Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment




1
Department of Medical Area, University of Udine, 33100 Udine, Italy
2
Department of Health Sciences, “Magna Graecia” University of Catanzaro, 88100 Catanzaro, Italy
3
Medical Genetics Institute, University Hospital of Udine, 33100 Udine, Italy
*
Authors to whom correspondence should be addressed.
Cancers 2019, 11(1), 61; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010061
Submission received: 13 December 2018
/
Revised: 30 December 2018
/
Accepted: 3 January 2019
/
Published: 9 January 2019
(This article belongs to the Special Issue Histone Modification in Cancer)
Abstract
:The epigenetic machinery deputed to control histone post-translational modifications is frequently dysregulated in cancer cells. With epigenetics being naturally reversible, it represents a good target for therapies directed to restore normal gene expression. Since the discovery of Bromodomain and Extra Terminal (BET) inhibitors, a great effort has been spent investigating the effects of chromatin readers’ inhibition, specifically the class of proteins assigned to bind acetylated and methylated residues. So far, focused studies have been produced on epigenetic regulation, dissecting a specific class of epigenetic-related proteins or investigating epigenetic therapy in a specific tumor type. In this review, recent steps toward drug discovery on the different classes of chromatin readers have been outlined, highlighting the pros and cons of current therapeutic approaches.
1. Introduction
Mammalian cells can maintain their specific phenotype and adapt to diverse environmental stimuli modifying their epigenome, which is built on a flexible set of mechanisms (creating the epigenetic code), mostly based on a fine cluster of chemical changes interplay, including DNA methylation and histone post-translational modifications (HPTMs) as well as histone variants assembly and 3D chromatin organization [1]. Acting in concert, these mechanisms are responsible for changes in local chromatin structure and dynamics, determining either the compactness or accessibility to specific loci. Moreover, beyond chromatin structure, epigenetic modifications represent a scaffold upon which transcriptional activators/repressors may directly affect gene expression [2]. Chromatin dynamic changes, which include nucleosome unwrapping, re-wrapping, sliding, assembly, and disassembly, involve the formation and/or disruption of interactions within the interfaces between the DNA, histones H3/H4, and H2A/H2B, which are the main components of the nucleosome [3]. Epigenetic modifications constitute a set of tags, which reflect the local state of chromatin; the map of histone covalent modifications is assembled to influence the interactions that stabilize the nucleosome structure by shifting the free energy difference between the fully wrapped nucleosome and altered nucleosome structures [4]. Altogether, these mechanisms build the epigenome, which is sensitive to environmental changes and shapes the flow of information from the genome to the proteome, defining the identity of a cell type [5].
In this review, we will outline recent steps toward drug discovery on the different classes of chromatin readers, highlighting the advantages and challenges in drug screening, focusing on the so-called ‘difficult targets’ and on the advantages of potential treatments.
2. Dissecting Chromatin Orthography
Epigenomic reprogramming plays a major role in triggering specific responses from early embryogenesis to the complete development of an organism [6]. Besides DNA methylation, which represents the most well-known reaction to endogenous and environmental stimuli, histone modifications are fundamental for epigenomic reprogramming [7]. Histones may undergo different chemical post-translational modifications (PTMs) to regulate the stable inheritance of cellular memory during mitotic division and fulfill specific regulatory pathways in cell-signaling networks [4]. A discrete number of HPTMs has been discovered, but thus far the main studied ones remain acetylation, methylation, phosphorylation, and ubiquitination. Since the cardinal role of HPTMs both in physiological cell maintenance and in disease states, a wide range of enzymes assigned to their regulation have been discovered and deeply investigated [8]. Three major functional classes of proteins are involved in the regulation and turnover of PTMs: Chromatin writers, which are designated to add a specific PTM; chromatin erasers, which remove them from a certain location within DNA; and chromatin readers, which recognize specific PTMs to accomplish their implicit meaning [4]. The most studied HPTMs and their related enzymes are listed in Table 1.
Relying on the type of HPTMs and their position within the chromatin, histone-modifying enzymes regulate a complex network that can either foster or hinder DNA packaging, in response to specific cellular stimuli [9]. This, in turn, can modify the DNA accessibility to i.e., DNA repair and transcription enzymes, thereby influencing the cellular phenotype without necessarily modifying the genotype. Positively charged histone lysines, which contribute to the tight interaction between DNA and nucleosomes, are among the most frequently modified amino acids: Acetylation of these residues can promote transcription of target genes by modulating the positive–negative charges allowing DNA access, while methylation of specific lysines or arginines has been associated to the opposite effect [10,11]. Undoubtedly, this is not a general statement, since acetylation and methylation of histone tails, depending on both residues and positions, have been related either to transcriptional activation or repression [12].
3. Interpreting the “Chromatin Tale”
To activate a specific cellular program, the meaning implied in a peculiar set of histone tags must be disclosed. To this aim, readers display an affinity for specific PTMs and are essential to interpret the rapid interchange between different cellular states. They are usually found in large multi-protein complexes, which include also writers and/or erasers, able to integrate different signaling pathways at the chromatin level [13]. Furthermore, the specificity of a chromatin reader to its cognate PTM is built not only on the direct interaction between the modified residues and the reader’s binding pocket, but also on secondary contact deriving from the flanking histone sequences surrounding the modified residue [14]. For example, a conserved asparagine residue located in the proximity of the bromodomains (BRDs) binding pocket interacts with the acetyl-lysine (Kac) through an anchoring hydrogen bond. Moreover, a conserved tyrosine residue associates the Kac with a water mediated hydrogen bond [15]. Indeed, co-crystal structures with peptidic substrates demonstrated that the Kac is recognized by a central deep hydrophobic cavity, where it is anchored by a hydrogen bond to a fundamental asparagine residue [16]. Modifications in these highly conserved features results in defective BRD-Kac recognition.
Readers can identify different residues and also diverse modification of the same residue. The complexity is augmented when the same amino acid could undergo several degrees of modification: For example, lysines could exist as mono-methylated, di-methylated, or tri-methylated [13]. For these reasons, chromatin readers are divided into families containing specific binding motifs (Figure 1). Well-characterized examples of reader domains include: Bromodomains (BRDs) typically binding acetyl-lysine; chromatin organization modifier (chromodomains, CRDs), malignant brain tumor (MBT), proline–tryptophan–tryptophan–proline motif (PWWP) as well as Tudor domains generally associated with methyl-lysine, and plant homeodomain (PHD) associated to multiple substrates [17]. Even if 17 types of histone modifications have been assessed, besides acetylation and methylation ones, little is known about the possible reader domains deputed to decipher their role in transcriptional activation and/or repression. Recently, the YEATS domain has been associated to control cell transcription rates by associating with crotonylated lysine residues in active promoters and/or enhancers [18].
The primary readers of N-acetylation of lysine residues is a family of proteins containing an evolutionary conserved domain known as bromodomain. There are more than 40 human proteins containing bromodomains, the most studied one is the Bromodomain and Extra-terminal (BET) family. It comprises four members (BRD2, BRD3, BRD4, and BRDT), which share common structural features with two N-terminal tandem bromodomains. They play a fundamental role in transcriptional elongation and cell-cycle progression, interacting mostly with H4K5/8/12/16ac [19]. Besides BET proteins, BAF180 is a reader protein containing six bromodomains and displaying a distinct pattern of affinity for H3Ksac, which is involved in genome stability, proliferation, and DNA repair [20]. The chemical methylation of lysine residues is engaged by two major families called the Royal Family (accounting for Tudor, Chromo-, and Malignant Brain Tumor (MBT) domains) and the PHD fingers [21]. MBT domains selectively recognize mono and dimethyl-lysine and have been functionally associated with the repression of gene expression while their dysregulation has been linked to several disease states. L3MBTL3, for instance, is found to be deleted in homozygosis in patients with medulloblastoma [22]. Moreover, the Tudor-containing protein 3 (TDRD3) is known to bind methyl-arginine residues and its overexpression has a strong predictive value for poor prognosis in estrogen receptor-negative breast cancers [23].
