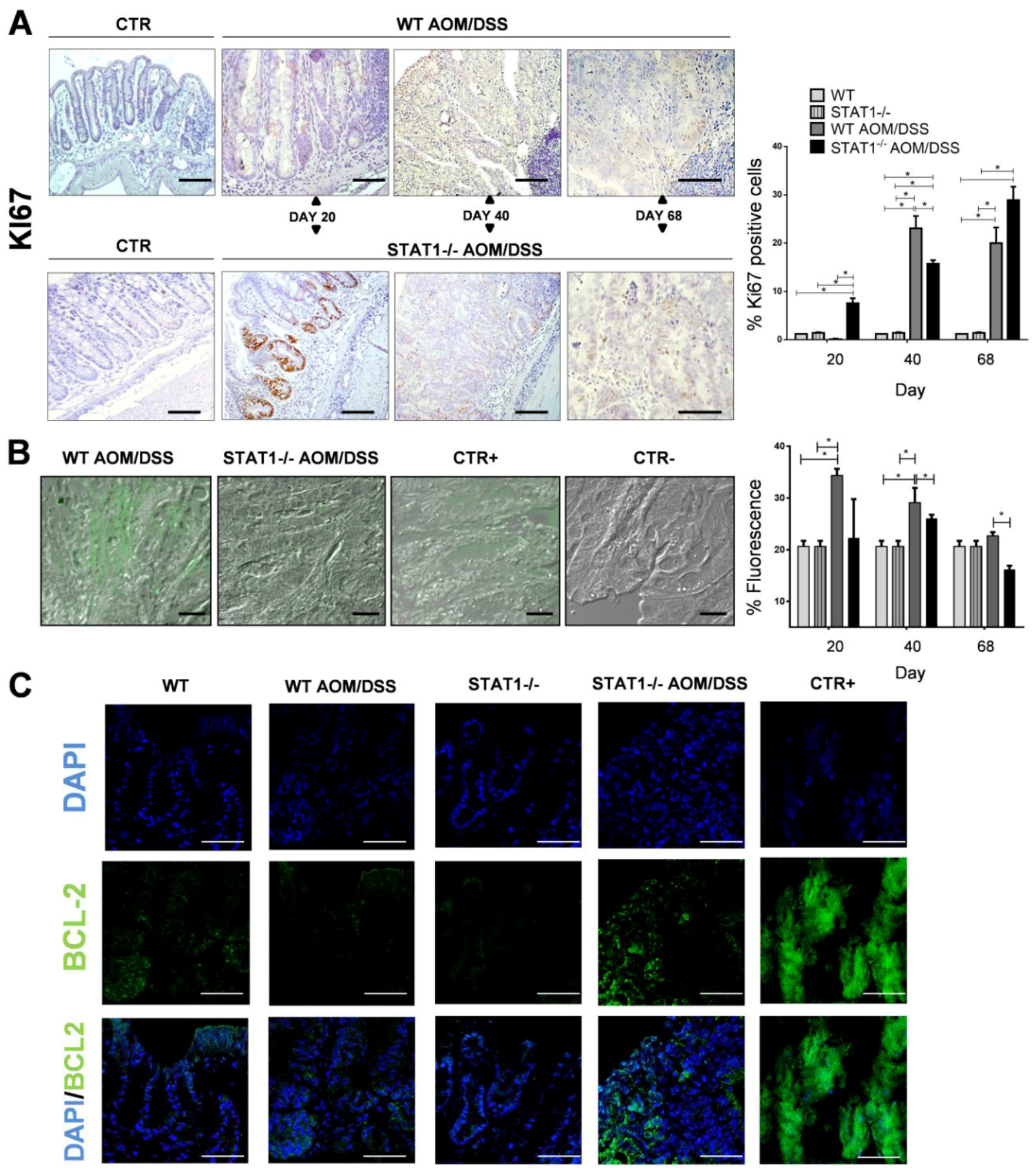
2.4. STAT1 Deficiency Increases Cell Proliferation and Reduces Apoptosis during Early CAC Development
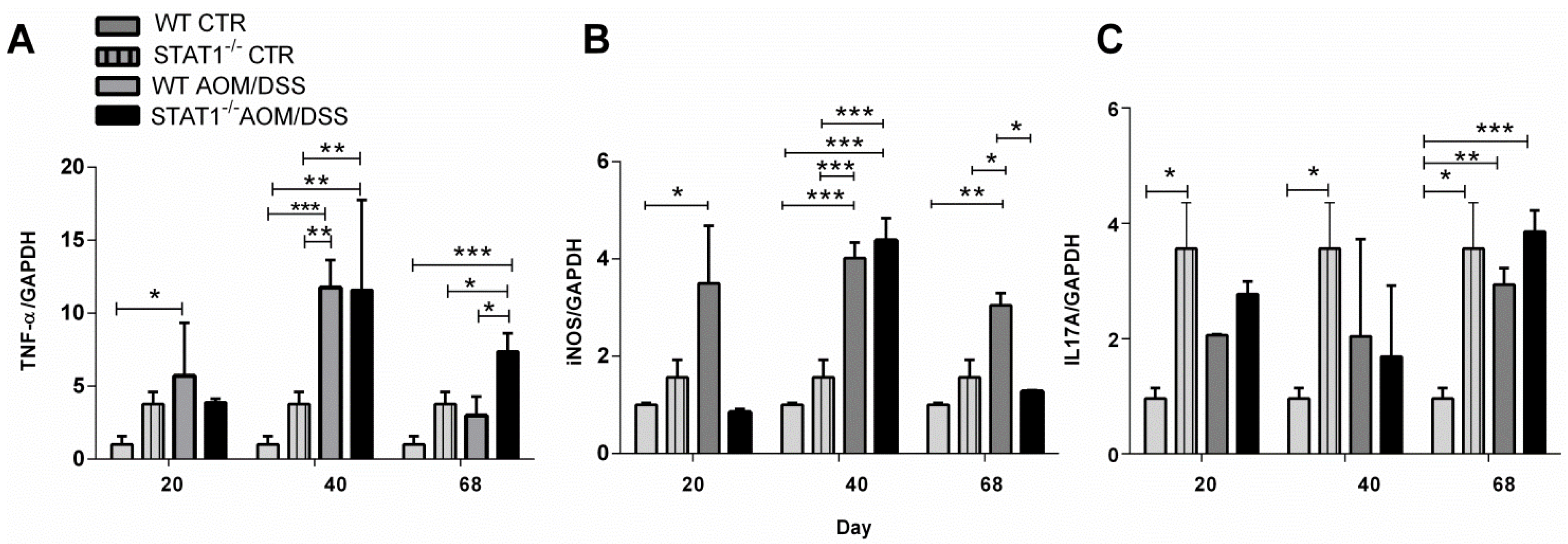
2.5. STAT1 Deficiency Alters the Expression of Proinflammatory Mediators in the Colon
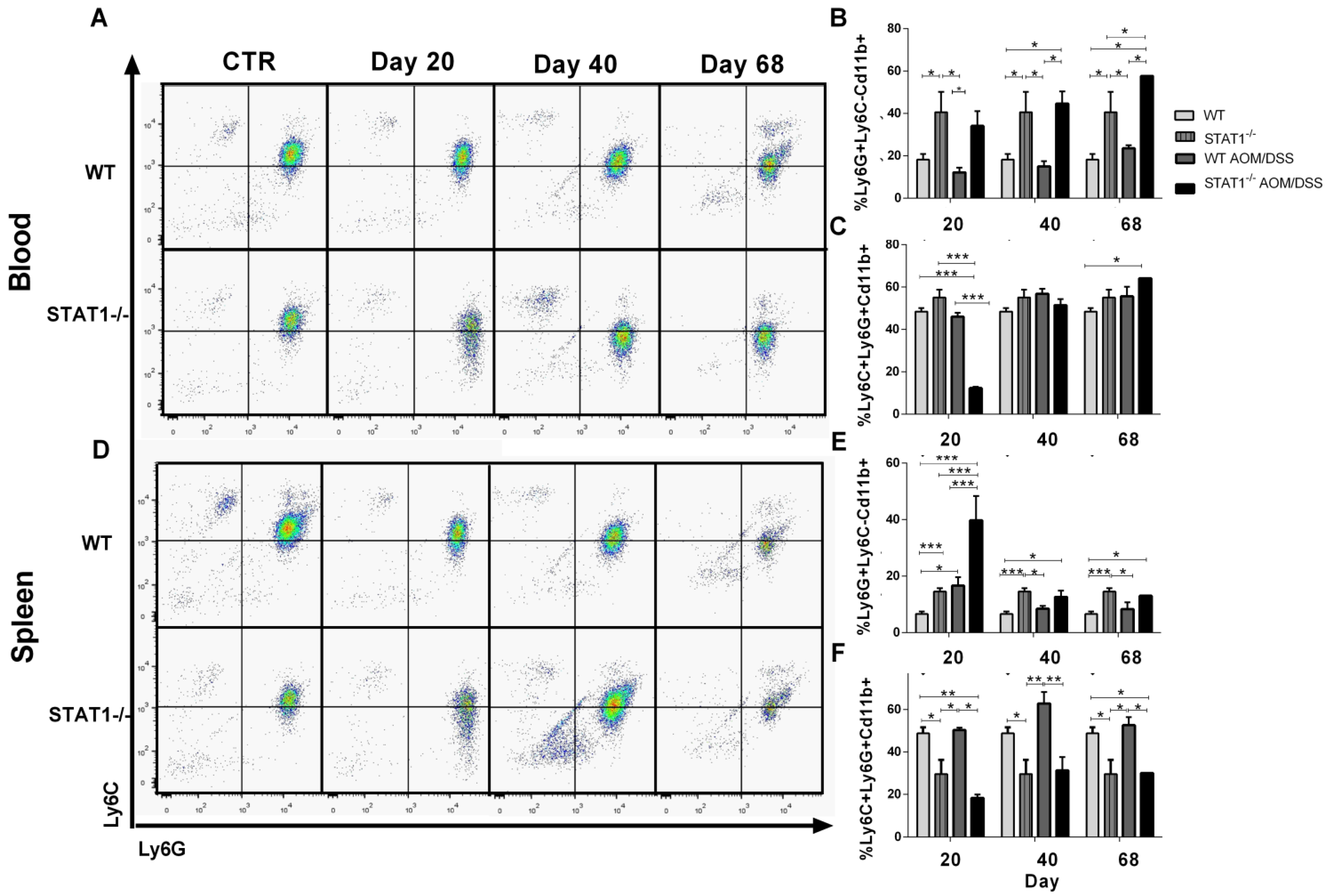
2.6. The Recruitment of Inflammatory Monocytes and Granulocytes during CAC Is STAT1 Dependent
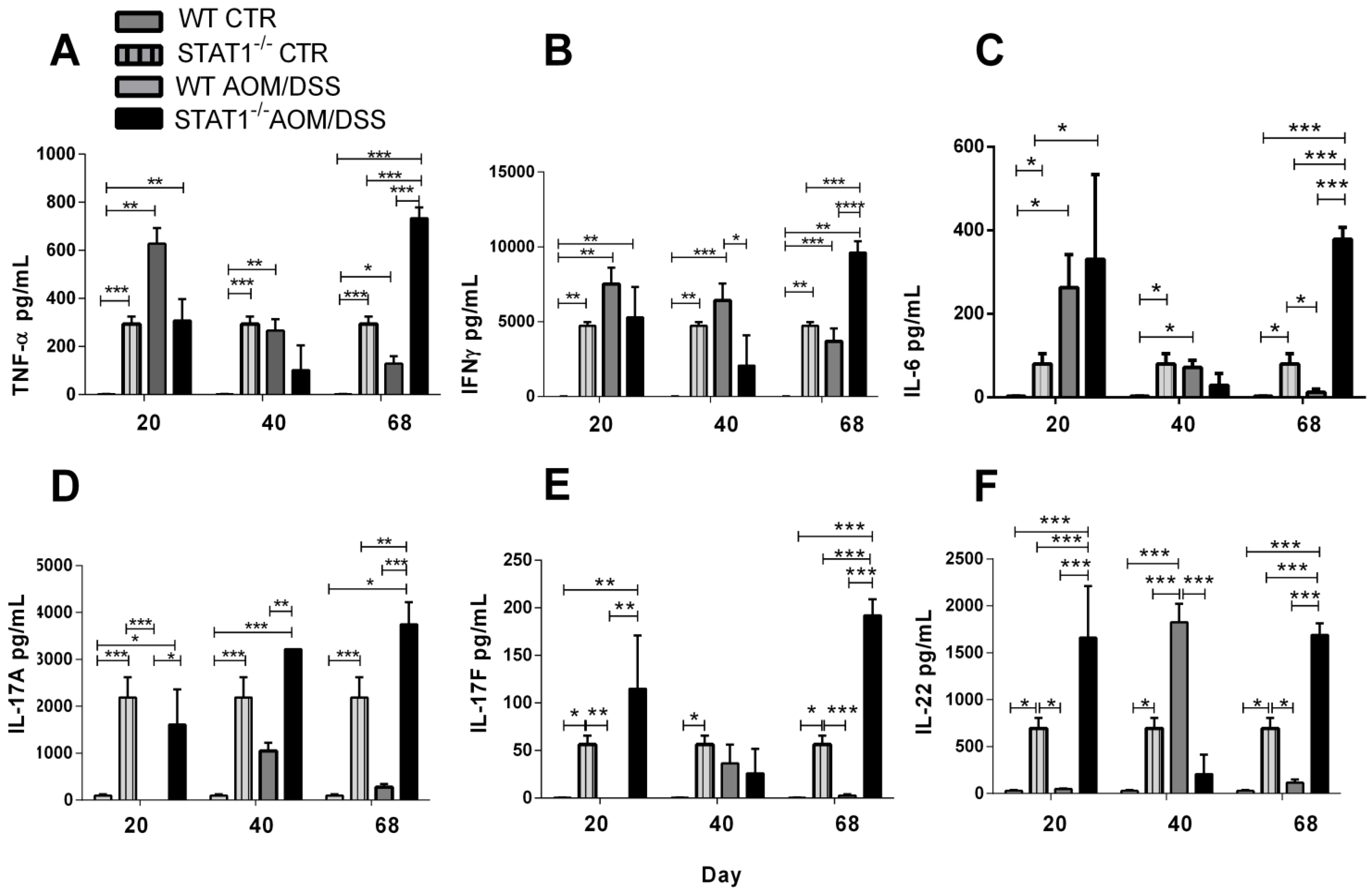
2.7. IL-17A, IL-17F and IL-22 Cytokine Production Is Increased during STAT1 Deficiency
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Murine Colitis Associated Colorectal Cancer (CAC)
4.3. Histological Analysis
4.4. RNA Extraction and RT-PCR
4.5. Quantification of Cytokines
4.6. Immunoblotting
4.7. Flow Cytometry
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon. Rectal. Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; De, F.F.; Zarantonello, L.; Ruffolo, C.; Ferraro, G.A.; Zanus, G.; Giordano, A.; Bassi, N.; Cillo, U. From Inflammation to Cancer in Inflammatory Bowel Disease: Molecular Perspectives. Anticancer Res. 2016, 36, 1447–1460. [Google Scholar] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Castano-Milla, C.; Chaparro, M.; Gisbert, J.P. Systematic review with meta-analysis: The declining risk of colorectal cancer in ulcerative colitis. Aliment. Pharmacol. Ther. 2014, 39, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Konishi, T.; Kishimoto, J.; Kotake, K.; Muto, T.; Sugihara, K. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: A nationwide Japanese study. Inflamm. Bowel. Dis. 2011, 17, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, A.F.; Pierre, J.F.; Kudsk, K.A. JAK-STAT and intestinal mucosal immunology. JAKSTAT 2013, 2, e25530. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Kadlubar, S.A.; Bondurant, K.L.; Wolff, R.K. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol. Carcinog. 