A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer

 , , , ,
, , , ,
Abstract
:1. Introduction
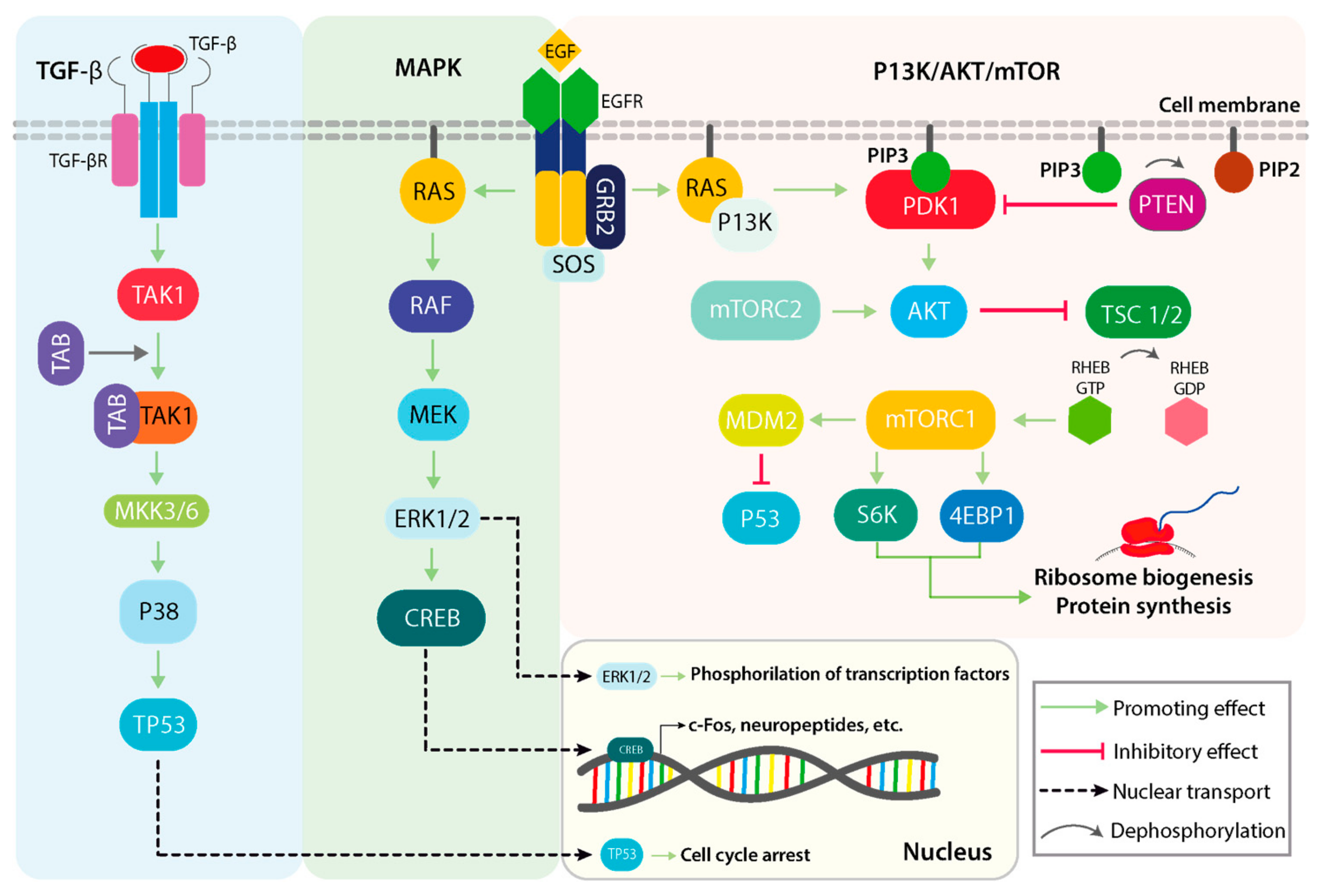
2. Physiological Roles of the MAPK Signaling Pathway
3. MAPK-Signaling Crosstalk and Pathologic Deregulations in Cancer
4. Implications of the Tumor Microenvironment in Regulating MAPK Signaling Pathway
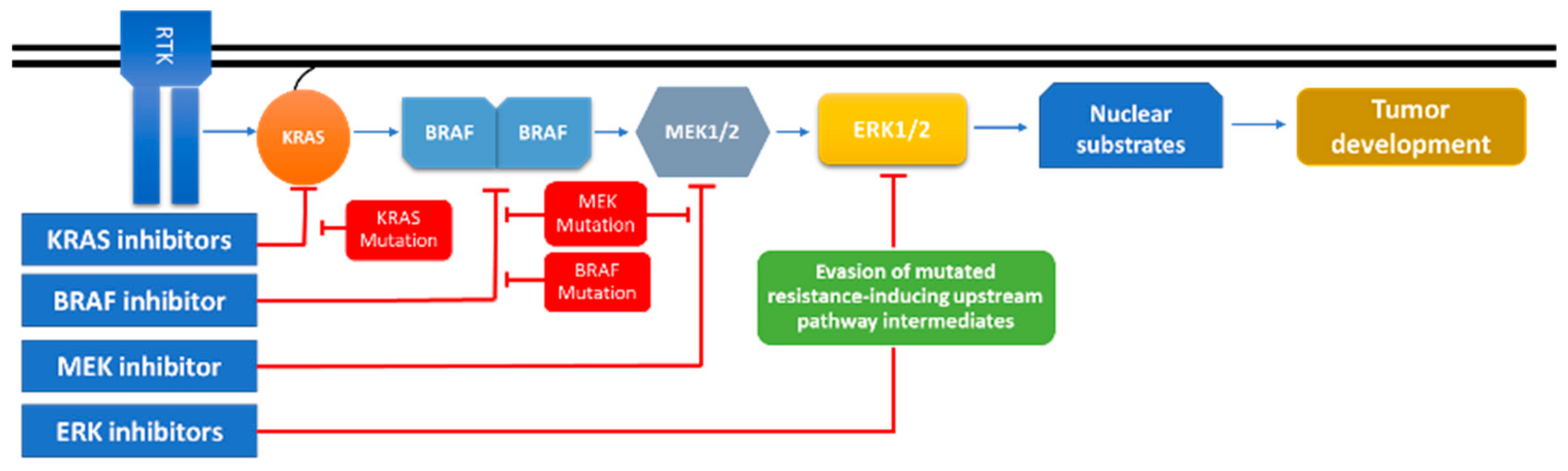
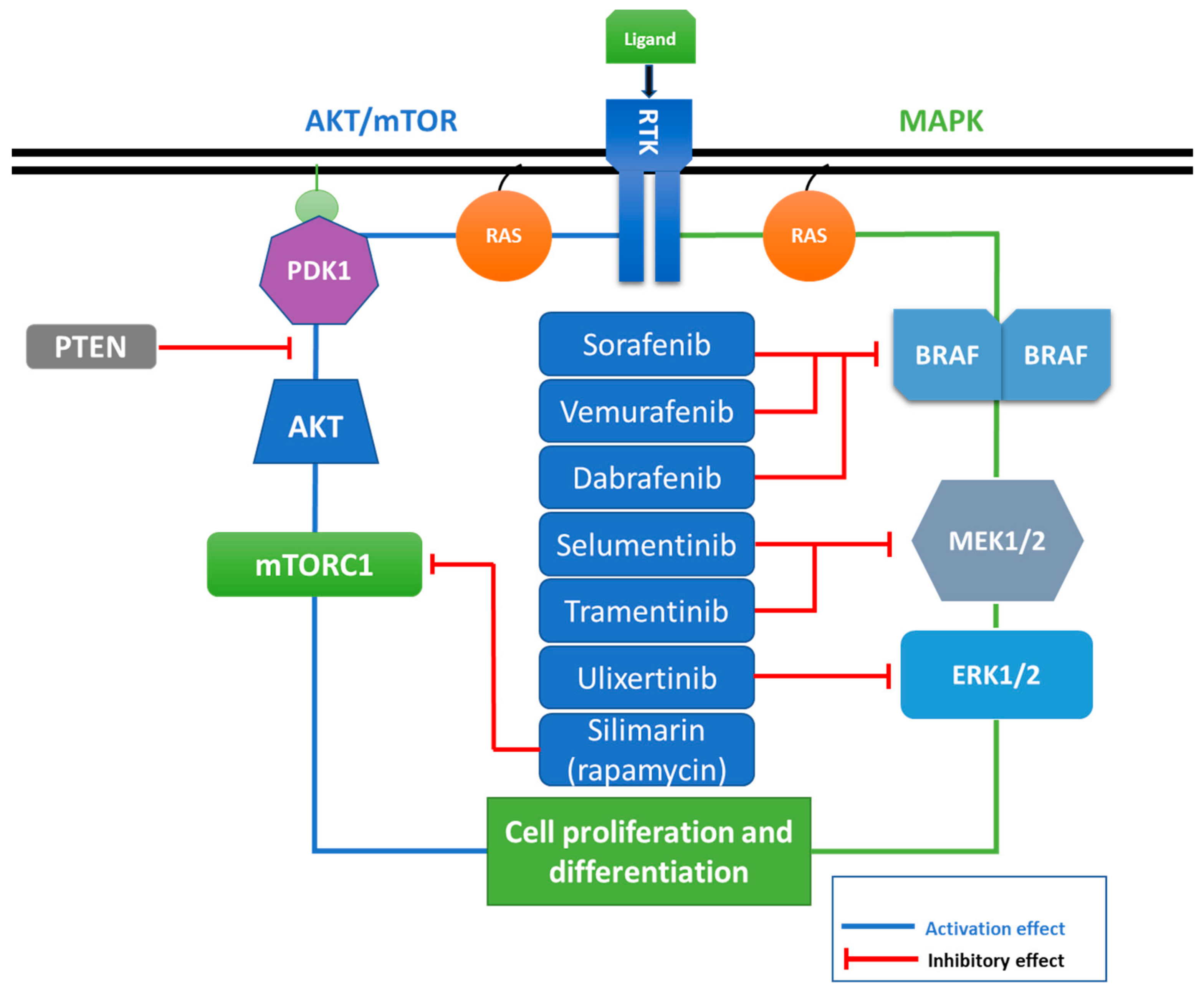
5. MAPK, Crosstalking Pathways, and Drug Resistance
6. MAPK Inhibitors, and Preclinical and Clinical Trial Molecules
7. MAPK and Natural Bioactive Compounds in Chemoprevention and Chemotherapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cancer Statistics. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 15 October 2019).
- Cainap, C.; Nagy, V.; Seicean, A.; Gherman, A.; Laszlo, I.; Lisencu, C.; Nadim, A.H.; Constantin, A.M.; Cainap, S. Results of third-generation epirubicin/cisplatin/xeloda adjuvant chemotherapy in patients with radically resected gastric cancer. J. BU ON 2016, 21, 349–359. [Google Scholar]
- Braicu, C.; Pileczki, V.; Irimie, A.; Berindan-Neagoe, I. p53siRNA therapy reduces cell proliferation, migration and induces apoptosis in triple negative breast cancer cells. Mol. Cell. Biochem. 2013, 381, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Irimie, A.I.; Braicu, C.; Cojocneanu-Petric, R.; Berindan-Neagoe, I.; Campian, R.S. Novel technologies for oral squamous carcinoma biomarkers in diagnostics and prognostics. Acta Odontol. Scand. 2015, 73, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, R.; Braicu, C.; Berindan-Neagoe, I. Another review on triple negative breast cancer. Are we on the right way towards the exit from the labyrinth? Breast 2013, 22, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Burz, C.; Aziz, B.Y.; Balacescu, L.; Lelutiu, L.; Buiga, R.; Samasca, G.; Irimie, A.; Lisencu, C. Tumor markers used in monitoring the tumor recurrence in patients with colorectal cancer. Clujul Med. 2016, 89, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Braicu, C.; Irimie, A.; Craciun, L.; Berindan-Neagoe, I. Nanopharmacology in translational hematology and oncology. Int. J. Nanomed. 2014, 9, 3465–3479. [Google Scholar]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sini. B 2018, 8, 552–562. [Google Scholar] [CrossRef]
- Bochis, O.V.; Irimie, A.; Pichler, M.; Berindan-Neagoe, I. The role of Skp2 and its substrate CDKN1B (p27) in colorectal cancer. J. Gastrointest. Liver Dis. 2015, 24, 225–234. [Google Scholar]
- Seles, M.; Hutterer, G.C.; Kiesslich, T.; Pummer, K.; Berindan-Neagoe, I.; Perakis, S.; Schwarzenbacher, D.; Stotz, M.; Gerger, A.; Pichler, M. Current Insights into Long Non-Coding RNAs in Renal Cell Carcinoma. Int. J. Mol. Sci. 2016, 17, 573. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Catana, C.; Calin, G.A.; Berindan-Neagoe, I. NCRNA combined therapy as future treatment option for cancer. Curr. Pharm. Des. 2014, 20, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Braicu, C.; Zimta, A.A.; Harangus, A.; Iurca, I.; Irimie, A.; Coza, O.; Berindan-Neagoe, I. The Function of Non-Coding RNAs in Lung Cancer Tumorigenesis. Cancers 2019, 11, 605. [Google Scholar] [CrossRef] [PubMed]
- Jurj, A.; Braicu, C.; Pop, L.A.; Tomuleasa, C.; Gherman, C.D.; Berindan-Neagoe, I. The new era of nanotechnology, an alternative to change cancer treatment. Drug Des. Dev. Ther. 2017, 11, 2871–2890. [Google Scholar] [CrossRef]
- Ganapathi, M.K.; Jones, W.D.; Sehouli, J.; Michener, C.M.; Braicu, I.E.; Norris, E.J.; Biscotti, C.V.; Vaziri, S.A.; Ganapathi, R.N. Expression profile of COL2A1 and the pseudogene SLC6A10P predicts tumor recurrence in high-grade serous ovarian cancer. Int. J. Cancer 2016, 138, 679–688. [Google Scholar] [CrossRef]
- Plotnikov, A.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6, 6685. [Google Scholar] [CrossRef]
- Chapnick, D.A.; Warner, L.; Bernet, J.; Rao, T.; Liu, X. Partners in crime: The TGFβ and MAPK pathways in cancer progression. Cell Biosci. 2011, 1, 42. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Johnson, D.S.; Chen, Y.H. Ras Family of Small GTPases In Immunity And Inflammation. Curr. Opin. Pharmacol. 2012, 12, 458–463. [Google Scholar] [CrossRef]