Phosphorylation is usually linked to transcriptional regulation by extracellular signals, chromatin condensation during mitosis, and DNA damage, and to date, only the 14-3-3 family members and the breast cancer-associated protein carboxy-terminal (BRCT)-containing proteins are known to bind phosphorylated residues on histone tails [24]. Histone ubiquitylation is historically associated to the activation of DNA damage repair (DDR) mechanisms, following mostly double strand brakes (DSBs). The major domains deputed to interact with H2A and H2B ubiquitylated lysines are the ubiquitin-binding domain (UBD) of the UIM-, MIU-, and UBZ-types [25]. RAP80 is an H2AK63 ubiquitylated reader involved in the regulation of DSBs mediated by the homologous recombination (HR) pathway. Specifically, it recruits BRCA1 and other components of the multimeric BRCA1-A complex to the damaged chromatin [26]. Involved in the same pathway, RAD18 promotes HR-mediated DSB repair through the interaction with RAD51C and the SMC5/6 cohesion complex. Since it is scarcely studied, lysines crotonylation has been associated to transcriptional regulation comparable to acetylation. Four YEATS family proteins exist in humans, namely AF9 (encoded by the MLLT3—Mixed-Lineage Leukemia Translocated To Chromosome 3—gene), ENL (encoded by the MLLT1—Mixed-Lineage Leukemia Translocated To Chromosome 1—gene), glioma amplified sequence 41 (GAS41), and YEATS2/4, which participate with different chromatin-associated complexes involved in transcription elongation, histone modification, and chromatin remodeling [18].
Furthermore, multi-domain protein readers exist, containing two or more different domains through which they can bind different modified residues simultaneously. An example is represented by the Tripartite Motif-containing protein 24 (TRIM24) that can directly associate with chromatin via its tandem PHD-bromodomain modulating gene expression [27]. TRIM24 can bind to euchromatin juxtaposing the two reader domains, creating a single docking site able to interact with H3K23ac through the BRD and to unmethylated H3K4 by the PHD-domain, allowing, for example, estrogen receptor (ER) binding to distal estrogen-response elements [27]. In addition, the complexity of these structures could be extended when different regulatory domains co-exist in the same protein sequence: 53BP1 and RNF168 both contain an ubiquityl-lysine reader and a writer domain able to regulate the damaged site, stimulating either HR or the non-homologous end joining (NHEJ) repair pathways [25].
4. Epigenetic Alterations are a Common Place in Cancer
As epigenetic regulation plays a crucial role in normal cellular homeostasis, any variation on this theme could result in abnormal gene expression of key proteins, leading to the onset or the progression of a wide range of diseases, and particularly in cancer [28].
As chromatin constitutes the primary form of protein-DNA assembly in the nucleus, epigenetic modifications can influence all DNA-associated processes, such as transcription, replication, and DNA repair. All these mechanisms are intimately connected to the faithful interpretation and inheritance of the genetic material and therefore are central to inducing and maintaining cell fate choices. Hence, any aberration in this fine-tuned regulation might potentially lead to the accumulation of genomic lesions and ultimately to the loss of cell identity; these consequences are typically associated with the development of neoplasia. Cancer cells, in fact, frequently bear aberrations at readers’ expenses, such as point mutations, translocations, amplifications, and deletions [29]. These alterations can both transform and foster disease progression directly or by affecting gene expression controlled by dysregulated signaling pathways or oncogenic transcription factors [17]. For example, mutations in ING family members (PHD finger containing proteins) have been associated with malignances, such as melanoma and breast cancer [30]. Moreover, the six tandem bromodomain containing protein, BAF180, was found to undergo a loss of function mutations in clear cell renal cell carcinoma (CCRCC) [31]. Fusion of Nucleoporin 98 (NUP98) with the PHD containing part of JARID1A (or KDM5A - Lysine Demethylase 5A) drove alterations in leukemia [32]. In NUT-midline carcinoma, a translocation that fuses the BRD3 or BRD4 protein to the NUT transcriptional regulator creates an oncoprotein that, through binding to acetylated histones, is thought to promote transcription of proliferation genes (e.g., MYC proto-oncogene) [33]. As for lysine-crotonylated readers, GAS41 is an oncogene frequently amplified in human gliomas while AF9 and ENL are two general fusion partners translocated in human mixed lineage leukemia (MLL) [18].
Not only genetic alterations involving chromatin readers occur during neoplastic transformation, but also dysregulation in their expression has been assessed in diverse neoplasms. TRIM24 over-expression correlates with poor survival outcomes in breast cancer patients due to its oncogenic potential in driving ER activity [27]. The YEATS Domain Containing 4 (YEATS4), instead, is found to be overexpressed in several human cancers, such as lung adenocarcinoma, glioblastoma, and colorectal cancer (CRC), promoting cell proliferation through the inhibition of senescence and an increase in multipolar mitotic spindle formation [34]. Despite progresses in the understanding of the molecular events that can drive the epigenetic abnormalities underneath the cancer epigenome, how mammalian cells normally package their genomes for proper gene expression and maintenance of chromosome integrity are questions that remain to be elucidated. Tightly connected is the topic of better characterization of cardinal players in epigenetic programming. As shown in Table 1, most phosphorylation, ADP-ribosylation, and ubiquitylation readers are still elusive, preventing, on one side, a full understanding of the consequences related to HPTMs reading and, on the other side, exploitation of this knowledge for therapeutic purposes.
5. Druggable Epigenome
Unlike the genetic mutations, epigenetic changes are naturally reversible and thus catalyzed researchers’ attention as good targets for therapies directed to restore normal gene expression profile, hindering regulators’ dysfunction. Moreover, targeting the epigenome as a viable drug strategy has been fostered and supported by the U.S. Food and Drug Administration (FDA), who, in 2004, granted regular approval to the DNA methyltransferases inhibitor, 5-azacytidine (Vidaza©), for the treatment of all subtypes of myelodysplastic syndrome (MDS) [35]. A couple of years later, Vorinostat (Zolinza©) or suberolyanilidehydroxamic acid (SAHA), a histone deacetylase (HDAC) inhibitor, received FDA approval for the treatment of cutaneous manifestations of cutaneous T-cell lymphoma (CTCL) in patients with progressive, persistent, or recurrent disease [36]. From that instant, epigenetic therapy gained momentum and several chemical agents (epidrugs) mostly targeting chromatin-related enzymes (i.e., writers and erasers) have been developed, entering preclinical studies. Novel and more specific DNMT and HDAC inhibitors were screened in vitro in experimental models of multiple cancer types, such as melanoma, breast cancer, CRC, osteosarcoma, and more.
EZH2 is a methyltransferase catalyzing H3K27 tri-methylation, an HPTM essential for chromatin compaction and gene silencing [37]. Due to its frequent overexpression and gain-of-function mutations [37,38] in solid cancers and lymphomas, several pharmaceutical companies have embarked on high-throughput screening campaigns, leading to the discovery of small-molecule compounds, inhibiting specifically its methyltransferase activity [39]. EZH2 inhibitors, EPZ-6438 and GSK2816126, for instance, showed anti-neoplastic activity in acute myeloid leukemia (AML) and are currently in clinical trials of lymphoma treatment (Table 2). Moreover, several LSD1 (histone demethylase with specificity toward H3K4 mono/di-methylation) inhibitors are now in clinical trials in refractory AML displaying no PML-RARα rearrangement (Table 2).
