Cetuximab-Mediated Protection from Hypoxia- Induced Cell Death: Implications for Therapy Sequence in Colorectal Cancer

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
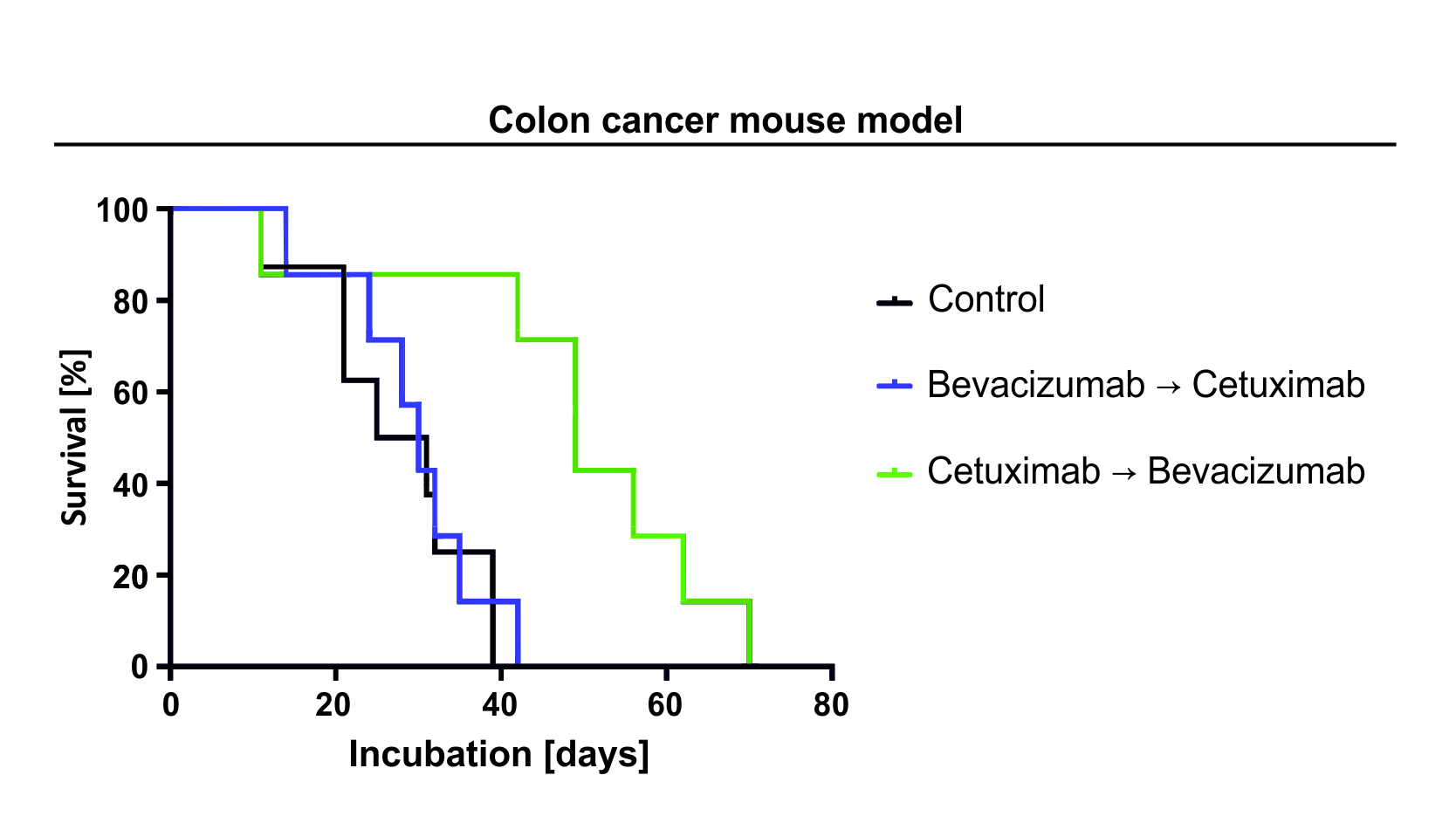
2.1. Cetuximab Does Not Influence Cell Growth or Viability in Normoxia
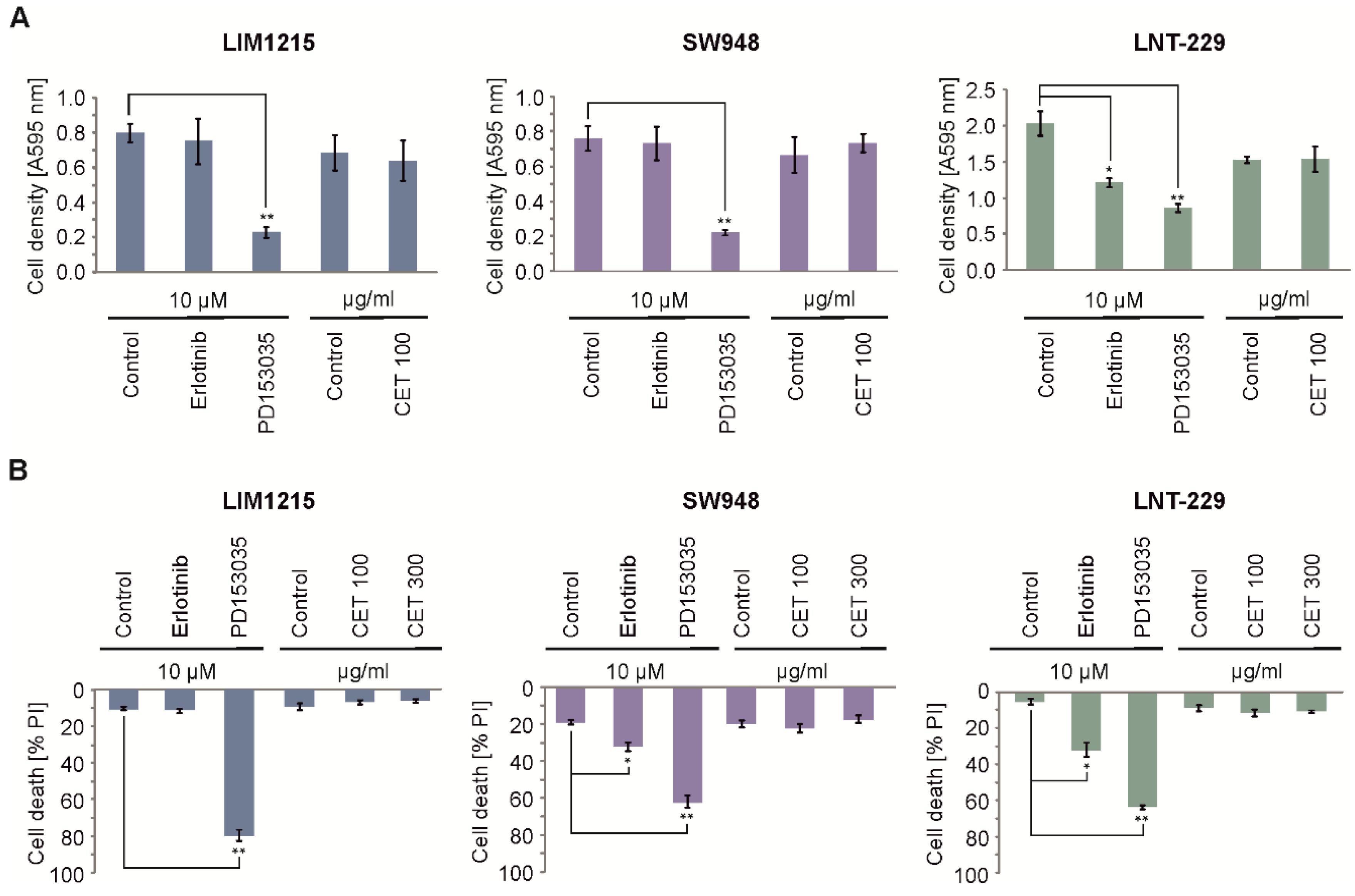
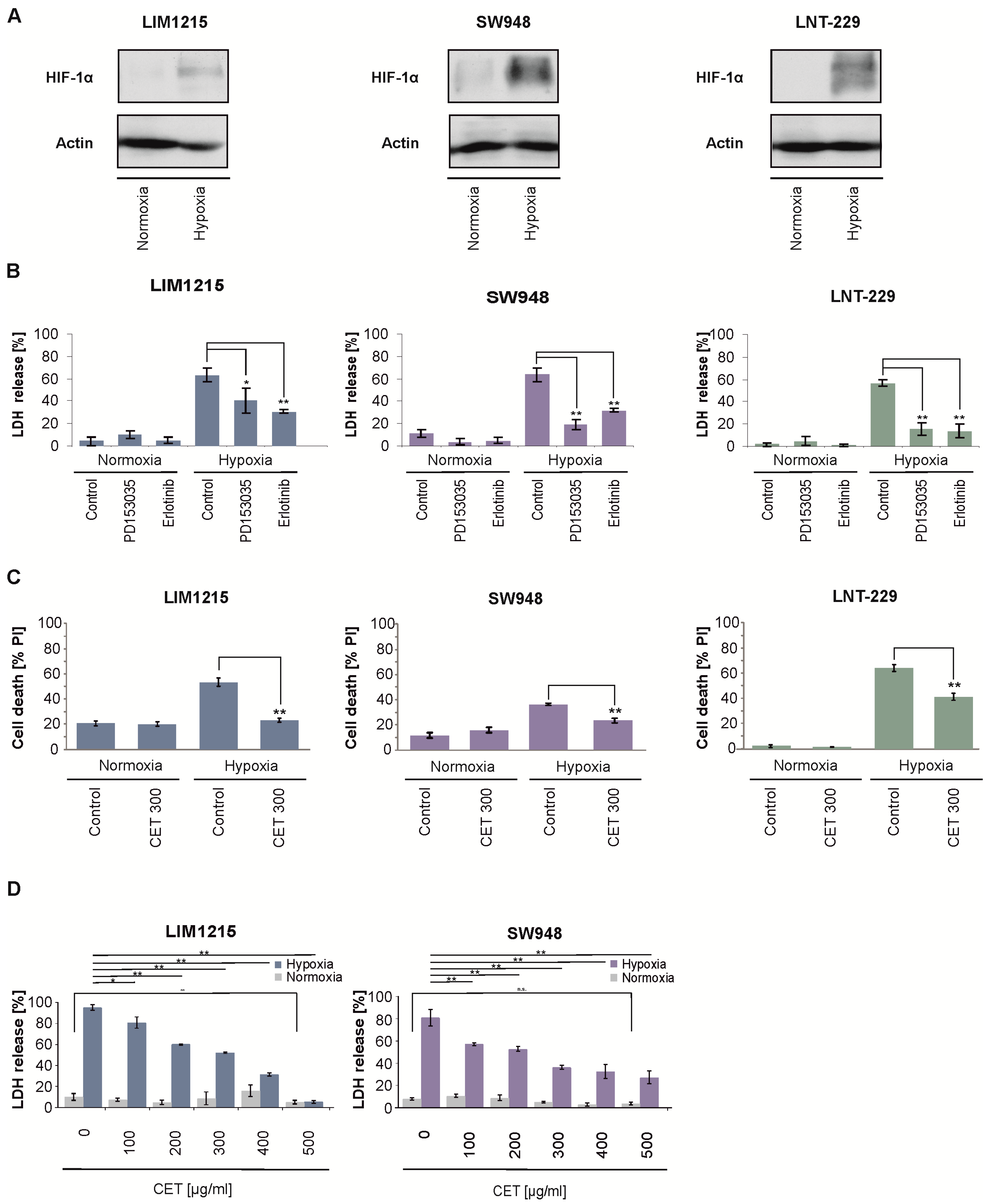
2.2. Cetuximab Protects Colon Carcinoma Cells From Hypoxia-Induced Cell Death
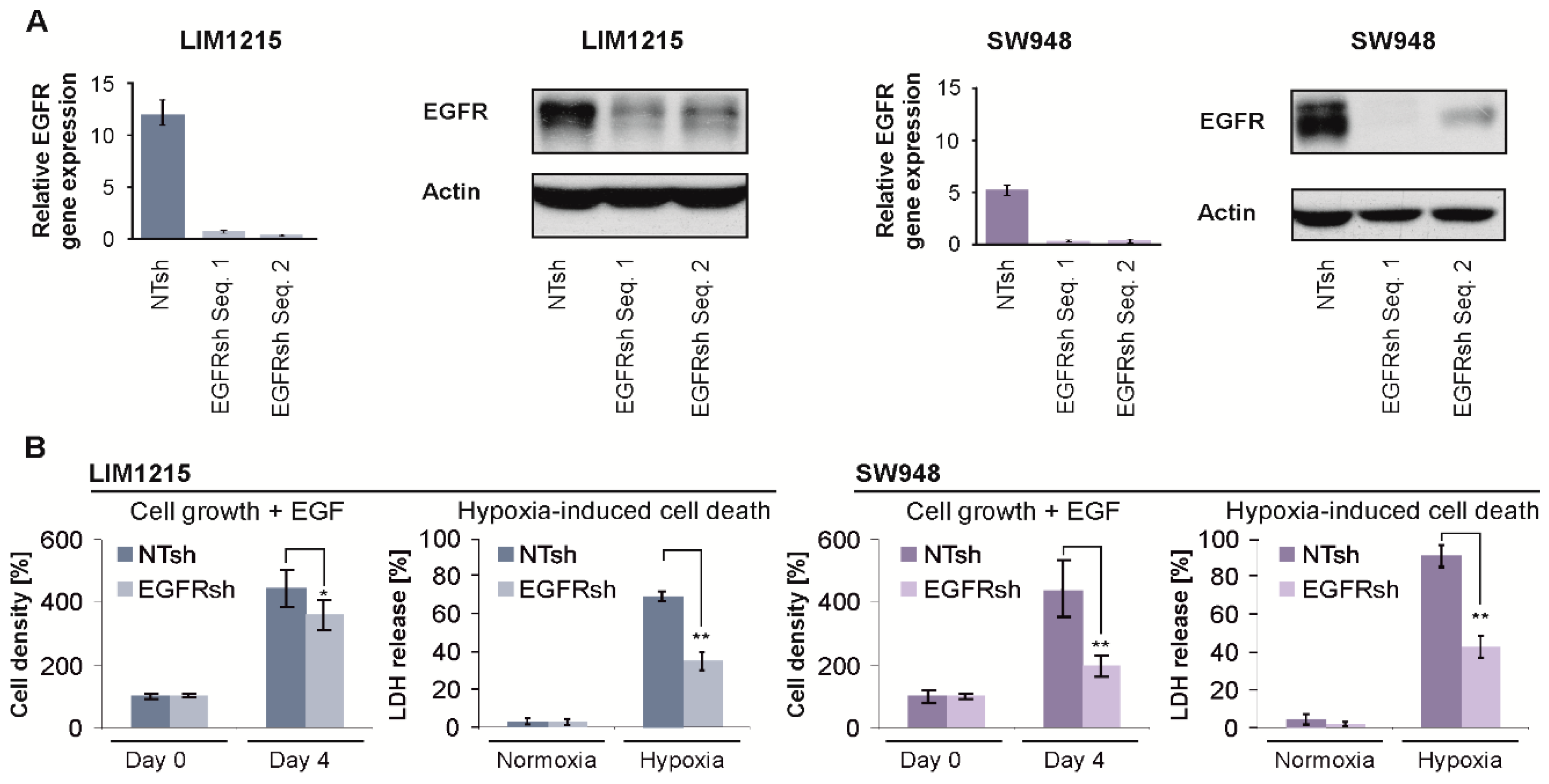
2.3. EGFR Gene Suppression Impairs Cell Growth and Protects From Hypoxia-Induced Cell Death
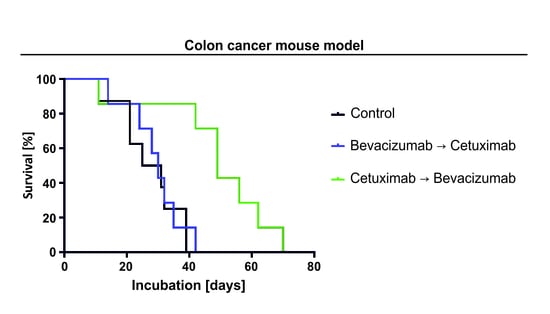
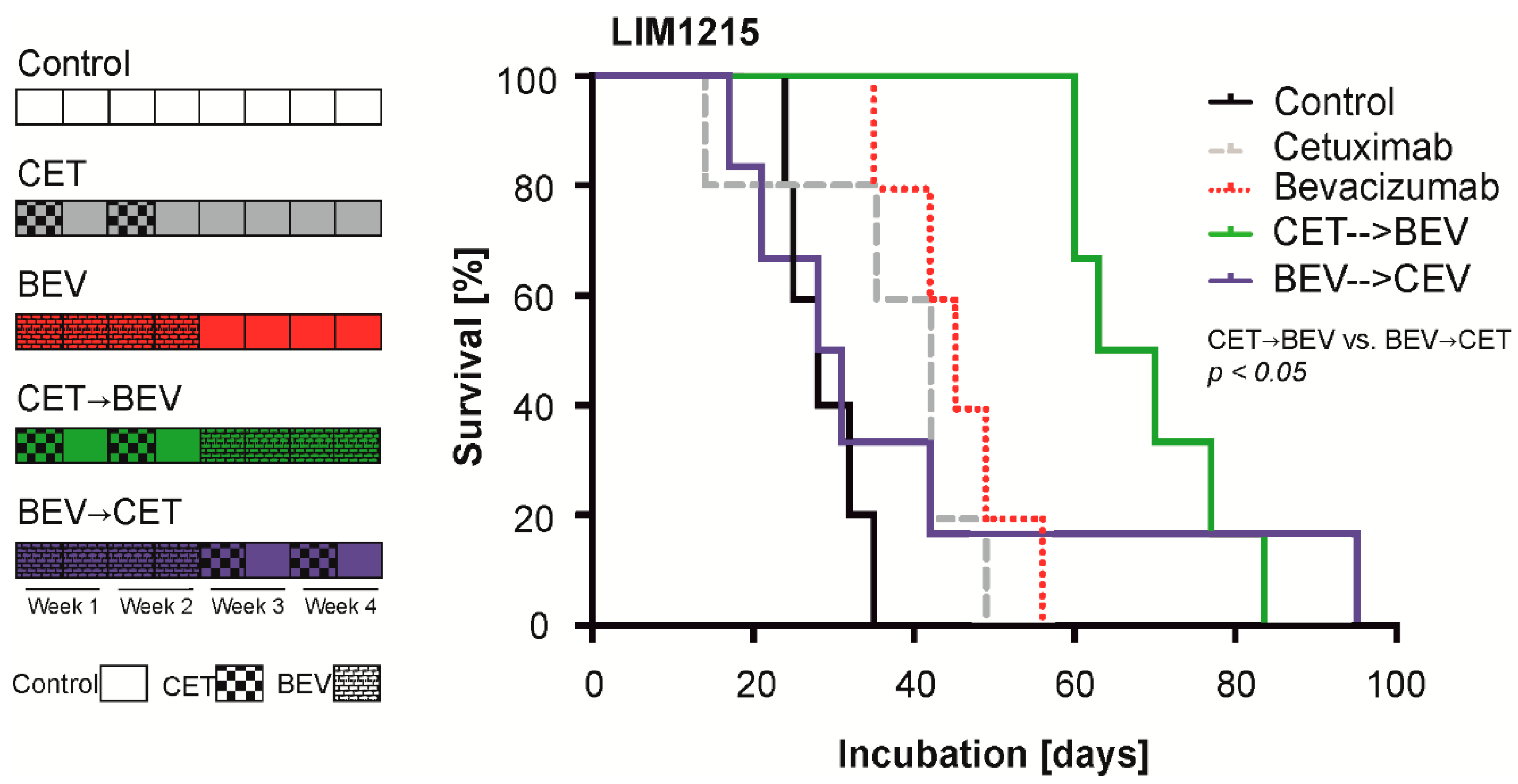
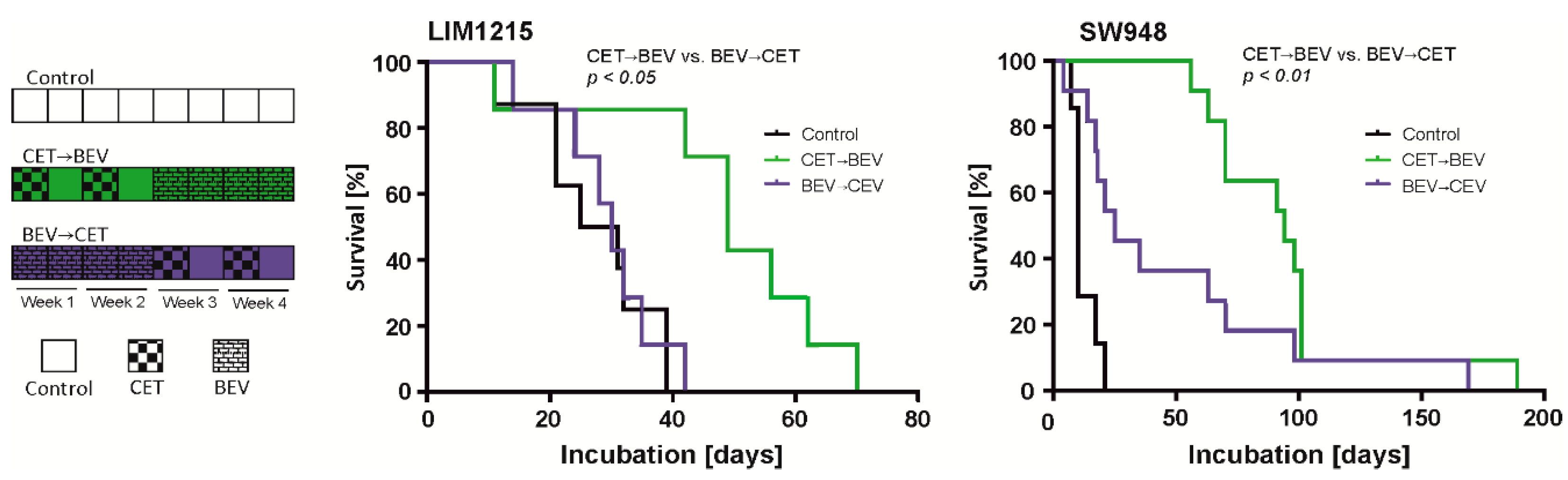
2.4. The Sequence of Cetuximab and Bevacizumab Administration Influences Outcome in a LIM1215 and a SW948 Subcutaneous Mouse Model
