CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer

 ,
,
Abstract
:1. Introduction
2. Results
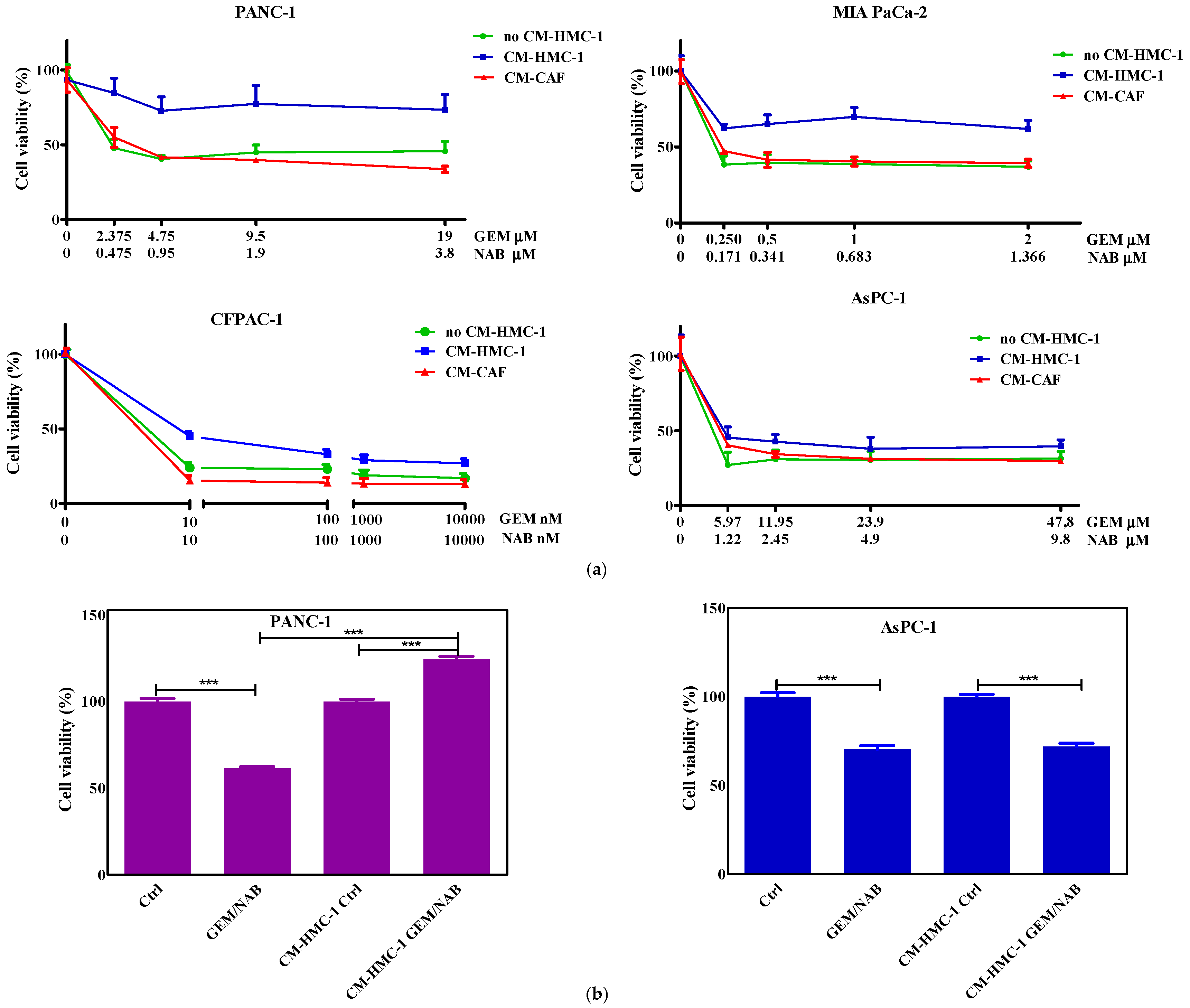
2.1. Pancreatic Cells’ Sensitivity to Gemcitabine and Nabpaclitaxel
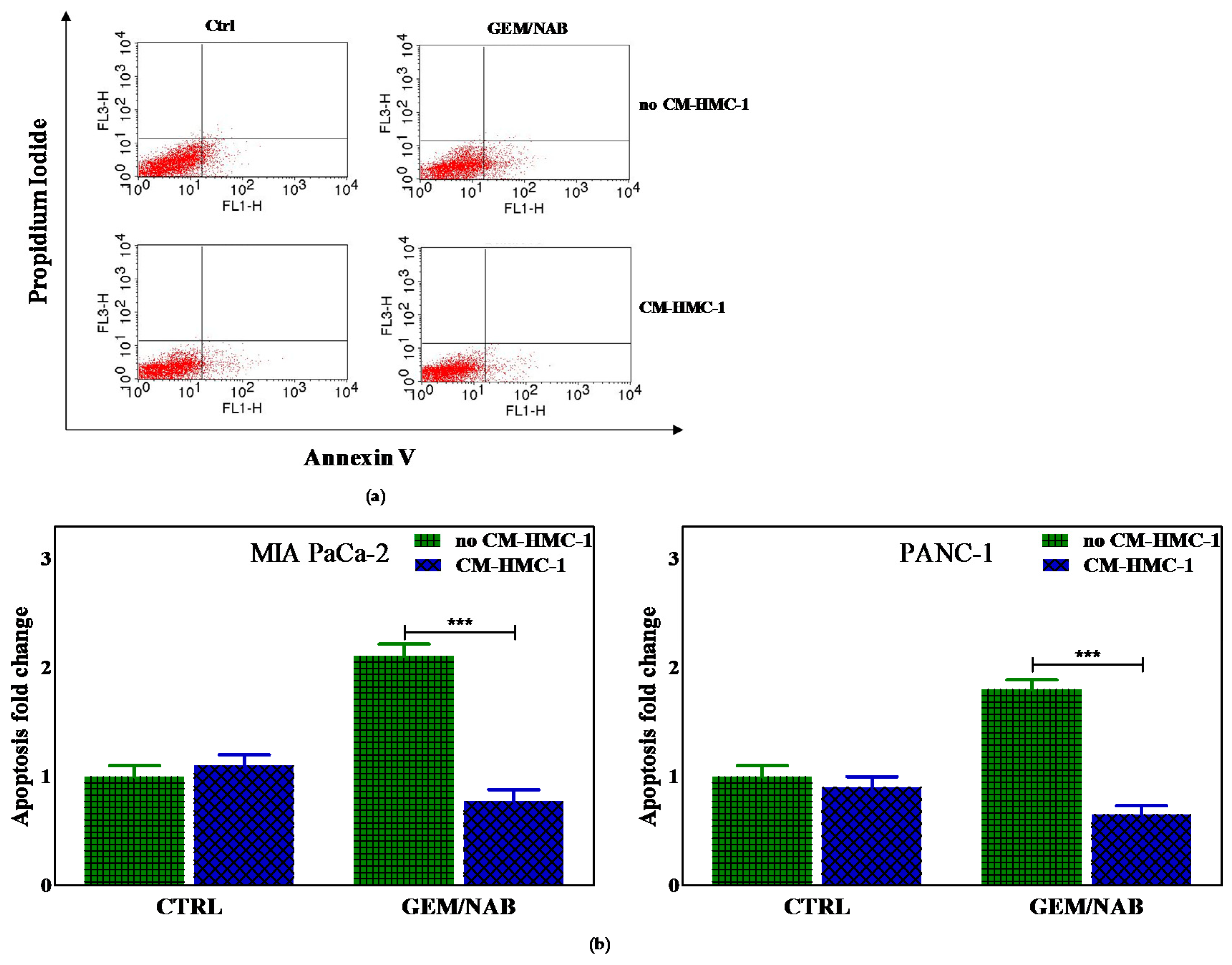
2.2. Effectiveness of Gemcitabine/Nabpaclitaxel (GEM/NAB) on Cell Viability and Apoptosis Induction Was Reduced with CM-HMC-1 but Not with CM-CAF
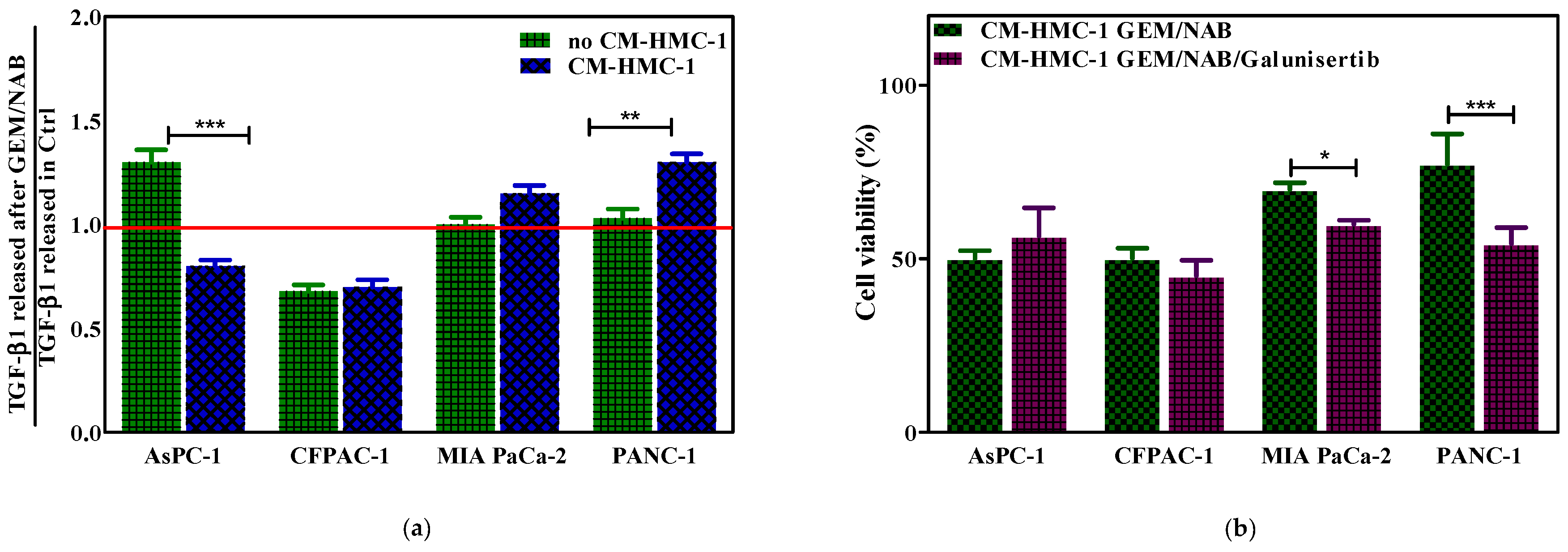
2.3. CM-HMC-1 Induced Resistance to GEM/NAB through the Activation of TGF-β Signalling
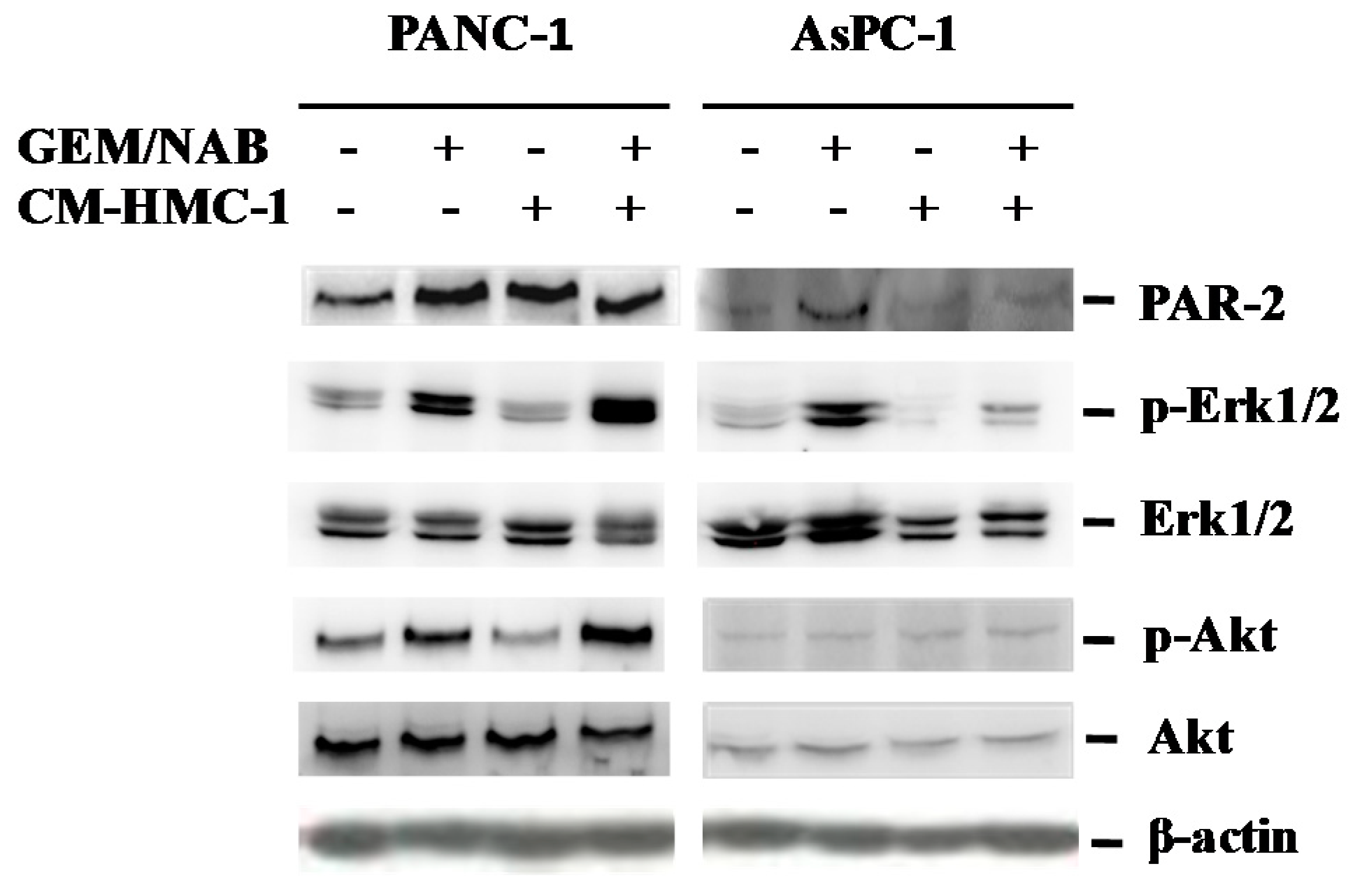
2.4. CM-HMC-1 Induces the Expression of PAR-2 and Activated Erk1/2 and Akt in TGF-β1 Activated Pancreatic Cells
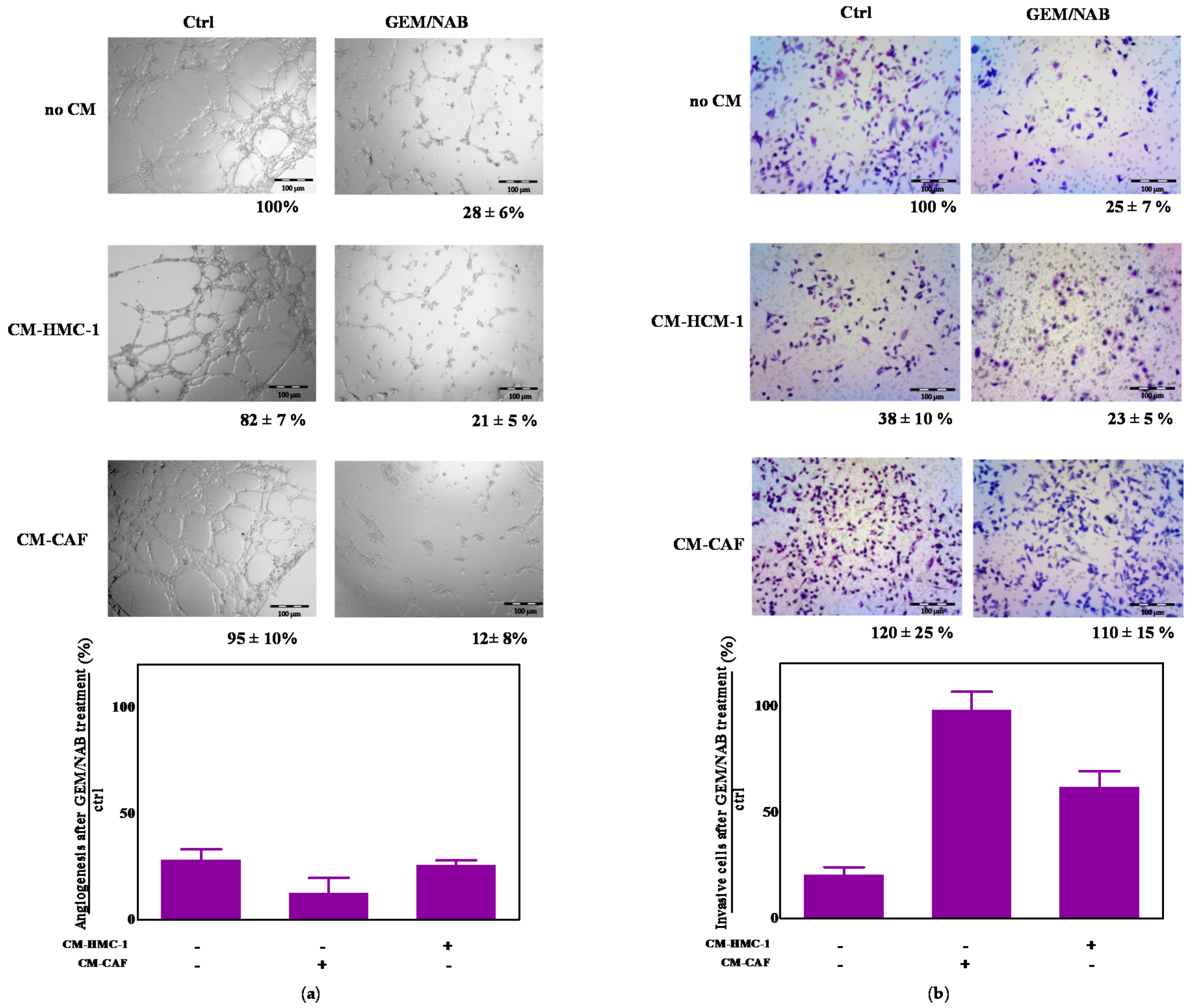
2.5. CM-HMC-1 and CM-CAF Do Not Affect GEM/NAB-Dependent Inhibition of Tumor Angiogenesis but They Reduce GEM/NAB-Dependent Inhibition of Tumor Invasion
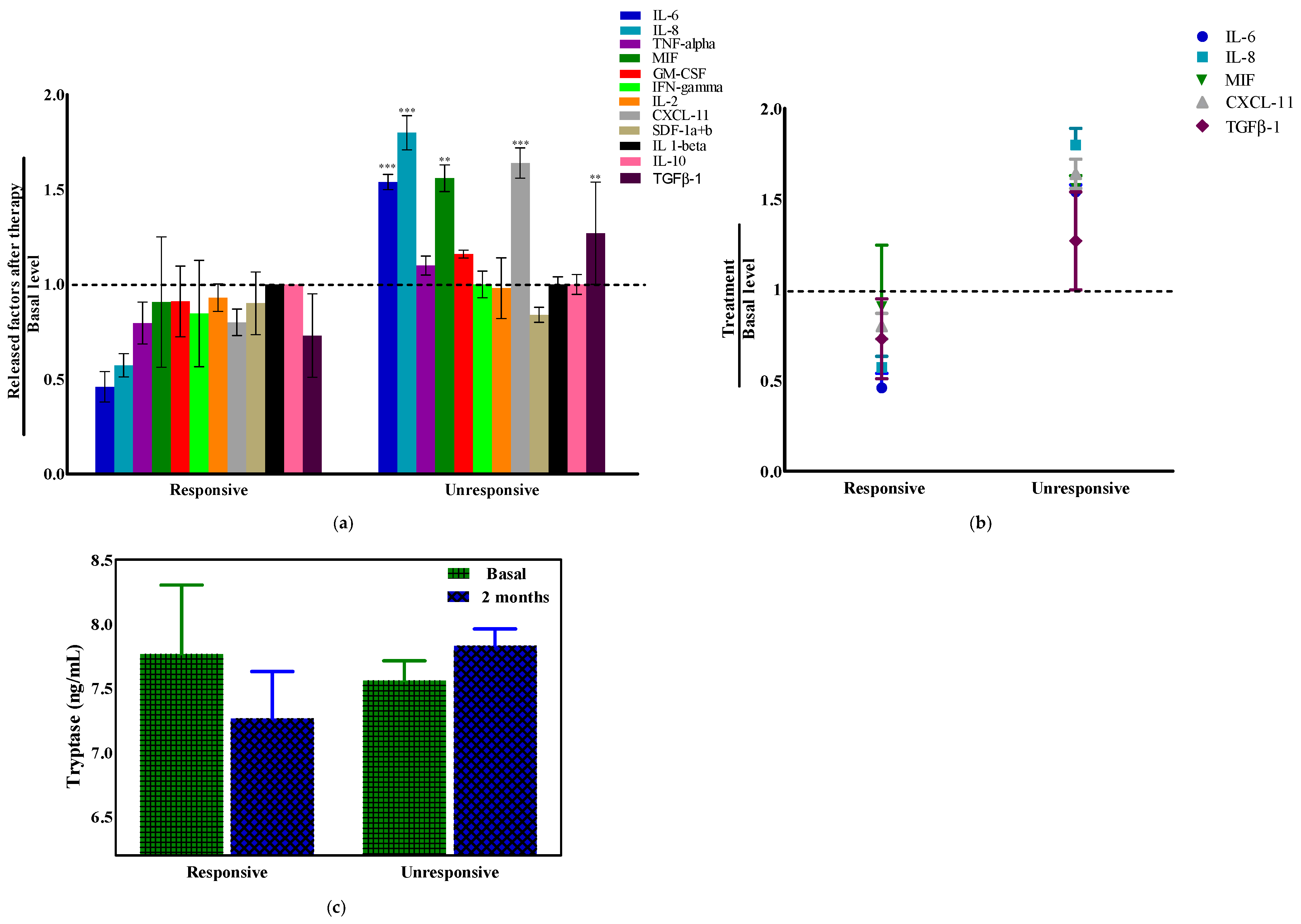
2.6. Tryptase and Cytokines’ Modulation in Pancreatic Ductal Adenocarcinoma (PDAC) Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. 3D Cell Culture
4.3. Cell Proliferation Assay
4.3.1. IC50 Determination
4.3.2. Combination Study
4.3.3. 3D Characterization
4.4. Cell Cycle Analysis
4.5. Cell Apoptosis Assay
4.6. Detection of Mast Cells-Released Tryptase and TGF-β1 by Enzyme-Linked Immunosorbent Assay (ELISA) Kit
4.7. Bio-PlexProTM Human Chemokine Assay
4.8. Western Blotting (WB) Analysis