2013, 52, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Ramana, C.V.; Gil, M.P.; Schreiber, R.D.; Stark, G.R. STAT1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002, 23, 96–101. [Google Scholar] [CrossRef]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Klampfer, L. The role of signal transducers and activators of transcription in colon cancer. Front. Biosci. 2008, 13, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E.; Sexl, V. The tumor suppressor function of STAT1 in breast cancer. JAKSTAT 2013, 2, e23353. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, J.; Boudny, V.; Kocak, I.; Lauerova, L.; Fait, V.; Vagundova, M. Malignant melanoma associates with deficient IFN-induced STAT 1 phosphorylation. Int. J. Mol. Med. 2003, 12, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.A.; Al-Attar, A.; Watson, N.F.; Scholefield, J.H.; Ilyas, M.; Durrant, L.G. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 2010, 59, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Gordziel, C.; Bratsch, J.; Moriggl, R.; Knösel, T.; Friedrich, K. Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br. J. Cancer 2013, 109, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yang, S.; Sun, N.; Chen, J. Differential expression of STAT1 and p21 proteins predicts pancreatic cancer progression and prognosis. Pancreas 2014, 43, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Molavi, O.; Su, M.; Lai, R. The clinical and biological significance of STAT1 in esophageal squamous cell carcinoma. BMC Cancer 2014, 14, 791. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Rosenstiel, P.; Hampe, J.; Nikolaus, S.; Groessner, B.; Schottelius, A.; Kühbacher, T.; Hämling, J.; Fölsch, U.R.; Seegert, D. Activation of signal transducer and activator of transcription (STAT) 1 in human chronic inflammatory bowel disease. Gut 2002, 51, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudter, J.; Weigmann, B.; Bartsch, B.; Kiesslich, R.; Strand, D.; Galle, P.R.; Lehr, H.A.; Schmidt, J.; Neurath, M.F. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am. J. Gastroenterol. 