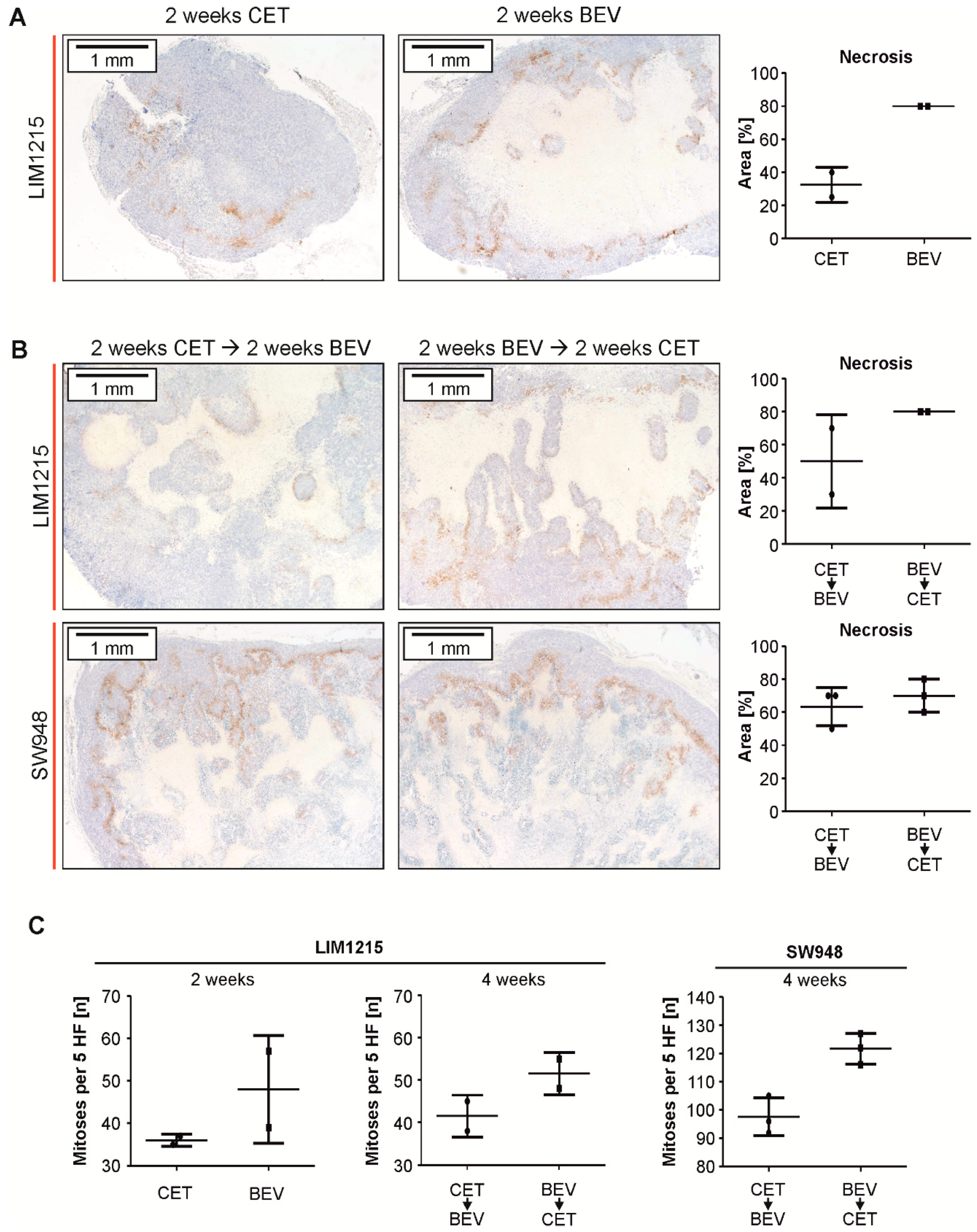
2.5. Subcutaneous Tumor Xenografts Exhibit Larger Areas of Necrosis When Treated With Bevacizumab
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Generation of EGFR Gene-Suppressed Cells
4.3. Reagents
4.4. Induction of Hypoxia
4.5. Cell Density and Cell Viability Assays
4.6. RNA Extraction and Quantitative Reverse Transcription-PCR Analysis
4.7. Immunoblot Analysis
4.8. Animal Experiment, Tumor Histology, and Immunohistochemistry
4.9. Statistics
4.10. Ethics Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edwards, M.S.; Chadda, S.D.; Zhao, Z.; Barber, B.L.; Sykes, D.P. A systematic review of treatment guidelines for metastatic colorectal cancer. Colorectal Dis. 2012, 14, e31–e47. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Gundin, J.; Fernandez-Carballido, A.M.; Martinez-Valdivieso, L.; Barreda-Hernandez, D.; Torres-Suarez, A.I. New Trends in the Therapeutic Approach to Metastatic Colorectal Cancer. Int. J. Med. Sci. 2018, 15, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N.; et al. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017, 317, 2392–2401. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet. Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Aljehani, M.A.; Morgan, J.W.; Guthrie, L.A.; Jabo, B.; Ramadan, M.; Bahjri, K.; Lum, S.S.; Selleck, M.; Reeves, M.E.; Garberoglio, C.; et al. Association of Primary Tumor Site With Mortality in Patients Receiving Bevacizumab and Cetuximab for Metastatic Colorectal Cancer. JAMA Surg. 2018, 153, 60–67. [Google Scholar] [CrossRef]
- Hecht, J.R.; Mitchell, E.; Chidiac, T.; Scroggin, C.; Hagenstad, C.; Spigel, D.; Marshall, J.; Cohn, A.; McCollum, D.; Stella, P.; et al. A randomized phase IIIB trial of chemotherapy, bevacizumab, and panitumumab compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Tol, J.; Koopman, M.; Cats, A.; Rodenburg, C.J.; Creemers, G.J.; Schrama, J.G.; Erdkamp, F.L.; Vos, A.H.; van Groeningen, C.J.; Sinnige, H.A.; et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modest, D.P.; Stintzing, S.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; et al. Impact of Subsequent Therapies on Outcome of the FIRE-3/AIO KRK0306 Trial: First-Line Therapy With FOLFIRI Plus Cetuximab or Bevacizumab in Patients With KRAS Wild-Type Tumors in Metastatic Colorectal Cancer. J. Clin. Oncol. 2015, 33, 3718–3726. [Google Scholar] [CrossRef]