As inhibition of specific enzymes represents a classical drug developing protocol, more challenging is the inhibition of the binding pocket of a histone-modified reader. Great effort was spent on this subject until researchers’ attention has tremendously been influenced by the discovery of selective inhibitors targeting the BET family of acetyl-lysine readers. Two distinct experimental approaches have led to the development of the first two BET inhibitors (BETis), the benzodiazepine I-BET762 (also known as GSK525762) and the thienodiazepine JQ1, which have been shown to be effective in the downregulation of the MYC oncogene in multiple cancer subtypes [29,40]. BET inhibition efficacy is based on the disruption of BET proteins’ interaction with acetylated histones by binding the acetyl-lysine recognition pocket of these chromatin readers. In addition to MYC downregulation, BET inhibitors’ biological effects resulted by modulating the expression of cell growth-related proteins. For example, BET inhibition induced a decrease in cell viability that turns out to be specifically related to cell cycle-linked genes’ downregulation (i.e., MCM5) in thyroid cancer [41]. Moreover, diverse studies have highlighted how BRD4 is able to regulate DNA damage repair, opening a novel theme on BET-inhibitors related therapeutic approaches [42,43]. BET inhibition results indeed in disproportionately large changes in gene expression, putatively explained by the association of BRD4 with exceptionally large enhancer elements, called “super-enhancers,” which are involved in lineage-specific gene regulation [44].
Overall, BET inhibition interferes with cancer cell cycle progression and DNA repair, hindering tumor progression both in vitro and in vivo. The reversal of cancer cell phenotype (i.e., the promotion of differentiation and growth impairment) because of BET inhibition provided the first proof of concept that readers can act as a potential therapeutic target for cancer treatment [45]. Since JQ1 and GSK525762 development, multiple BETis have been developed showing significant therapeutic effect in a wide range of human diseases [33]. Given that, BET inhibitors have entered clinical trials, proving their anti-neoplastic effects in diverse sub-types of hematological malignancies and solid tumors [46].
A second step in epigenetic readers-based drug discovery has been made by targeting PHD finger-containing proteins that harbor a “reading” specific site for H3K4 methylation. Disulfiram, an acetaldehyde dehydrogenase inhibitor previously FDA-approved in the treatment of alcoholism, and amiodarone-derived compounds were found to inhibit JARID1A PHD finger bound to H3K4me, possibly through physical alterations, causing the ejection of a structural zinc [47,48]. Both compounds showed anticancer effects in preclinical models of AML in which methyl-readers frequently undergo genetic fusions, causing aberrant transactivation of the developmental genes required to maintain the myeloid progenitor state [21,30,47]. However, their potency and selectivity seems poor and ligand-competitive chemicals remain to be developed for these PHD-containing oncoproteins. Additionally, the small molecule, UNC1215, was reported to inhibit L3MBTL3A activity, an MBT containing protein that selectively recognized mono- and di-methyl-lysines, competing with its endogenous targets, such as BCL2 Associated Transcription Factor 1 (BCLAF1), in a comparable way as JQ1 binds to BRD4 [22]. This potent and selective chemical probe represents a promising tool in the field of epigenetic therapy of histone methylation dysregulation, but further knowledge should be developed until UNC1215 may enter human testing.
Furthermore, next generation sequencing (NGS)-based approaches have highlighted how multi-alteration processes underpin tumor development and progression. These high-throughput techniques have great benefits for intensifying the list of putative targets or effectors in drug-based approaches, shedding light on tumor identity and highlighting further therapeutic targets.
6. New Challenges
While acetylation readers have been successfully exploited as therapeutic targets in different cancer models, little breakthrough has been made in targeting other chromatin readers. Few recent studies have focused on the dysregulation of YEATS4, known to hinder senescence and to foster cell proliferation, highlighting that its inhibition by means of endogenous or exogenous RNA interference induced apoptosis of CRC cells [34]. These data delineate it as a putative effective target for the clinical treatment of CRC, but, nowadays, no chemical probes targeting YEATS-containing proteins have been synthesized yet. Even if rapid progress has been made in the development of new small-molecule tools targeting the PHD finger domains, selective inhibitors, i.e., targeting TRIM24, frequently overexpressed in breast cancer and associated with poor overall survival and tumor progression, have not yet been developed [48,49].
From a therapeutic point of view, BET inhibitors have paved the way for chromatin readers’ inhibition in clinical trials. Over the past decade, BET inhibitors efficacy has been confirmed in several preclinical cancer models, including prostate, breast, colon pancreas, liver, thyroid, and brain carcinoma [33], evaluating a high level of BETi-related cancer sensitivity. To date, 26 clinical trials employing BET inhibitors exist (Table 3), 12 of which are combination trials with hormonal therapy (i.e., fulvestrant, enzalutamide), target-specific antibodies (i.e., anti-PD-L1, anti-JAK2, anti-BCL2), or other epidrugs (i.e., DNA methylation inhibitors).
Combination therapies involving BET inhibitors are a promising therapeutic approach since, besides the biological advantage in multi-target inhibition, they are meant to overcome the impermanent cytotoxic effects proved as single agents [39,46,50]. In fact, despite the promising results collected both in vitro and in vivo, early clinical trials with BET inhibitors have had sundry results, with responses that tend to be short-lived. Side effects included fatigue (50%), thrombocytopenia (29%), decreased appetite (21%), diarrhea (18%), dysgeusia (14%), nausea (14%), neutropenia (11%), and vomiting (11%) [46,51].
Notwithstanding, clinical research on epidrugs is still ongoing, focusing on optimizations for solid tumors treatments, firstly, to fully target the tumor burden in toto. Recently, novel approaches employing carriers, such as multimeric proteins, synthetic or biological nanoparticles, and microbubbles, have shown promising results in preclinical settings [52]. To this aim, HDACi have been encapsulated to enhance tumor targeting hindering side effects, showing an augmented pro-apoptotic effect when combined with radiotherapy [53]. Moreover, combination therapies could benefit from this nanotechnological approach, enclosing multiple small molecules in one nanoparticle targeting cancer cells, reducing side effects coming from single drugs itself, and directing the therapeutic potential right on the tumor burden. To this aim, recently, JQ1 has been inserted in a functionalized nanoparticle together with temozolomide to cross blood-brain barrier and deliver this combination therapy to glioblastoma [54].
Besides epidrugs, epigenetic therapy might rest on small RNA delivery or gene therapy (Figure 2). Adenoviral vectors (AV) used to carry therapeutic genes are common vectors used worldwide within clinical trials and account for most gene therapy-based approaches. AVs are well-tolerated and for decades have been used to induce a local antitumoral immune response; moreover, FDA approved AVs are used in combination with chemotherapy in squamous cell carcinoma of the head and neck (HNSCC) [55]. Possessing mild side effects (i.e., immune system-based reactions, long-term activity), AVs are valuable candidate effects of cancer treatments. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system has surely revolutionized the field of gene editing and represents a cutting-edge strategy for gene disruption in the host genome [56,57]. Briefly, the CRISPR/Cas9 approach is based on a synthetic fused chimeric single guide RNA (sgRNA) owing a sequence that is designed to be complementary to a target DNA site, introducing or deleting specific sequences or mutations [57,58]. For instance, gene editing with CRISPR/Cas9 could be employed to correct for oncogene deriving from fusion proteins. As an example, BRD4-NUT translocation is the primary determinant of NMC, in which BRD4 protein to the NUT transcriptional regulator drives the expression of BRD4, promoting transcription of cancer progression-related genes. The aforementioned techniques might aid for the formation of the oncoprotein, hindering tumor expansion.
Besides viral vectors, a huge slice of biotechnological research has produced several polymeric vesicles for delivery within the tumor mass of either targeted drugs or RNA-interference-based approaches (i.e., siRNAs, shRNAs, miRNAs) [59,60,61]. Nowadays, dozens of miRNAs hindering writers, erasers, and readers have been characterized. Indeed, miR-1340 and miR-608 directly suppressed BRD4 by binding to its coding sequence, reducing in vivo tumor growth in a xenograft mouse model of NMC and hepatocellular carcinoma, respectively [62,63]. MiR-124a inhibited PHF19 over-expression and hindered cell growth in human glioma [64], while miR-19a and miR-19b down-regulated the expression of BARD1 in leukemia [65]. Further, synthetic RNA interference-based approaches have been extensively used in preclinical settings to dissect molecular consequences derived from readers’ inhibition, such as PHF19 in melanoma [66], PHF20L1 (Tudor domain-containing protein) in breast cancer [67], and WDR5 in leukemia and pancreatic cancer [68,69], all inhibiting tumor progression in vitro and/or in mouse models.