- Vo, U.; Vajpai, N.; Flavell, L.; Bobby, R.; Breeze, A.L.; Embrey, K.J.; Golovanov, A.P. Monitoring Ras Interactions with the Nucleotide Exchange Factor Son of Sevenless (Sos) Using Site-specific NMR Reporter Signals and Intrinsic Fluorescence. J. Biol. Chem. 2016, 291, 1703–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf Family Kinases: Old Dogs Have Learned New Tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCain, J. The MAPK (ERK) Pathway: Investigational Combinations for the Treatment of BRAF-Mutated Metastatic Melanoma. Pharm. Ther. 2013, 38, 96–108. [Google Scholar]
- Fanger, G.R.; Johnson, N.L.; Johnson, G.L. MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J. 1997, 16, 4961–4972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.F. Keeping Tumors Out of the MAPK Fitness Zone. Cancer Discov. 2018, 8, 20–23. [Google Scholar] [CrossRef] [Green Version]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Johne, C.; Matenia, D.; Li, X.-y.; Timm, T.; Balusamy, K.; Mandelkow, E.M. Spred1 and TESK1—Two New Interaction Partners of the Kinase MARKK/TAO1 That Link the Microtubule and Actin Cytoskeleton. Mol. Biol. Cell 2008, 19, 1391–1403. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- COSMIC, Catalogue of Somatic Mutations in Cance. Available online: http://sanger.ac.uk/cosmic (accessed on 16 October 2019).
- Fernández-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef]
- Setia, S.; Nehru, B.; Sanyal, S.N. Upregulation of MAPK/Erk and PI3K/Akt pathways in ulcerative colitis-associated colon cancer. Biomed. Pharmacother. 2014, 68, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, M.K.; Curioni-Fontecedro, A.; Samaras, P.; Atrott, K.; Cosin-Roger, J.; Lang, S.; Scharl, M.; Rogler, G. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget 2015, 6, 42183–42196. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.W.; Pratilas, C.A.; Poulikakos, P.I.; Tadi, M.; Wang, W.; Taylor, B.S.; Halilovic, E.; Persaud, Y.; Xing, F.; Viale, A.; et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl. Acad. Sci. USA 2010, 107, 14903–14908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousova, K.; Jirku, M.; Bumba, L.; Bednarova, L.; Sulc, M.; Franek, M.; Vyklicky, L.; Vondrasek, J.; Teisinger, J. PIP2 and PIP3 interact with N-terminus region of TRPM4 channel. Biophys. Chem. 2015, 205, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xiang, Z.; Malaviarachchi, P.A.; Yan, Y.; Baltz, N.J.; Emanuel, P.D.; Liu, Y.L. PTEN is indispensable for cells to respond to MAPK inhibitors in myeloid leukemia. Cell. Signal. 2018, 50, 72–79. [Google Scholar] [CrossRef]
- Linthicum, W.; Thanh, M.H.; Vitolo, M.I.; Wen, Q. Effects of PTEN Loss and Activated KRAS Overexpression on Mechanical Properties of Breast Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 1613. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef]
- Mooz, J.; Oberoi-Khanuja, T.K.; Harms, G.S.; Wang, W.; Jaiswal, B.S.; Seshagiri, S.; Tikkanen, R.; Rajalingam, K. Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci. Signal. 2014, 7, 73. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef]
- Speth, Z.; Islam, T.; Banerjee, K.; Resat, H. EGFR signaling pathways are wired differently in normal 184A1L5 human mammary epithelial and MDA-MB-231 breast cancer cells. J. Cell Commun. Signal. 2017, 11, 341–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gschwantler-Kaulich, D.; Grunt, T.W.; Muhr, D.; Wagner, R.; Kolbl, H.; Singer, C.F. HER Specific TKIs Exert Their Antineoplastic Effects on Breast Cancer Cell Lines through the Involvement of STAT5 and JNK. PLoS ONE 2016, 11, e0146311. [Google Scholar] [CrossRef] [PubMed]
- Gkouveris, I.; Nikitakis, N.; Karanikou, M.; Rassidakis, G.; Sklavounou, A. JNK1/2 expression and modulation of STAT3 signaling in oral cancer. Oncol. Lett. 2016, 12, 699–706. [Google Scholar] [CrossRef] [PubMed]
- El-Habr, E.A.; Levidou, G.; Trigka, E.A.; Sakalidou, J.; Piperi, C.; Chatziandreou, I.; Spyropoulou, A.; Soldatos, R.; Tomara, G.; Petraki, K.; et al. Complex interactions between the components of the PI3K/AKT/mTOR pathway, and with components of MAPK, JAK/STAT and Notch-1 pathways, indicate their involvement in meningioma development. Virchows Arch. 2014, 465, 473–485. [Google Scholar] [CrossRef]
- Wolf, A.; Eulenfeld, R.; Gabler, K.; Rolvering, C.; Haan, S.; Behrmann, I.; Denecke, B.; Haan, C.; Schaper, F. JAK2-V617F-induced MAPK activity is regulated by PI3K and acts synergistically with PI3K on the proliferation of JAK2-V617F-positive cells. Jak-Stat 2013, 2, e24574. [Google Scholar] [CrossRef] [Green Version]