4.9. Invasion Assay
4.10. In Vitro Capillary Morphogenesis Assay
4.11. Study Population
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel Plus Gemcitabine for Metastatic Pancreatic Cancer: Long-Term Survival From a Phase III Trial. JNCI J. Natl. Cancer Inst. 2015, 107, dju413. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Corbo, V.; Tortora, G.; Scarpa, A. Molecular Pathology of Pancreatic Cancer: From Bench-to-Bedside Translation. Curr. Drug Targets 2012, 13, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signalling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef] [PubMed]
- Strouch, M.J.; Cheon, E.C.; Salabat, M.R.; Krantz, S.B.; Gounaris, E.; Melstrom, L.G.; Dangi-Garimella, S.; Wang, E.; Munshi, H.G.; Khazaie, K.; et al. Crosstalk between Mast Cells and Pancreatic Cancer Cells Contributes to Pancreatic Tumor Progression. Clin. Cancer Res. 2010, 16, 2257–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zhai, L.; Xue, R.; Shi, J.; Zeng, Q.; Gao, C. Mast cell tryptase contributes to pancreatic cancer growth through promoting angiogenesis via activation of angiopoietin-1. Int. J. Mol. Sci. 2016, 17, 834. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ullrich, S.E. Intratumoral mast cells promote the growth of pancreatic cancer. Oncoimmunology 2013, 2, e25964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroblewski, M.; Bauer, R.; Cubas