- Peeters, M.; Forget, F.; Karthaus, M.; Valladares-Ayerbes, M.; Zaniboni, A.; Demonty, G.; Guan, X.; Rivera, F. Exploratory pooled analysis evaluating the effect of sequence of biological therapies on overall survival in patients with RAS wild-type metastatic colorectal carcinoma. ESMO Open 2018, 3, e000297. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Bianco, R.; Caputo, R.; Damiano, V.; Troiani, T.; Melisi, D.; De Vita, F.; De Placido, S.; Bianco, A.R.; Tortora, G. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin. Cancer Res. 2004, 10, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Viloria-Petit, A.; Crombet, T.; Jothy, S.; Hicklin, D.; Bohlen, P.; Schlaeppi, J.M.; Rak, J.; Kerbel, R.S. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: A role for altered tumor angiogenesis. Cancer Res. 2001, 61, 5090–5101. [Google Scholar]
- Huijbers, E.J.; van Beijnum, J.R.; Thijssen, V.L.; Sabrkhany, S.; Nowak-Sliwinska, P.; Griffioen, A.W. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist. Updates 2016, 25, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, N.; De, P.; Brian, L.J. Evading anti-angiogenic therapy: Resistance to anti-angiogenic therapy in solid tumors. Am. J. Transl. Res. 2015, 7, 1675–1698. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, J.P.; Klumpp, A.; Wolburg, H.; Weller, M. Inhibition of epidermal growth factor receptor signaling protects human malignant glioma cells from hypoxia-induced cell death. Cancer Res. 2004, 64, 1575–1578. [Google Scholar] [CrossRef] [Green Version]