Nanoparticles are attractive carries because of their non-immunogenic nature and the possibility of being tissue specific with the loading of specific biomarkers on their surface [70,71,72]. All these cutting-edge technologies could be employed to package epidrugs and small RNAs that are not currently usable in vivo, i.e., unstable in plasma, or to prevent systemic side-effects, typical issues in epigenetic therapies. Nonetheless, to date, epigenetic modulation through small molecules (i.e., epidrugs) is by far the most used strategy in clinical workflows, notwithstanding the presence of systemic side effects or little therapeutic benefit in some cancer sub-types.
A deeper understanding of the interconnections between different readers-dependent epigenetic pathways in physiological and pathological conditions will surely help in managing epigenetic therapy.
7. Conclusions
Even if a lot of effort has been made, few chemical probes targeting epigenetic readers have been identified to date. Further evaluation must be accomplished to better characterize both readers’ dysfunction in cancer and chemical compounds able to specifically inhibit their functions. Moreover, small molecules targeting epigenetic-related proteins (i.e., writers, eraser and readers) still hold pros and cons. HDAC inhibitors therapy, for instance, lacks in therapeutic effectiveness in ovarian cancer, glioblastoma, and hepatocellular carcinoma [50,73,74]. Although potent as drugs for clinical use, BET inhibitors are not able to singularly bind a bromodomain containing family member, a feature that could certainly reduce therapy-related side effects [13,50].
A step forward in epigenetic therapy has been made with drug combination between epigenetic drugs and chemotherapy (platinum salts) or signaling pathway inhibitors (tyrosine kinases inhibitors or endocrine therapy) [50]. These combination therapies are, nowadays, entering numerous clinical trials, as summarized in Table 3. Surely, better models of the prediction of adverse events are needed to hinder side effects related to epidrugs (either alone or in combination). Another criticism is based on pharmacokinetics (i.e., half-life) that could prevent the use of reader’s inhibitors in vivo. Some small molecules, indeed, possess a short half-life (i.e., JQ1 stability in plasma is about 1 h), impeding their use in clinical trials. Vesicles or carriers could partially solve this issue, allowing drugs to penetrate the tumor burden, releasing the chemical probe within the mass, and avoiding its rapid degradation within the plasma. Moreover, most of the drug screening made so far has been based on focused libraries, but this has been revealed as an unsuccessful strategy for the so-called “difficult targets”, in which the binding domain is shallowed, or crystallography evaluations have classified them as undruggable. A different approach based on small-peptide binging, mirroring an RNA interference approach, may be a better strategy [17,55,59,60]. Ultimately, epidrugs-treated patients sometimes experience resistance or tumor relapses due to epigenetic reprogramming of tumor cells, which no-more rely on the pathway targeted by that epidrug or for the persistence of cancer stem cells (CSCs), hardly embedded within the tumor burden and unattainable by epidrugs.
In conclusion, investigation leading toward the identification of small-molecules that specifically disrupt dysregulated chromatin-binding proteins, such as those defined by expressed translocations or inappropriate over-expression, could shed light on how chromatin dynamics regulate gene expression. Moreover, dissecting readers’ biology and mechanisms coupled to the newest genomic analysis based on next-generation sequencing could reveal epigenetic addiction underpinning tumorigenic transformation and cancer progression, making precision medicine even closer.
Author Contributions
C.M., data acquisition, analysis and interpretation, manuscript preparation; S.B., data acquisition, editing and critical reading of the manuscript; D.R., editing and critical reading of the manuscript; G.D., editing and critical reading of the manuscript. All authors approved the final version of the manuscript.
Funding
The study was financially supported by the Department of Health Sciences, “Magna Graecia” University of Catanzaro, and by a grant to GD from Italian Minister of Foreign Affairs and International Cooperation (MAECI) (Progetti Grande Rilevanza Italia-Serbia No. PGR00223).
Conflicts of Interest
The authors declare no conflict of interest.
References
- El Kennani, S.; Crespo, M.; Govin, J.; Pflieger, D. Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes 2018, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Abedin, S.M.; Boddy, C.S.; Munshi, H.G. BET inhibitors in the treatment of hematologic malignancies: Current insights and future prospects. Oncotargets 2016, 9, 5943–5953. [Google Scholar]
- Bowman, G.D.; Poirier, M.G. Post-Translational Modifications of Histones That Influence Nucleosome Dynamics. Chem. Rev. 2015, 115, 2274–2295. [Google Scholar] [CrossRef]
- Celano, M.; Mio, C.; Sponziello, M.; Verrienti, A.; Bulotta, S.; Durante, C.; Damante, G.; Russo, D. Targeting post-translational histone modifications for the treatment of non-medullary thyroid cancer. Mol. Cell. Endocrinol. 2018, 409, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Bartke, T.; Borgel, J.; DiMaggio, P.A. Proteomics in epigenetics: New perspectives for cancer research. Brief. Funct. Genom. 2013, 12, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tollefsbol, T.O. Age-related epigenetic drift and phenotypic plasticity loss: Implications in prevention of age-related human diseases. Epigenomics 2016, 8, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Filip, M. A Comprehensive View of the Epigenetic Landscape. Part II: Histone Post-translational Modification, Nucleosome Level, and Chromatin Regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197. [Google Scholar] [CrossRef]