- Wee, S.; Jagani, Z.; Xiang, K.X.; Loo, A.; Dorsch, M.; Yao, Y.M.; Sellers, W.R.; Lengauer, C.; Stegmeier, F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009, 69, 4286–4293. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Im, E.; von Lintig, F.C.; Chen, J.; Zhuang, S.; Qui, W.; Chowdhury, S.; Worley, P.F.; Boss, G.R.; Pilz, R.B. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene 2002, 21, 6356–6365. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, K.E.; Cullinane, C.; Hannan, K.M.; Wall, M.; Chan, J.; Barber, F.; Foo, J.; Cameron, D.; Neilsen, A.; Ng, P.; et al. Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. Eur. J. Cancer 2013, 49, 3936–3944. [Google Scholar] [CrossRef]
- Li, J.P.; Yang, Y.X.; Liu, Q.L.; Pan, S.T.; He, Z.X.; Zhang, X.; Yang, T.; Chen, X.W.; Wang, D.; Qiu, J.X.; et al. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des. Dev. Ther. 2015, 9, 1627–1652. [Google Scholar]
- Kotani, H.; Adachi, Y.; Kitai, H.; Tomida, S.; Bando, H.; Faber, A.C.; Yoshino, T.; Voon, D.C.; Yano, S.; Ebi, H. Distinct dependencies on receptor tyrosine kinases in the regulation of MAPK signaling between BRAF V600E and non-V600E mutant lung cancers. Oncogene 2018, 37, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ge, C.C.; Wang, J.; Wu, X.X.; Li, X.M.; Li, W.; Wang, S.S.; Liu, T.; Hou, J.Z.; Sun, H.; et al. MEK inhibitor, PD98059, promotes breast cancer cell migration by inducing beta-catenin nuclear accumulation. Oncol. Rep. 2017, 38, 3055–3063. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-β Signalling. J. Signal Transduct. 2012, 2012, 294097. [Google Scholar] [CrossRef] [PubMed]
- Stramucci, L.; Pranteda, A.; Bossi, G. Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed]
- Slobodnyuk, K.; Radic, N.; Ivanova, S.; Llado, A.; Trempolec, N.; Zorzano, A.; Nebreda, A.R. Autophagy-induced senescence is regulated by p38alpha signaling. Cell Death Dis. 2019, 10, 376. [Google Scholar] [CrossRef]
- Peng, W.X.; Huang, J.G.; Yang, L.; Gong, A.H.; Mo, Y.Y. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol. Cancer 2017, 16, 161. [Google Scholar] [CrossRef]
- Han, D.; Wang, M.; Yu, Z.; Yin, L.; Liu, C.; Wang, J.; Liu, Y.; Jiang, S.; Ren, Z.; Yin, J. FGF5 promotes osteosarcoma cells proliferation via activating MAPK signaling pathway. Cancer Manag. Res. 2019, 11, 6457–6466. [Google Scholar] [CrossRef]
- Jilaveanu, L.B.; Zito, C.R.; Aziz, S.A.; Conrad, P.J.; Schmitz, J.C.; Sznol, M.; Camp, R.L.; Rimm, D.L.; Kluger, H.M. C-Raf is Associated with Disease Progression and Cell Proliferation in a Subset of Melanomas. Clin. Cancer Res. 2009, 15, 5704–5713. [Google Scholar] [CrossRef]
- Wang, T.; Seah, S.; Loh, X.; Chan, C.W.; Hartman, M.; Goh, B.C.; Lee, S.C. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget 2016, 7, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Van Gijn, S.E.; Wierenga, E.; van den Tempel, N.; Kok, Y.P.; Heijink, A.M.; Spierings, D.C.J.; Foijer, F.; van Vugtk, M.A.T.M.; Fehrmann, R.S.N. TPX2/Aurora kinase A signaling as a potential therapeutic target in genomically unstable cancer cells. Oncogene 2019, 38, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Huang, O.; Zhang, W.; Zhi, Q.; Xue, X.; Liu, H.; Shen, D.; Geng, M.; Xie, Z.; Jiang, M. Teriflunomide, an immunomodulatory drug, exerts anticancer activity in triple negative breast cancer cells. Exp. Biol. Med. 2015, 240, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Dubey, N.K.; Peng, B.Y.; Lin, C.M.; Wang, P.D.; Wang, J.R.; Chan, C.H.; Wei, H.J.; Deng, W.P. NSC 95397 Suppresses Proliferation and Induces Apoptosis in Colon Cancer Cells through MKP-1 and the ERK1/2 Pathway. Int. J. Mol. Sci. 2018, 19, 1625. [Google Scholar] [CrossRef]
- Liu, P.C.; Lu, G.; Deng, Y.; Wang, C.D.; Su, X.W.; Zhou, J.Y.; Chan, T.M.; Hu, X.; Poon, W.S. Inhibition of NF-kappaB Pathway and Modulation of MAPK Signaling Pathways in Glioblastoma and Implications for Lovastatin and Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL) Combination Therapy. PLoS ONE 2017, 12, e0171157. [Google Scholar]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Smith, M.P.; Sanchez-Laorden, B.; O’Brien, K.; Brunton, H.; Ferguson, J.; Young, H.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef]
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell Biol. 2012, 24, 600–606. [Google Scholar] [CrossRef]
- Ionescu, C.; Braicu, C.; Chiorean, R.; Cojocneanu Petric, R.; Neagoe, E.; Pop, L.; Chira, S.; Berindan-Neagoe, I. TIMP-1 expression in human colorectal cancer is associated with SMAD3 gene expression levels: A pilot study. J. Gastrointest. Liver Dis. 2014, 23, 413–418. [Google Scholar]