Córdova, M.; Udonta, F.; Ben-Batalla, I.; Legler, K.; Hauser, C.; Egberts, J.; Janning, M.; Velthaus, J.; et al. Mast cells decrease efficacy of anti-angiogenic therapy by secreting matrix-degrading granzyme B. Nat. Commun. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, G.; Blom, T.; Kusche-Gullberg, M.; Kjellen, L.; Butterfield, J.H.; Sundström, C.; Nilsson, K.; Hellman, L. Phenotypic Characterization of the Human Mast-Cell Line HMC-1. Scand. J. Immunol. 1994, 39, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, H.; Ohnishi, H.; Hama, K.; Shinozaki, S.; Kita, H.; Osawa, H.; Yamamoto, H.; Sato, K.; Tamada, K.; Sugano, K. Cyclooxygenase-2 is required for activated pancreatic stellate cells to respond to proinflammatory cytokines. Am. J. Physiol. Cell Physiol. 2006, 292, C259–C268. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Hill, C.S. Smad4 Dependency Defines Two Classes of Transforming Growth Factor β (TGF-β) Target Genes and Distinguishes TGF-β-Induced Epithelial-Mesenchymal Transition from Its Antiproliferative and Migratory Responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor β receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Witte, D.; Rauch, B.H.; Settmacher, U.; Lehnert, H.; Gieseler, F.; Kaufmann, R. Proteinase-activated receptor 2 may drive cancer progression by facilitating TGF-β signalling. Int. J. Mol. Sci. 2017, 18, 2494. [Google Scholar] [CrossRef] [PubMed]