2005, 100, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.K.; de la Motte, C.A.; Kessler, S.P.; Hascall, V.C.; Hill, D.R.; Strong, S.A. Hyaluronan-mediated leukocyte adhesion and dextran sulfate sodium-induced colitis are attenuated in the absence of signal transducer and activator of transcription 1. Am. J. Pathol. 2008, 173, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Guo, W.; Wu, L.; Gu, Y.; Gu, L.; Xu, S.; Wu, X.; Shen, Y.; Ke, Y.; Tan, R.; et al. Selective sequestration of STAT1 in the cytoplasm via phosphorylated SHP-2 ameliorates murine experimental colitis. J. Immunol. 2012, 189, 3497–3507. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Nishimoto, S.; Muto, G.; Sekiya, T.; Tamiya, T.; Kimura, A.; Morita, R.; Asakawa, M.; Chinen, T.; Yoshimura, A. SOCS1 is essential for regulatory T cell functions by preventing loss of Foxp3 expression as well as IFN-{gamma} and IL-17A production. J. Exp. Med. 2011, 208, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Kobayashi, T.; Chinen, T.; Saeki, K.; Takaki, H.; Koga, K.; Minoda, Y.; Sanada, T.; Yoshioka, T.; Mimata, H.; et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med. 2006, 203, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Owusu, B.Y.; Augenlicht, L.; Klampfer, L. The Role of STAT1 for Crosstalk between Fibroblasts and Colon Cancer Cells. Front. Oncol. 2014, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Fu, X.Y.; Plate, J.; Chong, A.S. IFN-gamma induces cell growth inhibition by Fas-mediated apoptosis: Requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998, 58, 2832–2837. [Google Scholar] [PubMed]
- Huang, S.; Bucana, C.D.; Van Arsdall, M.; Fidler, I.J. STAT1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene 2002, 21, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Tobelaim, W.S.; Beaurivage, C.; Champagne, A.; Pomerleau, V.; Simoneau, A.; Chababi, W.; Yeganeh, M.; Thibault, P.; Klinck, R.; Carrier, J.C.; et al. Tumour-promoting role of SOCS1 in colorectal cancer cells. Sci. Rep. 2015, 5, 14301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfer, L.; Huang, J.; Corner, G.; Mariadason, J.; Arango, D.