- Tudoran, O.; Soritau, O.; Balacescu, O.; Balacescu, L.; Braicu, C.; Rus, M.; Gherman, C.; Virag, P.; Irimie, F.; Berindan-Neagoe, I. Early transcriptional pattern of angiogenesis induced by EGCG treatment in cervical tumour cells. J. Cell. Mol. Med. 2012, 16, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Longmate, W.; DiPersio, C.M. Beyond adhesion: emerging roles for integrins in control of the tumor microenvironment. F1000 Res. 2017, 6, 1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potempa, S.; Ridley, A.J. Activation of Both MAP Kinase and Phosphatidylinositide 3-Kinase by Ras Is Required for Hepatocyte Growth Factor/Scatter Factor–induced Adherens Junction Disassembly. Mol. Biol. Cell 1998, 9, 2185–2200. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.D.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Curry, J.M.; Ko, Y.H.; Lin, Z.; Tuluc, M.; Cognetti, D.; Birbe, R.C.; Pribitkin, E.; Bombonati, A.; Pestell, R.G.; et al. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFkappaB target stromal MCT4. Cell Cycle 2013, 12, 2580–2597. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Sang, N.; Stiehl, D.P.; Bohensky, J.; Leshchinsky, I.; Srinivas, V.; Caro, J. MAPK Signaling Up-regulates the Activity of Hypoxia-inducible Factors by Its Effects on p300. J. Biol. Chem. 2003, 278, 14013–14019. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, J.; Karlsson, M.C. TGF-beta-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin. Cancer Biol. 2012, 22, 455–461. [Google Scholar] [CrossRef]
- Turley, E.A.; Veiseh, M.; Radisky, D.C.; Bissell, M.J. Mechanisms of Disease: Epithelial–mesenchymal transition—does cellular plasticity fuel neoplastic progression? Nat. Rev. Clin. Oncol. 2008, 5, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Cullis, J.; Das, S.; Bar-Sagi, D. Kras and Tumor Immunity: Friend or Foe? Cold Spring Harb. Perspect. Med. 2018, 8, a031849. [Google Scholar] [CrossRef] [PubMed]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed]
- Gulei, D.; Mehterov, N.; Ling, H.; Stanta, G.; Braicu, C.; Berindan-Neagoe, I. The “good-cop bad-cop” TGF-beta role in breast cancer modulated by non-coding RNAs. Biochim. Biophys. Acta 2017, 1861, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Pulai, J.; Loeser, R.F. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002, 46, 2368–2376. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, H.X.; Wright, J.; Holbeck, S.; Millin, M.D.; Tomaszewski, J.; Zweibel, J.; Collins, J.; Doroshow, J.H. Utilizing targeted cancer therapeutic agents in combination: Novel approaches and urgent requirements. Nat. Rev. Drug Discov. 2010, 9, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, R.W.; Brockway-Lunardi, L.M.; Bonk, D.T.; Dohoney, K.M.; Doroshow, J.H.; Meech, S.J.; Ratain, M.J.; Topalian, S.L.; Pardoll, D.M. Opportunities and challenges in the development of experimental drug combinations for cancer. J. Natl. Cancer Inst. 2011, 103, 1222–1226. [Google Scholar] [CrossRef]
- Alspach, E.; Flanagan, K.C.; Luo, X.; Ruhland, M.K.; Huang, H.; Pazolli, E.; Donlin, M.J.; Marsh, T.; Piwnica-Worms, D.; Monahan, J.; et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 2014, 4, 716–729. [Google Scholar] [CrossRef]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38alpha MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Solit, D.B. Resistance to MEK inhibitors: Should we co-target upstream? Sci. Signal. 2011, 4, 16. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockorny, B.; Rusan, M.; Chen, W.; Liao, R.G.; Li, Y.; Piccioni, F.; Wang, J.; Tan, L.; Thorner, A.R.; Li, T.; et al. RAS-MAPK Reactivation Facilitates Acquired Resistance in FGFR1-Amplified Lung Cancer and Underlies a Rationale for Upfront FGFR-MEK Blockade. Mol. Cancer Ther. 2018, 17, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Hinton, S.D. The role of pseudophosphatases as signaling regulators. Biochim. Biophys. Acta 2019, 1866, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Pathria, G.; Garg, B.; Borgdorff, V.; Garg, K.; Wagner, C.; Superti-Furga, G.; Wagner, S.N. Overcoming MITF-conferred drug resistance through dual AURKA/MAPK targeting in human melanoma cells. Cell Death Dis. 2016, 7, e2135. [Google Scholar] [CrossRef] [PubMed]