- Russo, D.; Durante, C.; Bulotta, S.; Puppin, C.; Puxeddu, E.; Filetti, S.; Damante, G. Targeting histone deacetylase in thyroid cancer. Expert Opin. Targets 2013, 17, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Choi, S. Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets. Genes 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharm. 2015, 172, 2716–2732. [Google Scholar] [CrossRef] [PubMed]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharm. 2016, 106, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lori, L.; Pasquo, A.; Lori, C.; Petrosino, M.; Chiaraluce, R.; Tallant, C.; Knapp, S.; Consalvi, V. Effect of BET Missense Mutations on Bromodomain Function, Inhibitor Binding and Stability. PLoS ONE 2016, 11, e0159180. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Greschik, H.; Schüle, R.; Günther, T. Selective targeting of epigenetic reader domains. Expert Opin. Drug Discov. 2017, 12, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, Y.; Xiong, X.; Chen, Z.; Li, H. YEATS Domain—A Histone Acylation Reader in Health and Disease. J. Mol. Biol. 2017, 429, 1994–2002. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, P.M.; Chambers, A.L.; Oliver, A.W.; Downs, J.A. Cancer and the bromodomains of BAF180. Biochem. Soc. Trans. 2012, 40, 364–369. [Google Scholar] [CrossRef]
- Wagner, T.; Robaa, D.; Sippl, W.; Jung, M. Mind the Methyl: Methyllysine Binding Proteins in Epigenetic Regulation. ChemMedChem 2014, 9, 466–483. [Google Scholar] [CrossRef] [PubMed]
- James, L.I.; Barsyte-Lovejoy, D.; Zhong, N.; Krichevsky, L.; Korboukh, V.K.; Herold, M.J.; MacNevin, C.J.; Norris, J.L.; Sagum, C.A.; Tempel, W.; et al. Discovery of a chemical probe for the L3MBTL3 methyl-lysine reader domain. Nat. Chem. Biol. 2013, 9, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Goulet, I.; Boisvenue, S.; Mokas, S.; Mazroui, R.; Côté, J. TDRD3, a novel Tudor domain-containing protein, localizes to cytoplasmic stress granules. Hum. Mol. Genet. 2008, 17, 3055–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicka, A.; Seiser, C. Sensing core histone phosphorylation—A matter of perfect timing. Biochim. Biophys. Acta 2014, 1839, 711–718. [Google Scholar] [CrossRef]
- Smeenk, G.; Mailand, N. Writers, Readers, and Erasers of Histone Ubiquitylation in DNA Double-Strand Break Repair. Front. Genet. 2016, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Matunis, M.J.; Wolberger, C. RAP80, ubiquitin and SUMO in the DNA damage response. J. Mol. Med. 2017, 95, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Appikonda, S.; Thakkar, K.N.; Barton, M.C. Regulation of gene expression in human cancers by TRIM24. Drug Discov. Today Technol. 2016, 19, 57–63. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.-M. Chemical modulators for epigenome reader domains as emerging epigenetic therapies for cancer and inflammation. Curr. Opin. Chem. Biol. 2017, 39, 116–125. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J.P. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. NUP98-PHF23 is a chromatin modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of PHD domain histone reader function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef]
- Brugarolas, J. PBRM1 and BAP1 as Novel Targets for Renal Cell Carcinoma. Cancer J. Sudbury Mass 2013, 19, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, T.; Chen, W.C.; Göllner, S.; Howell, L.; Jin, L.; Hebestreit, K.; Klein, H.-U.; Popescu, A.C.; Burnett, A.; Mills, K.; et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 2012, 18, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G.; Sahai, V.; Redig, A.J.; Collier, K.A.; Eckerdt, F.D.; Munshi, H.G. Targeting BET bromodomain proteins in solid tumors. Oncotarget 2016, 7, 53997–54009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, K.; Yang, J.; Hu, Y.; Deng, A. Knockdown of YEATS4 inhibits colorectal cancer cell proliferation and induces apoptosis. Am. J. Transl. Res. 2015, 7, 616–623. [Google Scholar] [PubMed]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Azacitidine (5-azacytidine, VidazaTM) for Injectable Suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoy, G.; DiMaggio, P.A.; Chan, E.Y.; Zee, B.M.; Blanco, M.A.; Bryant, B.; Flaniken, I.Z.; Liu, S.; Kang, Y.; Trojer, P.; et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 2013, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Sponziello, M.; Durante, C.; Boichard, A.; Dima, M.; Puppin, C.; Verrienti, A.; Tamburrano, G.; Di Rocco, G.; Redler, A.; Lacroix, L.; et al. Epigenetic-related gene expression profile in medullary thyroid cancer revealed the overexpression of the histone methyltransferases EZH2 and SMYD3 in aggressive tumours. Mol. Cell. Endocrinol. 2014, 392, 8–13. [Google Scholar] [CrossRef]
- Lu, R.; Wang, G.G. Pharmacologic Targeting of Chromatin Modulators as Therapeutics of Acute Myeloid Leukemia. Front. Oncol. 2017, 7, 241. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Mio, C.; Lavarone, E.; Conzatti, K.; Baldan, F.; Toffoletto, B.; Puppin, C.; Filetti, S.; Durante, C.; Russo, D.; Orlacchio, A.; et al. MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr. Relat. Cancer 2016, 23, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Loreto, C.D.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2018, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- Conticello, C.; Martinetti, D.; Adamo, L.; Buccheri, S.; Giuffrida, R.; Parrinello, N.; Lombardo, L.; Anastasi, G.; Amato, G.; Cavalli, M.; et al. Disulfiram, an old drug with new potential therapeutic uses for human hematological malignancies. Int. J. Cancer 2012, 131, 2197–2203. [Google Scholar] [CrossRef] [Green Version]
- Igoe, N.; Bayle, E.D.; Fedorov, O.; Tallant, C.; Savitsky, P.; Rogers, C.; Owen, D.R.; Deb, G.; Somervaille, T.C.P.; Andrews, D.M.; et al. Design of a Biased Potent Small Molecule Inhibitor of the Bromodomain and PHD Finger-Containing (BRPF) Proteins Suitable for Cellular and In Vivo Studies. J. Med. Chem. 2017, 60, 668–680. [Google Scholar] [CrossRef]
- Palmer, W.S. Development of small molecule inhibitors of BRPF1 and TRIM24 bromodomains. Drug Discov. Today Technol. 2016, 19, 65–71. [Google Scholar] [CrossRef]