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Oncogenic Ki-ras inhibits the expression of interferon-responsive genes through inhibition of STAT1 and STAT2 expression. J. Biol. Chem. 2003, 278, 46278–46287. [Google Scholar] [CrossRef] [PubMed]
- Clapper, M.L.; Cooper, H.S.; Chang, W.C. Dextran sulfate sodium-induced colitis-associated neoplasia: A promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007, 28, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.H.; Zheng, Q.Y.; Li, G.Q.; Hu, X.B.; Feng, S.L.; Xu, G.L.; Zhang, K.Q. STAT1-mediated down-regulation of Bcl-2 expression is involved in IFN-gamma/TNF-alpha-induced apoptosis in NIT-1 cells. PLoS ONE 2015, 10, e0120921. [Google Scholar]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toor, S.M.; Syed Khaja, A.S.; El Salhat, H.; Bekdache, O.; Kanbar, J.; Jaloudi, M.; Elkord, E. Increased Levels of Circulating and Tumor-Infiltrating Granulocytic Myeloid Cells in Colorectal Cancer Patients. Front. Immunol. 2016, 7, 560. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Neurath, M.F. Integrating Immunologic Signaling Networks: The JAK/STAT Pathway in Colitis and Colitis-Associated Cancer. Vaccines 2016, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Leon-Cabrera, S.A.; Molina-Guzman, E.; Delgado-Ramirez, Y.G.; Vázquez-Sandoval, A.; Ledesma-Soto, Y.; Pérez-Plasencia, C.G.; Chirino, Y.I.; Delgado-Buenrostro, N.L.; Rodríguez-Sosa, M.; Vaca-Paniagua, F.; et al. Lack of STAT6 Attenuates Inflammation and Drives Protection against Early Steps of Colitis-Associated Colon Cancer. Cancer Immunol. Res. 2017, 5, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.; Bothwell, A.L.M. Stat6 Promotes Intestinal Tumorigenesis in a Mouse Model of Adenomatous Polyposis by Expansion of MDSCs and Inhibition of Cytotoxic CD8 