- Haq, R.; Yokoyama, S.; Hawryluk, E.B.; Jönsson, G.B.; Frederick, D.T.; McHenry, K.; Porter, D.; Tran, T.-N.; Love, K.T.; Langer, R.; et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc. Natl. Acad. Sci. USA 2013, 110, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Najem, A.; Krayem, M.; Sales, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur. J. Cancer 2017, 83, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Ciccarelli, C.; Zani, B.M. Biological Rationale for Targeting MEK/ERK Pathways in Anti-Cancer Therapy and to Potentiate Tumour Responses to Radiation. Int. J. Mol. Sci. 2019, 20, 2530. [Google Scholar] [CrossRef]
- Del Curatolo, A.; Conciatori, F.; Cesta Incani, U.; Bazzichetto, C.; Falcone, I.; Corbo, V.; D’Agosto, S.; Eramo, A.; Sette, G.; Sperduti, I.; et al. Therapeutic potential of combined BRAF/MEK blockade in BRAF-wild type preclinical tumor models. J. Exp. Clin. Cancer Res. 2018, 37, 140. [Google Scholar] [CrossRef]
- Sato, H.; Yamamoto, H.; Sakaguchi, M.; Shien, K.; Tomida, S.; Shien, T.; Ikeda, H.; Hatono, M.; Torigoe, H.; Namba, K.; et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Cancer Sci. 2018, 109, 3183. [Google Scholar] [CrossRef]
- Mali, A.V.; Joshi, A.A.; Hegde, M.V.; Kadam, S.S. Enterolactone modulates the ERK/NF-kappaB/Snail signaling pathway in triple-negative breast cancer cell line MDA-MB-231 to revert the TGF-beta-induced epithelial-mesenchymal transition. Cancer Biol. Med. 2018, 15, 137–156. [Google Scholar]
- Lu, H.; Liu, S.; Zhang, G.; Wu, B.; Zhu, Y.; Frederick, D.T.; Hu, Y.; Zhong, W.; Randell, S.; Sadek, N.; et al. PAK Signaling Drives Acquired Drug Resistance to MAPK Inhibitors in BRAF-mutant Melanomas. Nature 2017, 550, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, X.; Qu, J.; Straubinger, R.M.; Jusko, W.J. Multi-Scale Network Model Supported by Proteomics for Analysis of Combined Gemcitabine and Birinapant Effects in Pancreatic Cancer Cells. CPT 2018, 7, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Maverakis, E.; Cornelius, L.A.; Bowen, G.M.; Phan, T.; Patel, F.B.; Fitzmaurice, S.; He, Y.; Burrall, B.; Duong, C.; Kloxin, A.M.; et al. Metastatic melanoma—A review of current and future treatment options. Acta Derm. Venereol. 2015, 95, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Qin, H.; Xu, J. The Role of Autophagy in the Resistance to BRAF Inhibition in BRAF-Mutated Melanoma. Target. Oncol. 2018, 13, 437–446. [Google Scholar] [CrossRef]
- Ahern, T.P.; Lash, T.L.; Damkier, P.; Christiansen, P.M.; On behalf of the Danish Breast Cancer Cooperative Group; Cronin-Fenton, D.P. Statins and breast cancer prognosis: Evidence and opportunities. Lancet Oncol. 2014, 15, e461–e468. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00334542 (accessed on 16 October 2019).
- Furberg, C.D.; Friedman, L.M. Approaches to data analyses of clinical trials. Prog. Cardiovasc. Dis. 2012, 54, 330–334. [Google Scholar] [CrossRef]
- Polivka, J., Jr.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Carter, C.A.; Rajan, A.; Keen, C.; Szabo, E.; Khozin, S.; Thomas, A.; Brzezniak, C.; Guha, U.; Doyle, L.A.; Steinberg, S.M.; et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann. Oncol. 2016, 27, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A First-in-Human Phase I Study of the Oral p38 MAPK Inhibitor, Ralimetinib (LY2228820 Dimesylate), in Patients with Advanced Cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L.; et al. Dabrafenib plus trametinib in patients with BRAF(V600)-mutant melanoma brain metastases (COMBI-MB): A multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Blumenschein, G.R., Jr.; Smit, E.F.; Planchard, D.; Kim, D.W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Andrlova, H.; Zeiser, R.; Meiss, F. Cobimetinib (GDC-0973, XL518). Recent Results Cancer Res. 2018, 211, 177–186. [Google Scholar]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms shaping the role of ERK1/2 in cellular senescence. Mol. Med. Rep. 2019, 19, 759–770. [Google Scholar]
- Tomuleasa, C.; Cristea, V.; Irimie, A. Sorafenib for advanced-stage hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2012, 24, 346–347. [Google Scholar] [CrossRef]
- Pall, E.; Groza, I.; Cenariu, M.; Soritau, O.; Gocza, E.; Tomuleasa, C. Establishment of an embryonic stem cell line from blastocyst stage mouse embryos. Rom. J. Morphol. Embryol. 2011, 52, 1005–1010. [Google Scholar] [PubMed]
- Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT01271803 (accessed on 15 October 2019).