- Ronnekleiv-Kelly, S.M.; Sharma, A.; Ahuja, N. Epigenetic therapy and chemosensitization in solid malignancy. Cancer Treat. Rev. 2017, 55, 200–208. [Google Scholar] [CrossRef]
- Aftimos, P.G.; Bechter, O.; Awada, A.; Jungels, C.; Dumez, H.; Huyvaert, N.; Costermans, J.; Lee, C.; Meeus, M.-A.; Burkard, U.; et al. Phase I first-in-man trial of a novel bromodomain and extra-terminal domain (BET) inhibitor (BI 894999) in patients (Pts) with advanced solid tumors. J. Clin. Oncol. 2017, 35, 2504. [Google Scholar] [CrossRef]
- Cramer, S.A.; Adjei, I.M.; Labhasetwar, V. Advancements in the delivery of epigenetic drugs. Expert Opin. Drug Deliv. 2015, 12, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.M.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.-L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Holkers, M.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A.F.V. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci. Rep. 2014, 4, 5105. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current Progress of siRNA/shRNA Therapeutics in Clinical Trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, G.E.; Maggisano, V.; Celano, M.; Cosco, D.; Mignogna, C.; Baldan, F.; Lepore, S.M.; Allegri, L.; Moretti, S.; Durante, C.; et al. Anti-hTERT siRNA-Loaded Nanoparticles Block the Growth of Anaplastic Thyroid Cancer Xenograft. Mol. Cancer 2018, 17, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Tonouchi, E.; Gen, Y.; Muramatsu, T.; Hiramoto, H.; Tanimoto, K.; Inoue, J.; Inazawa, J. miR-3140 suppresses tumor cell growth by targeting BRD4 via its coding sequence and downregulates the BRD4-NUT fusion oncoprotein. Sci. Rep. 2018, 8, 4482. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Meng, D.; Zhang, S.-H.; Zhang, Y.; Deng, Z.; Kong, L.-B. microRNA-608 inhibits human hepatocellular carcinoma cell proliferation via targeting the BET family protein BRD4. Biochem. Biophys. Res. Commun. 2018, 501, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ji, H.; Tang, H.; Xu, Z. microRNA-124a suppresses PHF19 over-expression, EZH2 hyper-activation, and aberrant cell proliferation in human glioma. Biochem. Biophys. Res. Commun. 2018, 503, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Lepore, I.; Dell’Aversana, C.; Pilyugin, M.; Conte, M.; Nebbioso, A.; De Bellis, F.; Tambaro, F.P.; Izzo, T.; Garcia-Manero, G.; Ferrara, F.; et al. HDAC Inhibitors Repress BARD1 Isoform Expression in Acute Myeloid Leukemia Cells via Activation of miR-19a and/or b. PLoS ONE 2013, 8, e83018. [Google Scholar] [CrossRef] [PubMed]
- Ghislin, S.; Deshayes, F.; Middendorp, S.; Boggetto, N.; Alcaide-Loridan, C. PHF19 and Akt control the switch between proliferative and invasive states in melanoma. Cell Cycle 2012, 11, 1634–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Liu, L.; Shan, W.; Yang, Z.-Q. An integrated genomic analysis of Tudor domain–containing proteins identifies PHD finger protein 20-like 1 (PHF20L1) as a candidate oncogene in breast cancer. Mol. Oncol. 2016, 10, 292–302. [Google Scholar] [CrossRef]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754. [Google Scholar] [CrossRef] [Green Version]
- Carugo, A.; Genovese, G.; Seth, S.; Nezi, L.; Rose, J.L.; Bossi, D.; Cicalese, A.; Shah, P.K.; Viale, A.; Pettazzoni, P.F.; et al. In Vivo Functional Platform Targeting Patient-Derived Xenografts Identifies WDR5-Myc Association as a Critical Determinant of Pancreatic Cancer. Cell Rep. 2016, 16, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Paolino, D.; Cosco, D.; Gaspari, M.; Celano, M.; Wolfram, J.; Voce, P.; Puxeddu, E.; Filetti, S.; Celia, C.; Ferrari, M.; et al. Targeting the thyroid gland with thyroid-stimulating hormone (TSH)-nanoliposomes. Biomaterials 2014, 35, 7101–7109. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Shi, K.; Qu, Y.; Chu, B.; Qian, Z. Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor. Mol. Methods Clin. Dev. 2018, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Chen, R.; Chen, X.; Zhang, H.; Song, L.; Cui, W.; Zhang, J.; Ye, D.; Zhang, Y.; Wang, Z. Oridonin-loaded and GPC1-targeted gold nanoparticles for multimodal imaging and therapy in pancreatic cancer. Int. J. Nanomed. 2018, 13, 6809–6827. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.-T.; Bai, B.; Zhu, J.; Di, C.-X.; Li, X.; Zhou, W.-C. Epigenetic actions of environmental factors and promising drugs for cancer therapy. Oncol. Lett. 2018, 15, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef] [PubMed]
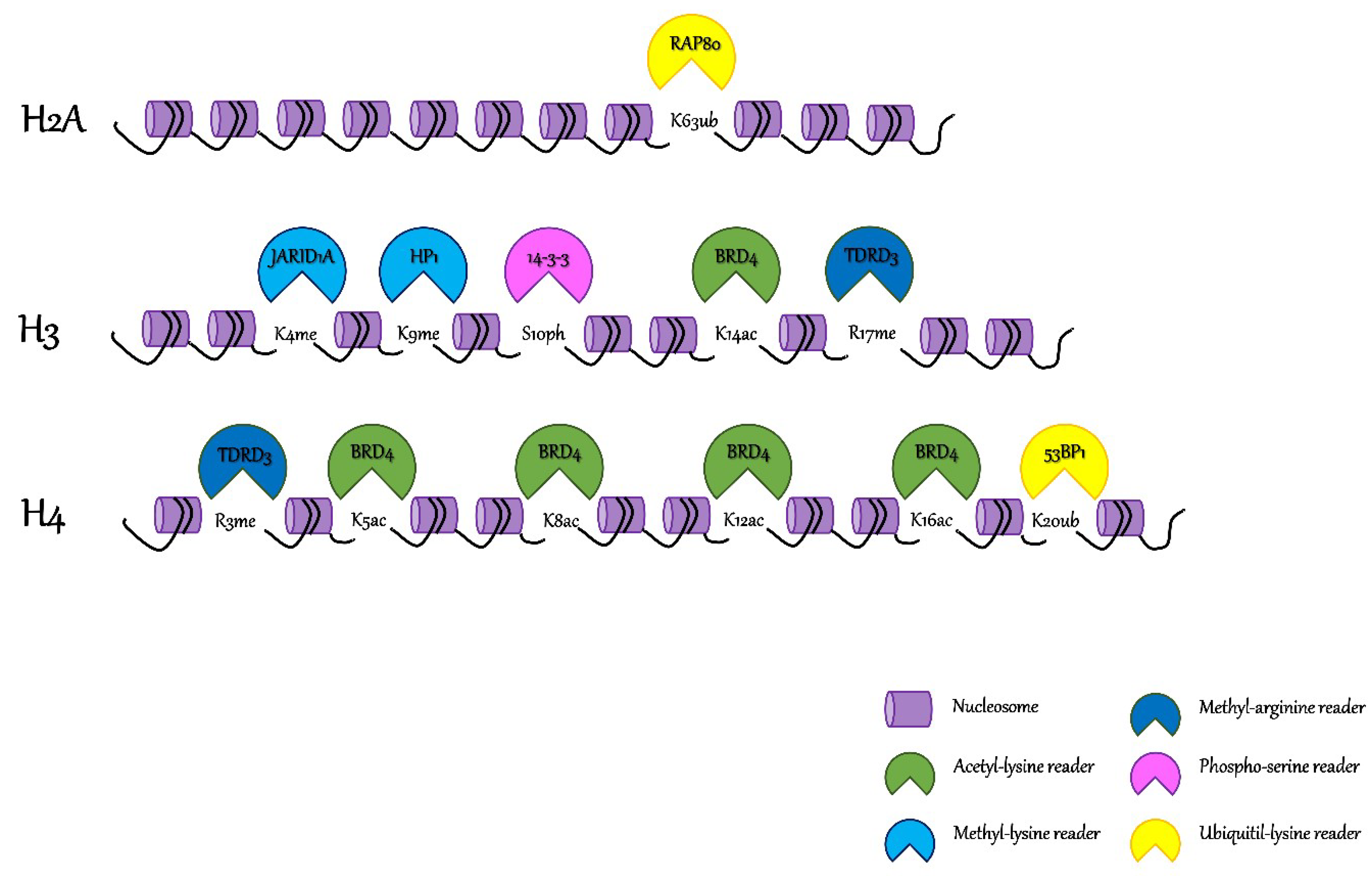
Figure 1.
Post-translational modifications (PTMs) on histones tails and their related chromatin readers. Eukaryotic DNA is packaged and subdivided into functional units called nucleosomes (here represented by a purple cylinder). Readers of lysine acetylation (i.e., Bromodomain-containing protein 4 (BRD4)) are represented in green, readers of lysine methylation (i.e., heterochromatin protein 1 (HP1) and Lysine-specific demethylase 5A (JARID1A)) are represented in light blue, readers of arginine methylation (i.e., Tudor domain-containing protein 3 (TDRD3)) are represented in dark blue, readers of lysine ubiquitylation (i.e., Tumor suppressor p53-binding protein 1 (53BP1) and receptor associated protein 80 (RAP80)) are represented in yellow, and readers of serine phosphorylation (i.e., 14-3-3) are represented in pink.
Figure 1.
Post-translational modifications (PTMs) on histones tails and their related chromatin readers. Eukaryotic DNA is packaged and subdivided into functional units called nucleosomes (here represented by a purple cylinder). Readers of lysine acetylation (i.e., Bromodomain-containing protein 4 (BRD4)) are represented in green, readers of lysine methylation (i.e., heterochromatin protein 1 (HP1) and Lysine-specific demethylase 5A (JARID1A)) are represented in light blue, readers of arginine methylation (i.e., Tudor domain-containing protein 3 (TDRD3)) are represented in dark blue, readers of lysine ubiquitylation (i.e., Tumor suppressor p53-binding protein 1 (53BP1) and receptor associated protein 80 (RAP80)) are represented in yellow, and readers of serine phosphorylation (i.e., 14-3-3) are represented in pink.