Response. Neoplasia 2017, 19, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knösel, T.; Kenner, L.; et al. The ratio of STAT1 to STAT3 expression is a determinant of colorectal cancer growth. Oncotarget 2016, 7, 51096–51106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Hainzl, E.; Stockinger, S.; Rauch, I.; Heider, S.; Berry, D.; Lassnig, C.; Schwab, C.; Rosebrock, F.; Milinovich, G.; Schlederer, M.; et al. Intestinal Epithelial Cell Tyrosine Kinase 2 Transduces IL-22 Signals to Protect from Acute Colitis. J. Immunol. 2015, 195, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, W.J.; Gibson, T.F.; Zelterman, D.; Hao, L.; Henegariu, O.; Bothwell, A.L. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 5540–5544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, Y.S.; Han, D.S.; Lee, A.R.; Eun, C.S.; Youn, J.; Kim, H.Y. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastelein, R.A.; Hunter, C.A.; Cua, D.J. Discovery and biology of IL-23 and IL-27: Related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007, 25, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Gallo, E.; Abbas, A.K. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J. Immunol. 2010, 185, 6461–6471. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 2006, 16, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Varikuti, S.; Oghumu, S.; Elbaz, M.; Volpedo, G.; Ahirwar, D.K.; Alarcon, P.C.; Sperling, R.H.; Moretti, E.; Pioso, M.S.; Kimble, J.; et al. STAT1 gene deficient mice develop accelerated breast cancer growth and metastasis which is reduced by IL-17 blockade. Oncoimmunology 2017, 6, e1361088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braster, R.; Bögels, M.; Beelen, R.H.; van Egmond, M. The delicate balance of macrophages in colorectal cancer; their role in tumour development and therapeutic potential. Immunobiology 2017, 222, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, M.; Maeda, K.; Nagahara, H.; Ohtani, H.; Sakurai, K.; Yamazoe, S.; Kimura, K.; Toyokawa, T.; Amano, R.; Tanaka, H.; et al. Prognostic significance of the lymphocyte-to-monocyte ratio in patients with metastatic colorectal cancer. World J. Gastroenterol. 