- Oldford, S.A.; Marshall, J.S. Mast cells as targets for immunotherapy of solid tumors. Mol. Immunol. 2014, 63, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Yako, Y.Y.; Kruger, D.; Smith, M.; Brand, M. Cytokines as biomarkers of pancreatic ductal adenocarcinoma: A systematic review. PLoS ONE 2016, 11, e0154016. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Blain, S.W.; Lo, R.S. TGFβ Signalling in Growth Control, Cancer, and Heritable Disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Ikeda, O.; Egami, H.; Ishiko, T.; Ishikawa, S.; Kamohara, H.; Hidaka, H.; Mita, S.; Ogawa, M. Expression of proteinase-activated receptor-2 in human pancreatic cancer: A possible relation to cancer invasion and induction of fibrosis. Int. J. Oncol. 2003, 22, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Zeeh, F.; Witte, D.; Gädeken, T.; Rauch, B.H.; Grage-Griebenow, E.; Leinung, N.; Fromm, S.J.; Stölting, S.; Mihara, K.; Kaufmann, R.; et al. Proteinase-activated receptor 2 promotes TGF-β-dependent cell motility in pancreatic cancer cells by sustaining expression of the TGF-β type I receptor ALK5. Oncotarget 2016, 7, 41095–41109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funamizu, N.; Hu, C.; Lacy, C.; Schetter, A.; Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, M.B.; Ried, T.; Yfantis, H.G.; et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; He, P.; Wang, J.; Schetter, A.; Tang, W.; Funamizu, N.; Yanaga, K.; Uwagawa, T.; Satoskar, A.R.; Gaedcke, J.; et