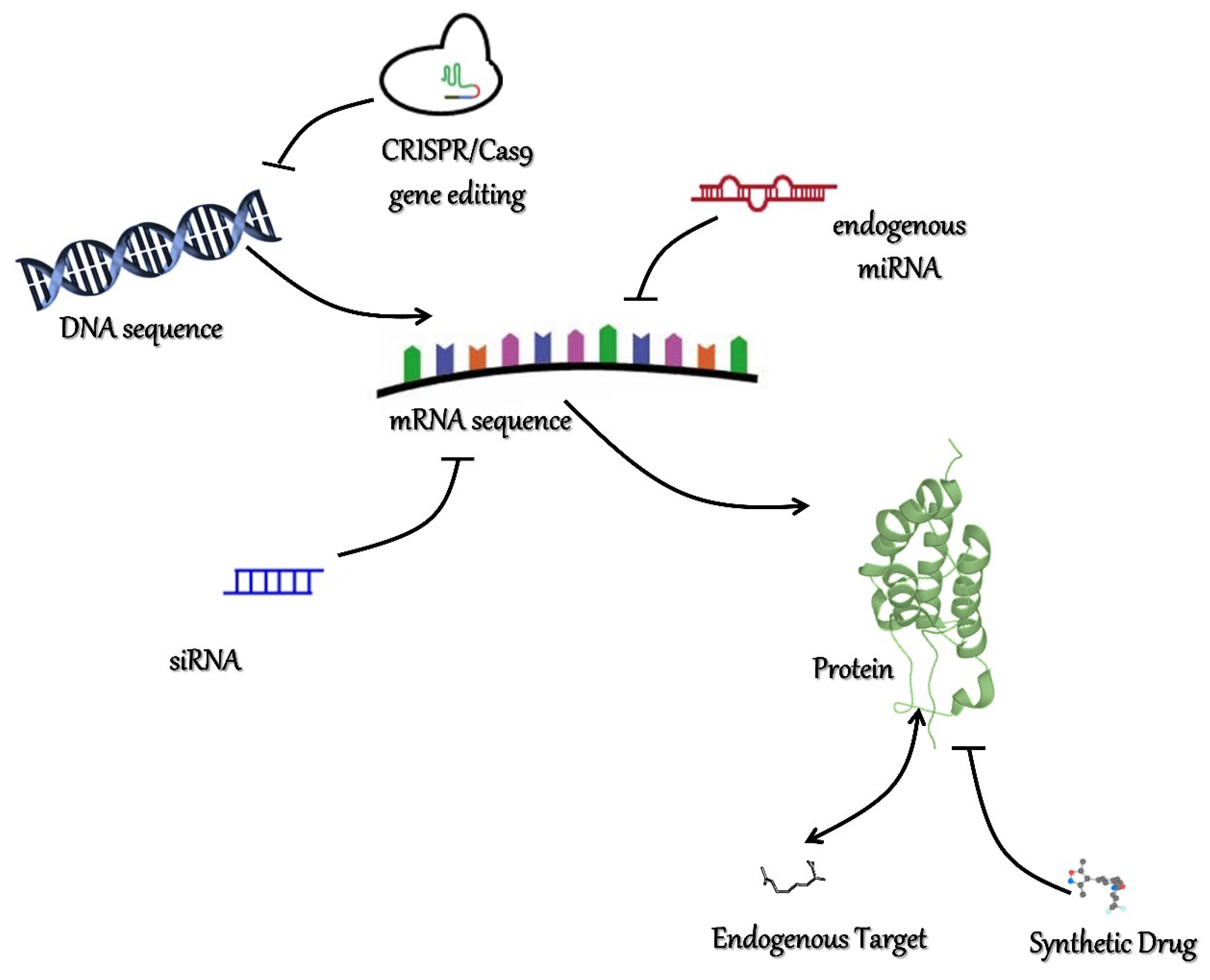
Figure 2.
Strategies in chromatin readers’ inhibition. Schematic representation of the diverse methods in readers’ inhibition. Mutations involving readers’ coding sequence or promoters/enhancers’ sequence and causing its mis-regulation could be corrected by the CRISPR/Cas9 gene editing technique. Aberrant gene expression might be suppressed by RNA interference techniques, such as endogenous miRNA targeting or synthetic siRNA delivery. Deregulation could also be hindered at protein levels by means of small molecules acting as exogenous competitors or target mimicking.
Figure 2.
Strategies in chromatin readers’ inhibition. Schematic representation of the diverse methods in readers’ inhibition. Mutations involving readers’ coding sequence or promoters/enhancers’ sequence and causing its mis-regulation could be corrected by the CRISPR/Cas9 gene editing technique. Aberrant gene expression might be suppressed by RNA interference techniques, such as endogenous miRNA targeting or synthetic siRNA delivery. Deregulation could also be hindered at protein levels by means of small molecules acting as exogenous competitors or target mimicking.


{kind=link}
{kind=link}
Table 1.
Summary of the main histone post-translational modifications (HPTMs) and their correlated regulatory enzymes.
Table 1.
Summary of the main histone post-translational modifications (HPTMs) and their correlated regulatory enzymes.
| Enzymes | Residues | Families | Components |
|---|---|---|---|
| Methylation | |||
| Writers | Lysine | Methyltransferases (HMTs) | EZH1/2, MLL1-5, SET1/7/9, SUV39h1/2 SUV40h1/2, G9a, EHMT1, NSD1/2, SMYD2, DOT1L |
| Arginine | Methyltransferases (HMTs) | PRMT1/2/4/5/6/7, CARM1 | |
| Erasers | Lysine | Demethylases (KDMs) | UTX, JMJD3, KDM1A/B, KDM2A/B, KDM4A/B/C/D, KDM5A/B/C/D, PHF2/8 JHDM1a/b, JHDM2a/b, JMJD2A/B/C/D |
| Arginine | Demethylases (RDMs) | JMJD6 | |
| Readers | Lysine | Chromodomains Tudor domains PWWP domains Ankyrin repeats | MOF, MRG15 MBT, PHF1/19, TDRD7 BRPF1, NSD1-3 G9a/GLP |
| Arginine | Tudor domains | WDR5, TDRD3, SMN1 | |
| Acetylation | |||
| Writers | Lysine | Acetyltransferases (HATs) | KAT2A/B, KAT3A/B, KAT6/5/7/8, Tip60, CREBBP, EP300, PCAF |
| Erasers | Lysine | Deacetylases | HDAC1/2/3/4/5/7/8/9/11, SIRT1/2/6/7 |
| Readers | Lysine | Bromodomains | BRD2/3/4/T |
| Phosphorylation | |||
| Writers | Serine | Kinases | CDK1/2, MSK1/2, Mst1, ATR, ATM, RSK2, AMPK, IKK-alpha, AuroraB |
| Threonine | Kinases | Haspin/Gsg2, Dlk/Zip | |
| Tyrosine | Kinases | Mst1, WSTF | |
| Erasers | Serine | Phosphatases | PP2A1, PP1 |
| Threonine | Phosphatases | PPgamma | |
| Tyrosine | Phosphatases | EYA1/3 | |
| Readers | Serine | 14-3-3 proteins BRCT domain | 14-3-3β/γ/η/ε/μ |
| Threonine | BIR domain | XRCC1, NBS1, BARD1 | |
| Tyrosine | PTB domain | ||
| Ubiquitynation | |||
| Writers | Lysine | Ubiquitin-ligases | BRCA1-BARD1, RING1A/RING1B/BMI1 |
| Erasers | Lysine | Isopeptidases | OTUB1/2, BRCC36, USP3/16/26/44 |
| Readers | Lysine | 53BP1 | |
| ADP-ribosylation | |||
| Writers | Glutamate Arginine Glutamate | ADP-ribosyltransferases | PARP1 |
| Erasers | Glutamate Arginine Glutamate | ADP-ribosylhydrolases | PARG, MDO1/2, TARG |
| Readers | Glutamate Arginine Glutamate | Macrodomains PBZ WWE domain | RNF146 APLF, CHFR |
PWWP: Pro-Trp-Trp-Pro; BRCT: BRCA1 C Terminus; BIR: bacuolavirus IAP Repeat; PTB: Phosphotyrosine-binding; PBZ: PAR-binding zinc finger; WWE: Trp-Trp-Glu.
Table 2.
Some of the clinical trials investigating the antineoplastic effects of chromatin writers/erasers inhibitors.
Table 2.
Some of the clinical trials investigating the antineoplastic effects of chromatin writers/erasers inhibitors.