2015, 21, 9966–9973. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.M.; Hettinger, J.; Feuerer, M. Monocytes and macrophages in cancer: Development and functions. Cancer Microenviron. 2013, 6, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Wang, H.; Deng, L.; Hou, J.; Shi, R.; Yao, M.; Gao, Y.; Yao, A.; Wang, X.; Yu, L.; et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 2013, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Young, R.A.; Daley, G.Q. Hematopoietic remodeling in interferon-gamma-deficient mice infected with mycobacteria. Blood 1998, 91, 2914–2924. [Google Scholar] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, 60, 3678. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leon-Cabrera, S.; Vázquez-Sandoval, A.; Molina-Guzman, E.; Delgado-Ramirez, Y.; Delgado-Buenrostro, N.L.; Callejas, B.E.; Chirino, Y.I.; Pérez-Plasencia, C.; Rodríguez-Sosa, M.; Olguín, J.E.; et al. Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development. Cancers 2018, 10, 341. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10090341
Leon-Cabrera S, Vázquez-Sandoval A, Molina-Guzman E, Delgado-Ramirez Y, Delgado-Buenrostro NL, Callejas BE, Chirino YI, Pérez-Plasencia C, Rodríguez-Sosa M, Olguín JE, et al. Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development. Cancers. 2018; 10(9):341. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10090341
Chicago/Turabian StyleLeon-Cabrera, Sonia, Armando Vázquez-Sandoval, Emmanuel Molina-Guzman, Yael Delgado-Ramirez, Norma L. Delgado-Buenrostro, Blanca E. Callejas, Yolanda I. Chirino, Carlos Pérez-Plasencia, Miriam Rodríguez-Sosa, Jonadab E. Olguín, and et al. 2018. "Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development" Cancers 10, no. 9: 341. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10090341