al. A Novel MIF Signalling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer Res. 2016, 76, 3838–3850. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.-B.; Tong, M.-T.; Wang, J.; Hu, H.; Zhai, C.-Y.; Huang, C.-X.; Li, D. Suppression of IL-6 Gene by shRNA Augments Gemcitabine Chemosensitization in Pancreatic Adenocarcinoma Cells. BioMed Res. Int. 2018, 2018, 3195025. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1β can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, R.; Musteanu, M.; Garcia-Garcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Muñoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Gieseler, F.; Kaufmann, R.; Settmacher, U.; Lehnert, H.; Rauch, B.H. Signalling crosstalk of TGF-β/ALK5 and PAR2/PAR1: A complex regulatory network controlling fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 1568. [Google Scholar] [CrossRef] [PubMed]
- Rattenholl, A.; Seeliger, S.; Buddenkotte, J.; Schön, M.; Schön, M.P.; Ständer, S.; Vergnolle, N.; Steinhoff, M. Proteinase-activated receptor-2 (PAR2): A tumor suppressor in skin carcinogenesis. J. Investig. Dermatol. 2007, 127, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; DIaz, A.M.; Torres, C.; Mangan, R.J.; DeCant, B.; McKinney, R.; Tsao, M.S.; Lowy, A.; Munshi, H.G.; Jung, B.; et al. TGFβ engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 2017, 36, 4336–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signalling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Brunen, D.; Willems, S.M.; Kellner, U.; Midgley, R.; Simon, I.; Bernards, R. TGF-β: An emerging player in drug resistance. Cell Cycle 2013, 12, 2960–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Liao, Q.; Zhao, Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Osuga, Y.; Koga, K.; Yoshino, O.; Hirata, T.; Morimoto, C.; Harada, M.; Takemura, Y.; Nose, E.; Yano, T.; et al. The expression and possible roles of chemokine CXCL11 and its receptor CXCR3 in the human endometrium. J. Immunol. 2006, 177, 8813–8821. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, R.M.; Porcelli, L.; Lopedota, A.A.; Laquintana, V.; Lopalco, A.; Cutrignelli, A.; Altamura, E.; Di Fonte, R.; Azzariti, A.; Franco, M.; et al. Targeting human liver cancer cells with lactobionic acid-G(4)-PAMAM-FITC sorafenib loaded dendrimers. Int. J. Pharm. 2017, 528, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, L.; Iacobazzi, R.M.; Quatrale, A.E.; Bergamini, C.; Denora, N.; Crupi, P.; Antonacci, D.; Mangia, A.; Simone, G.; Silvestris, N.; et al. Grape seed extracts modify the outcome of oxaliplatin in colon cancer cells by interfering with cellular mechanisms of drug cytotoxicity. Oncotarget 2017, 8, 50845–50863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzariti, A.; Mancarella, S.; Porcelli, L.; Quatrale, A.E.; Caligiuri, A.; Lupo, L.; Dituri, F.; Giannelli, G. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/α3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology 2016, 64, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Serratì, S.; Chillà, A.; Laurenzana, A.; Margheri, F.; Giannoni, E.; Magnelli, L.; Chiarugi, P.; Dotor, J.; Feijoo, E.; Bazzichi, L.; et al. Systemic sclerosis endothelial cells recruit and activate dermal fibroblasts by induction of a connective tissue growth factor (CCN2)/transforming growth factor β-dependent mesenchymal-to-mesenchymal transition. Arthritis Rheum. 2013, 65, 258–269. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Fibbi, G.; Cinelli, M.; Guiducci, S.; Del Rosso, A.; Margheri, F.; Serratì, S.; Pucci, M.; Kahaleh, B.; Fan, P.; et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004, 50, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) 1 | ||
|---|---|---|
| Cell Line | GEM | NAB |
| AsPC-1 | 23.9 ± 2.1 | 4.9 ± 1.1 |
| PANC-1 | 9.5 ± 0.8 | 1.9 ± 0.4 |
| MIA PaCa-2 | 1 ± 0.15 | 0.683 ± 0.104 |
| CFPAC-1 | 0.007 ± 0.003 | 0.008 ± 0.005 |
| Patients Characteristics | Frequency (%) or Median (IQ) |
|---|---|
| Age | 64.7 |
| Gender | |
| Male | 7 (70%) |
| Female | 3 (30%) |
| Primary tumor location | |
| Head | 7 (70%) |