- Susman, S.; Rus-Ciuca, D.; Soritau, O.; Tomuleasa, C.; Buiga, R.; Mihu, D.; Pop, V.I.; Mihu, C.M. Pancreatic exocrine adult cells and placental stem cells co-culture. Working together is always the best way to go. Rom. J. Morphol. Embryol. 2011, 52, 999–1004. [Google Scholar] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Holen, K.D.; Northfelt, D.W.; Pitot, H.C.; Picus, J.; Flynn, P.J.; Erlichman, C. Phase 2 study of bevacizumab plus erlotinib in patients with advanced hepatocellular cancer. Cancer 2012, 118, 2424–2430. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Javle, M.; Bekaii-Saab, T.S.; Finn, R.S.; Wainberg, Z.A.; Laheru, D.A.; Weekes, C.D.; Tan, B.R.; Khan, G.N.; Zalupski, M.M.; et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br. J. Cancer 2017, 116, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasawa, M.M.G.; Mohan, C. Fruits as Prospective Reserves of bioactive Compounds: A Review. Nat. Prod. Bioprospect. 2018, 8, 335–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cojocneanu Petric, R.; Braicu, C.; Raduly, L.; Zanoaga, O.; Dragos, N.; Monroig, P.; Dumitrascu, D.; Berindan-Neagoe, I. Phytochemicals modulate carcinogenic signaling pathways in breast and hormone-related cancers. Onco Targets Ther. 2015, 8, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Catana, C.S.; Atanasov, A.G.; Berindan-Neagoe, I. Natural products with anti-aging potential: Affected targets and molecular mechanisms. Biotechnol. Adv. 2018, 36, 1649–1656. [Google Scholar] [CrossRef]
- Irimie, A.I.; Braicu, C.; Pasca, S.; Magdo, L.; Gulei, D.; Cojocneanu, R.; Ciocan, C.; Olariu, A.; Coza, O.; Berindan-Neagoe, I. Role of Key Micronutrients from Nutrigenetic and Nutrigenomic Perspectives in Cancer Prevention. Medicina 2019, 55, 283. [Google Scholar] [CrossRef]
- Budisan, L.; Gulei, D.; Zanoaga, O.M.; Irimie, A.I.; Sergiu, C.; Braicu, C.; Gherman, C.D.; Berindan-Neagoe, I. Dietary Intervention by Phytochemicals and Their Role in Modulating Coding and Non-Coding Genes in Cancer. Int. J. Mol. Sci. 2017, 18, 1178. [Google Scholar] [CrossRef]
- Braicu, C.; Mehterov, N.; Vladimirov, B.; Sarafian, V.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. Nutrigenomics in cancer: Revisiting the effects of natural compounds. Semin. Cancer Biol. 2017, 46, 84–106. [Google Scholar] [CrossRef]
- Budisan, L.; Gulei, D.; Jurj, A.; Braicu, C.; Zanoaga, O.; Cojocneanu, R.; Pop, L.; Raduly, L.; Barbat, A.; Moldovan, A.; et al. Inhibitory Effect of CAPE and Kaempferol in Colon Cancer Cell Lines-Possible Implications in New Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 1199. [Google Scholar] [CrossRef] [PubMed]
- Papademetrio, D.L.; Lompardia, S.L.; Simunovich, T.; Costantino, S.; Mihalez, C.Y.; Cavaliere, V.; Alvarez, E. Inhibition of Survival Pathways MAPK and NF-kB Triggers Apoptosis in Pancreatic Ductal Adenocarcinoma Cells via Suppression of Autophagy. Target. Oncol. 2016, 11, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Gherman, C.; Braicu, O.L.; Zanoaga, O.; Jurj, A.; Pileczki, V.; Maralani, M.; Drigla, F.; Braicu, C.; Budisan, L.; Achimas-Cadariu, P.; et al. Caffeic acid phenethyl ester activates pro-apoptotic and epithelial-mesenchymal transition-related genes in ovarian cancer cells A2780 and A2780cis. Mol. Cell. Biochem. 2016, 413, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Park, S.; Bazer, F.W.; Song, G. Apigenin Reduces Survival of Choriocarcinoma Cells by Inducing Apoptosis via the PI3K/AKT and ERK1/2 MAPK Pathways. J. Cell. Physiol. 2016, 231, 2690–2699. [Google Scholar] [CrossRef]
- Li, H.; Yoon, J.H.; Won, H.J.; Ji, H.S.; Yuk, H.J.; Park, K.H.; Park, H.Y.; Jeong, T.S. Isotrifoliol inhibits pro-inflammatory mediators by suppression of TLR/NF-kappaB and TLR/MAPK signaling in LPS-induced RAW264.7 cells. Int. Immunopharmacol. 2017, 45, 110–119. [Google Scholar] [CrossRef]
- Lim, W.; Jeong, M.; Bazer, F.W.; Song, G. Coumestrol Inhibits Proliferation and Migration of Prostate Cancer Cells by Regulating AKT, ERK1/2, and JNK MAPK Cell Signaling Cascades. J. Cell. Physiol. 2017, 232, 862–871. [Google Scholar] [CrossRef]
- Lim, W.; Yang, C.; Park, S.; Bazer, F.W.; Song, G. Inhibitory Effects of Quercetin on Progression of Human Choriocarcinoma Cells Are Mediated Through PI3K/AKT and MAPK Signal Transduction Cascades. J. Cell. Physiol. 2017, 232, 1428–1440. [Google Scholar] [CrossRef]
- Kim, G.D. Kaempferol Inhibits Angiogenesis by Suppressing HIF-1alpha and VEGFR2 Activation via ERK/p38 MAPK and PI3K/Akt/mTOR Signaling Pathways in Endothelial Cells. Prev. Nutr. Food Sci. 2017, 22, 320–326. [Google Scholar] [CrossRef]
- Malloy, K.M.; Wang, J.; Clark, L.H.; Fang, Z.; Sun, W.; Yin, Y.; Kong, W.; Zhou, C.; Bae-Jump, V.L. Novasoy and genistein inhibit endometrial cancer cell proliferation through disruption of the AKT/mTOR and MAPK signaling pathways. Am. J. Transl. Res. 2018, 10, 784–795. [Google Scholar]
- Cui, S.; Wang, J.; Wu, Q.; Qian, J.; Yang, C.; Bo, P. Genistein inhibits the growth and regulates the migration and invasion abilities of melanoma cells via the FAK/paxillin and MAPK pathways. Oncotarget 2017, 8, 21674–21691. [Google Scholar] [CrossRef]
- Borah, N.; Gunawardana, S.; Torres, H.; McDonnell, S.; Van Slambrouck, S. 5,6,7,3′,4′,5′-Hexamethoxyflavone inhibits growth of triple-negative breast cancer cells via suppression of MAPK and Akt signaling pathways and arresting cell cycle. Int. J. Oncol. 2017, 51, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Liu, Y.; Li, Q.; Guo, X.; Gu, L.; Ma, Z.G.; Zhu, Y.P. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed. Environ. Sci. 2013, 26, 902–911. [Google Scholar] [PubMed]