| Target | Intervention | Status | Condition | Study Type | Phase | NCT Number |
|---|---|---|---|---|---|---|
| EZH2 | SHR2554 | RECRUITING | AML and myelodysplastic syndromes | INTERVENTIONAL | I | NCT03603951 |
| EPZ-6438 | RECRUITING | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I/II | NCT01897571 | |
| CPI-1205 | RECRUITING | Advanced Solid Tumors | INTERVENTIONAL | I/II | NCT03525795 | |
| DOT1L | EPZ-5676 | COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | I | NCT02141828 |
| PRMT5 | JNJ-64619178 | RECTUITING | Advanced solid tumors | INTERVENTIONAL | I | NCT03573310 |
| GSK3326595 | RECTUITING | Advanced solid tumors | INTERVENTIONAL | I | NCT02783300 | |
| LSD1 | IMG-7289 | ACTIVE | AML and myelodysplastic syndromes | INTERVENTIONAL | I | NCT02842827 |
| INCB059872 | RECRUITING | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I/II | NCT02712905 | |
| HDAC | Panobinostat (LBH589) | COMPLETED | HL and MM | INTERVENTIONAL | III | NCT01034163 |
| ACTIVE | Hematologic Malignancies | INTERVENTIONAL | II | NCT01802879 | ||
| COMPLETED | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I | NCT00472368 | ||
| COMPLETED | CTCL | INTERVENTIONAL | II/III | NCT00425555 | ||
| Belinostat (PXD101) | COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | II | NCT00357032 | |
| COMPLETED | OC | INTERVENTIONAL | II | NCT00301756 | ||
| Vorinostat | COMPLETED | Advanced BC | I | NCT00719875 | ||
| COMPLETED | Advanced CTCL | INTERVENTIONAL | II | NCT00091559 | ||
| ACTIVE | Advanced NSCLC | I | NCT01059552 | |||
| CHR-3996 | COMPLETED | Advanced Solid Tumors | INTERVENTIONAL | I/II | NCT00697879 | |
| Givinostat | RECRUITING | Chronic myeloproliferative neoplasms | INTERVENTIONAL | II | NCT01761968 | |
| Romidepsin | COMPLETED | T cell lymphoma | INTERVENTIONAL | II | NCT00007345 | |
| KA2507 | RECRUITING | Advanced solid tumors | I | NCT03008018 | ||
| DNMT | SGI-110 | COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | I/II | NCT01261312 |
| Deoxycytidine (Aza TdC) | RECRUITING | Advanced solid tumors | INTERVENTIONAL | I | NCT03366116 | |
| Decitabine | COMPLETED | Metastatic PTC or FTC | INTERVENTIONAL | II | NCT00085293 | |
| COMPLETED | AML and myelodysplastic syndromes | INTERVENTIONAL | II | NCT00492401 | ||
| Disulfiram | COMPLETED | PC | INTERVENTIONAL | II | NCT01118741 | |
| RECRUITING | Metastatic BC | INTERVENTIONAL | II | NCT03323346 |
AML, acute myeloid leukemia; MM; multiple myeloma; HL, Hodgkin’s lymphoma; CTCL, cutaneous T cell lymphoma; OC, ovarian cancer; BC, breast cancer; NSCLC, non-small cell lung cancer; PTC, papillary thyroid cancer; FTC, follicular thyroid cancer; PC, prostate cancer.
Table 3.
Interventional clinical trials investigating the antineoplastic effects of Bromodomain and Extra Terminal (BET) inhibitors as single agent or in combination with other Food and Drug Administration (FDA)-approved drugs.
Table 3.
Interventional clinical trials investigating the antineoplastic effects of Bromodomain and Extra Terminal (BET) inhibitors as single agent or in combination with other Food and Drug Administration (FDA)-approved drugs.
| BET Inhibitors | Intervention | Status | Condition | Study Type | Phase | NCT Number |
|---|---|---|---|---|---|---|
| I-BET762 (GSK525762) | GSK525762 + FULVESTRANT vs. GSK525762 + PLACEBO | RECRUITING | ER and/or PR-positive/HER2-Negative Advanced or Metastatic Breast Cancer | INTERVENTIONAL | II | NCT02964507 |
| GSK525762 + ABIRATERONE/ENZALUTAMIDE +PREDNISONE | RECRUITING | Castration-resistant Prostate Cancer | INTERVENTIONAL | I | NCT03150056 | |
| GSK525762 monotherapy | RECRUITING | Relapsed Refractory Hematologic Malignancies | INTERVENTIONAL | I | NCT01943851 | |
| GSK525762 monotherapy | ACTIVE | NUT Midline Carcinoma | INTERVENTIONAL | I | NCT03702036 | |
| MK-8628 monotherapy | COMPLETED | Advanced Solid Tumor | INTERVENTIONAL | I | NCT02259114 | |
| MK-8628 monotherapy | COMPLETED | Hematologic Malignancies | INTERVENTIONAL | I | NCT01713582 | |
| MK-8628 monotherapy | ACTIVE | Hematologic Malignancies | INTERVENTIONAL | I | NCT02698189 | |
| FT-1101 | FT-1101 + AZACITIDINE vs. FT-1101 + PLACEBO | RECRUITING | Hematologic Malignancies | INTERVENTIONAL | I | NCT02543879 |
| CPI-0610 | CPI-0610 + Ruxolitinib vs. CPI-0610 + PLACEBO | RECRUITING | Hematologic Malignancies | INTERVENTIONAL | I-II | NCT02158858 |
| CPI-0610 monotherapy | COMPLETED | Multiple Myeloma | INTERVENTIONAL | I | NCT02157636 | |
| CPI-0610 monotherapy | ACTIVE | Lymphoma | INTERVENTIONAL | I | NCT01949883 | |
| INCB054329 | INCB054329 monotherapy | COMPLETED | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I-II | NCT02431260 |
| RO6870810 | RO6870810 + Atezolizumab vs. RO6870810 + PLACEBO | RECRUITING | Advanced Ovarian Cancer and TNBC | INTERVENTIONAL | I | NCT03292172 |
| RO6870810 and VENETOCLAX + RITUXIMAB vs. RO6870810 and VENETOCLAX + PLACEBO | RECRUITING | DLBCL | INTERVENTIONAL | I | NCT03255096 | |
| GSK2820151 | GSK2820151 monotherapy | ACTIVE | Advanced or Recurrent Solid Tumors | INTERVENTIONAL | I | NCT02630251 |
| ZEN003694 | ZEN003694 monotherapy | COMPLETED | Metastatic Castration-resistant Prostate Cancer | INTERVENTIONAL | I | NCT02705469 |
| ZEN003694 + ENZALUTAMIDE vs. ZEN003694 + PLACEBO | RECRUITING | Metastatic Castration-resistant Prostate Cancer | INTERVENTIONAL | I-II | NCT02711956 | |
| BMS-986158 | BMS-986158 and NIVOLUMAB | RECRUITING | Advanced Tumors | INTERVENTIONAL | I-II | NCT02419417 |
| ABBV-075 | ABBV-075 and VENETOCLAX | RECRUITING | Solid Tumors | INTERVENTIONAL | I | NCT02391480 |
| GS-5829 | GS-5829 + ENZALUTAMIDE vs. GS-5829 + PLACEBO | ACTIVE | Metastatic Castration-resistant Prostate Cancer | INTERVENTIONAL | I-II | NCT02607228 |
| GS-5829 + FULVESTRANT vs. GS-5829 + EXEMESTANE | COMPLETED | Advanced Solid Tumors and Lymphomas | INTERVENTIONAL | I | NCT02392611 | |
| PLX51107 | PLX51107 monotherapy | RECRUITING | Advanced Solid Tumors and Hematologic Malignancies | INTERVENTIONAL | I | NCT02683395 |
FULVESTRANT, anti-estrogen receptor; ABIROTERONE, ENZALUTAMIDE, anti-androgen; PREDNISONE, corticosteroid; AZACITADINE, DNA methylation inhibitor; RUXOLITINIB, JAK2 inhibitor; ATEZOLIZUMAB, anti-PD-L1 monoclonal antibody; VENETOCLAX, BCL2 inhibitor; RITUXIMAB, anti-CD20 monoclonal antibody; NIVOLUMAB, anti-PD1 monoclonal antibody; EXEMESTONE, anti-estrogen. BC, breast cancer; CR-PC, castration-resistant prostate cancer; NMC, NUT midline carcinoma; NSCLC, non-small cell lung cancer; TNBC, triple negative breast cancer; GBM, glioblastoma multiforme; MM; multiple myeloma; OC, ovarian cancer; DLBCL, diffuse large B-cell lymphoma.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mio, C.; Bulotta, S.; Russo, D.; Damante, G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers 2019, 11, 61. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010061
AMA Style
Mio C, Bulotta S, Russo D, Damante G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers. 2019; 11(1):61. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010061
Chicago/Turabian StyleMio, Catia, Stefania Bulotta, Diego Russo, and Giuseppe Damante. 2019. "Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment" Cancers 11, no. 1: 61. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11010061
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.