| Body/tail | 3 (30%) |
| Stage at diagnosis | |
| IV stage (TMN 7th edition) | 10 (100%) |
| Site of metastasis | |
| Liver | 6 (60%) |
| Lung | 1 (10%) |
| Nodes | 7 (70%) |
| Peritoneum | 2 (20%) |
| Other | 1 (10%) |
| ECOG PS | |
| 0 | 3 (30%) |
| 1 | 6 (60%) |
| 2 | 1 (10%) |
| First line therapy | |
| GEM/NAB | 10 (100%) |
| First line PFS | |
| <6 months | 4 (40%) |
| >6 months | 6 (60%) |
| Response to first-line CT | |
| Responders (CR, PR, SD) | 6 (60%) |
| Non Responders (Pro) | 4 (40%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porcelli, L.; Iacobazzi, R.M.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.G.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers 2019, 11, 330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030330
Porcelli L, Iacobazzi RM, Di Fonte R, Serratì S, Intini A, Solimando AG, Brunetti O, Calabrese A, Leonetti F, Azzariti A, et al. CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers. 2019; 11(3):330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030330
Chicago/Turabian StylePorcelli, Letizia, Rosa Maria Iacobazzi, Roberta Di Fonte, Simona Serratì, Angelica Intini, Antonio Giovanni Solimando, Oronzo Brunetti, Angela Calabrese, Francesco Leonetti, Amalia Azzariti, and et al. 2019. "CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer" Cancers 11, no. 3: 330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11030330