- Zhu, J.; Yu, W.; Liu, B.; Wang, Y.; Shao, J.; Wang, J.; Xia, K.; Liang, C.; Fang, W.; Zhou, C.; et al. Escin induces caspase-dependent apoptosis and autophagy through the ROS/p38 MAPK signalling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2017, 8, e3113. [Google Scholar] [CrossRef]
- Aldonza, M.B.D.; Hong, J.Y.; Bae, S.Y.; Song, J.; Kim, W.K.; Oh, J.; Shin, Y.; Lee, S.H.; Lee, S.K. Suppression of MAPK Signaling and Reversal of mTOR-Dependent MDR1-Associated Multidrug Resistance by 21α-Methylmelianodiol in Lung Cancer Cells. PLoS ONE 2015, 10, e0127841. [Google Scholar]
- Luo, W.; Liu, X.; Sun, W.; Lu, J.J.; Wang, Y.; Chen, X. Toosendanin, a natural product, inhibited TGF-beta1-induced epithelial-mesenchymal transition through ERK/Snail pathway. Phytother. Res. 2018, 32, 2009–2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | Cell line | Agent | Biological Relevance | Reference |
|---|---|---|---|---|
| Oral cancer | SCC9 and SCC25 | SP600125 | Affects cell viability and cell cycle progression via JNK/STAT3 | [45] |
| Melanoma | BRAF mutant cells | PLX4032 | Inhibits ERK signaling cascade in a mutant BRAF-selective mode | [35] |
| USAC, YUSOC, YUMAC, YUFIC, YUROB, YUGEN, YULAC, MEL501, MEL624, and MEL928 cell lines | GW5074 | Inhibition of cRAF without affecting BCL2 and pBad | [62] | |
| Lung and breast cancer cell lines | EGFR and KRAS-mutant cell lines | AZD6244 | Activation of PI3K/AKT, negative feedback on ERBB receptors, target ERK | [42] |
| Breast cancer | MCF-7 and MDA-MB-231 cell lines | PD98059 | Promotes invasion, ineffective in breast cancer models, targeting MEK | [56] |
| MCF-7, T47D, MDA-MB-231, and BT-549 cell lines | Simvastatin | Anti-tumoral effects by reversing metabolic products of the mevalonate pathway; inhibited MAPK by dephosphorylating sequential cascades of cRAF–MEK1/2–ERK1/2 | [63] | |
| MCF-7 and MDA-MB-231 cell lines | Alisertib | Promotes apoptosis and autophagy by targeting Aurora A via p38 p38/AKT/mTOR pathways | [64] | |
| MDA-MB-468, BT549, and MDA-MB-231 cell lines | Teriflunomide | Reduce cell proliferation, activation of apoptosis and inhibition of EMT via MAPK | [65] | |
| Colon cancer | SW480, SW620, and DLD-1 cell lines | NSC95397 | Reduces cell proliferation via Cdc25 and MKP-1 | [66] |
| Prostate cancer | Mutant mice with prostate specific deletion of Pten | PD325901 | Activation of RAS/MEK related to PTEN/PI3K/AKT, that conduct the activation of EMT and metastasis | [40] |
| Myeloid leukemia | TF-1 cells | PD98059 | Dual effects on MAPK and AKT pathways in hematopoietic cells | [38] |
| Glioblastoma | A172, M059J, M059K, and U87, and HEK293T cell lines and nude mice | Lovastatin | Inhibition of NF-κB and ERK but activates JNK; sensitizes TRAIL-induced apoptosis by upregulation of DR5 level via NF-κB inactivation | [67] |
| In Vitro and In Vivo Studies | Compounds | Biological Relevance | Reference |
|---|---|---|---|
| NCI-H1395, NCI-H1755, NCI-H1666, NCI-H508, and SKMEL-28 MRC-5 and 8505C HT-29 and CAL-12T HCC364 and xenograft mouse | Dabrafenib (BRAF inhibitor), vemurafenib (BRAF inhibitor), trametinib (MEK inhibitor), and selumetinib (MEK inhibitor) | Targets critical survival signals in lung cancer, BRAF non-V600E mutant cases | [55] |
| HCC827, HK2–6, HKE-3, and derivative NCI-H1299 cell lines and xenograft mouse | Dabrafenib, RAF265 (RAF/VEGF inhibitor), trametinib, and lapatinib (EGFR/HER2 inhibitor) | Prevents paradoxical MAPK activation and afford synergistic growth inhibition or additional EGFR blockade in lung adenocarcinoma | [100] |
| NCI-H2077, RT112, DMS114, and NCI-H520 cells and nude mice | Crizotinib (EML4-ALK) and Trametinib | Prevents drug resistance in in ALK-positive tumors | [94] |
| HCC827, HCC4006, and PC-9, gefitinib-resistant cells, and afatinib-resistant cells | Trametinib and taselisib (PIK3CA inhibitor) | Inhibition of MEK and PI3K signaling pathways prevent acquired resistance to EGFR TKIs | [101] |
| Cell lines sensitive and resistant to therapy and xenograft mouse | PF-04691502 (PI3K/mTOR inhibitor) and PF502 (PI3K/mTOR inhibitor) | RAS signaling as a key mediator of PF502 resistance | [53] |
| MDA-MB-231 | Enterolactone (phytoestrogen) | EMT regulation (inhibiting TGFβ-induced EMT by blocking ERK/NF-κB/Snail) | [102] |
| Metastatic melanoma cell lines and mice models | PLX4720 (BRAF V600E inhibitors) and PD0325901 (MEK inhibitor) | Drug resistance, via MEK and BRAF, PI3K signaling | [103] |
| A375, WM266-4, SKMel28, and SKMel2 cells | PD184352 (MEK inhibitor), selumetinib BMS-345541 (NF-κB inhibitor), and SC-514 (NF-κB inhibitor) | Inhibition of TNFα signaling using IκB inhibitors elevated the efficacy of MAPK pathway inhibitors by targeting tumor cell immune microenvironment | [70] |
| HMEL-B and HMEL-B/M cells | MLN8237 (AURKA inhibitor) and SB415286 (GSK3A inhibitor) | AURKA/BRAF- and AURKA/MEK-mediated resistance mechanism | [96] |
| Human primary melanocytes, WM1575 and WM3619, and nude mice | PLX4720 and obatoclax (BCL2 inhibitor) | Combined treatment prevents drug resistance and apoptosis | [97] |
| NRAS-mutant melanoma cells | Pimasertib (MEK inhibitor), ABT-199 (BCL-2 inhibitor), APR-246 (TP53 activator) | Prevent resistance in NRAS-mutant and TP53 mutant by targeting MEK and BCL-2 | [98] |
| PANC-1 | Gemcitabine (DNA synthesis inhibitor) and birinapant (IAP antagonist) | Prevent drug resistance activation via FAS and p38 | [104] |
| Clinical Model | Compound | Target Mechanism | Clinical Trial Phase | Observation | Reference |
|---|---|---|---|---|---|
| Myelodysplastic syndrome | ARRY-614 | p38/Tie2 | Phase I (NCT01496495), 2011–2014, completed | Well tolerated, had sufficient activity, and increased therapeutic efficacy | [123] |
| Solid tumors/multiple myeloma | Trametinib and afuresertib | pan-AKT kinase inhibitor and of MEK1/2 | Phase II (NCT01476137) 2011–2017, completed | Intermittent dose; displayed good tolerability | [113] |
| Advanced cancer (60 participants, non-randomized) | Ralimetinib (LY2228820 dimesylate) | p38 MAPK | Phase I (NCT01393990) 2011–2014, completed | Acceptable safety, tolerability, and pharmacokinetics | [115] |
| Advanced solid tumors (125 participants—melanoma and lung cancer) | Ulixertinib (BVD-523) | ERK1/2 | Phase I dose escalation (NCT01781429) 2013–2018, completed | Responses occurred in patients with NRAS-, BRAF V600-, and non-V600 BRAF-mutant tumors | [30,124] |
| KRAS-mutated and Wild type lung cancer | Selumetinib +/− erlotinib | MEK1/2 and EGFR inhibitor | Phase II (NCT01229150) 2010–2017, completed | No significant improvement related to overall survival | [114] |
| BRAF V600-mutated NSLC | Dabrafenib + trametinib | BRAF and MEK | Phase II (NCT01336634) 2010–2019, active, not recruiting | Important clinical benefit | [117] |
| BRAFV600-mutant melanoma brain metastases | Dabrafenib + trametinib | BRAF and MEK MAPK | Phase II (NCT02039947) 2010–2019, completed | Median duration of response was relatively short | [116] |
| Advanced melanoma BRAF V600 | Vemurafenib (PLX4032) versus facarbazine chemotherapy | BRAF and methylation agent | Phase 3 trial (NCT01006980) 2011–2016, completed | High rate of response for patient with activating BRAF mutations | [119] |
| Advanced melanoma BRAF V600 | Vemurafenib and cobimetinib | BRAF and MEK MAPK | Phase I (NCT01271803) 2011–2016, completed | Metabolic alterations rapid after initiation of therapy | [30,125] |
| Colorectal cancer, NSCLC | Prexasertib (LY2606368) and ralimetinib | Chk1 and P38 MAPK | Phase I (NCT02860780) 2016–2018, completed | Safety profile, target inhibition, and dose-proportional exposure | [126] |
| KRAS-mutant-positive NSCLC | Trametinib (GSK1120212) | MEK1/2 | Phase II (NCT01362296) 2011–2014, completed | Trametinib and docetaxel have similar profession free survival | [118] |
| Adult primary hepatocellular carcinoma | Erlotinib and bevacizumab | EGFR inhibitor and VEGF-A | Phase II (NCT00365391) 2006–2015, completed | Had minimal activity based on evaluated progression-free survival | [127] |
| Biliary cancer patients | Binimetinib (MEK162) | MEK1/2 inhibitor. | Phase 1 (NCT00959127) 2009–2013, completed | Safe and tolerable, anti-tumor activity in a dose escalation study | [128] |
| Compounds | Disease | Preclinical Model | Molecular Target | Biological Relevance | Reference |
|---|---|---|---|---|---|
| Caffeic acid phenethyl ester (CAPE) + U0126 | Pancreatic ductal adenocarcinoma | MIAPaCa-2 and PANC-1 | ↓MAPK and NF-κB expression level | Reduces cell growth by cell-type-specific activation of apoptosis (MIAPaCa-2 caspase-dependent and PANC-1 caspase-independent mode) | [136] |
| Apigenin | Choriocarcinoma | JAR and JEG3 | ↓PI3K/AKT and ERK1/2 expression level | Reduces cell viability and migratory capacity; increases apoptosis | [138] |
| Coumestrol | Prostate cancer | PC3 and LNCaP | ↑phosphorylation of ERK1/2, JNK, P90RSK, and P53; ↓phosphorylation of AKT proteins | Inhibits cell proliferation and migration; activates apoptosis | [140] |
| Quercetin | Choriocarcinoma | JAR and JEG3 cells | ↓phosphorylation of AKT, P70S6K and S6; ↑phosphorylation of ERK1/2, P38, JNK and P90RSK proteins | Inhibition of proliferation, cell-cycle progression and invasion; stimulation of ROS production | [141] |
| Kaempferol | Endometrial malignant transformation | HUVECs andEBM-2 | ↓phosphorylation of ERK and p38; ERK, p38, Akt; ↓HIF-1α and VEGFR2 proteins | Inhibits angiogenesis | [142] |
| Genistein | Melanoma | Murine melanoma cell line B16F10 | ↓ phosphorylation of FAK, paxillin, tensin-2, vinculin, p38, ERK, and JNK proteins | Inhibits the growth and regulates the migration and invasion | [144] |
| Novasoy and genistein | Endometrial cancer | ECC-1 and RL-95-2 cells | ↑phosphorylation of p42/44 in both cell line; ↓ phosphorylation of S6 only in RL-95-2 cells | Reduces cell proliferation and cell-cycle arrest in G2; induces apoptosis | [143] |
| Resveratrol | T-cell acute lymphoblastic leukemia | T-ALL cell lines, Molt-4 (glucocorticoid resistant) and Jurkat (glucocorticoid resistant) | ↓Akt/mTOR/p70S6K/4E-BP1; ↑p38-MAPK | Induces apoptosis and autophagy | [147] |
| Escine | Osteosarcoma | MNNG, Saos-2, MG-63, U-2OS | ↑ p38 expression level | Induces apoptosis and autophagy | [148] |
| Triterpenoids (21α-methylmelianodiol) | Lung cancer | A549 cells | ↓ ERK, p-ERK, JNK, p-JNK, p38, and no effect on p-p38 | Targets drug resistance via P-glycoprotein (P-gp)/MDR1-association | [149] |
| Toosendanin | Lung cancer | A549 and H1975 cells | ↓ phosphorylation of ERK; ↓Snail, TGFβ1 expression level | Inhibits TGFβ1-induced EMT and migration, invasion, and adhesion | [150] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101618
Braicu C, Buse M, Busuioc C, Drula R, Gulei D, Raduly L, Rusu A, Irimie A, Atanasov AG, Slaby O, et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers. 2019; 11(10):1618. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101618
Chicago/Turabian StyleBraicu, Cornelia, Mihail Buse, Constantin Busuioc, Rares Drula, Diana Gulei, Lajos Raduly, Alexandru Rusu, Alexandru Irimie, Atanas G. Atanasov, Ondrej Slaby, and et al. 2019. "A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer" Cancers 11, no. 10: 1618. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101618