- Hartel, I.; Ronellenfitsch, M.; Wanka, C.; Wolking, S.; Steinbach, J.P.; Rieger, J. Activation of AMP-activated kinase modulates sensitivity of glioma cells against epidermal growth factor receptor inhibition. Int. J. Oncol. 2016, 49, 173–180. [Google Scholar] [CrossRef]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.H.; Byun, D.S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008, 68, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronellenfitsch, M.W.; Brucker, D.P.; Burger, M.C.; Wolking, S.; Tritschler, F.; Rieger, J.; Wick, W.; Weller, M.; Steinbach, J.P. Antagonism of the mammalian target of rapamycin selectively mediates metabolic effects of epidermal growth factor receptor inhibition and protects human malignant glioma cells from hypoxia-induced cell death. Brain 2009, 132, 1509–1522. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.R.; Moore, D.F.; Hidalgo, M.; Doroshow, J.H.; Poplin, E.A.; Goodin, S.; Mauro, D.; Rubin, E.H. Pharmacokinetics of cetuximab after administration of escalating single dosing and weekly fixed dosing in patients with solid tumors. Clin. Cancer Res. 2006, 12, 6517–6522. [Google Scholar] [CrossRef] [Green Version]
- Heskamp, S.; Boerman, O.C.; Molkenboer-Kuenen, J.D.; Oyen, W.J.; van der Graaf, W.T.; van Laarhoven, H.W. Bevacizumab reduces tumor targeting of antiepidermal growth factor and anti-insulin-like growth factor 1 receptor antibodies. Int. J. Cancer 2013, 133, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Derangère, V.; Fumet, J.D.; Boidot, R.; Bengrine, L.; Limagne, E.; Chevriaux, A.; Vincent, J.; Ladoire, S.; Apetoh, L.; Rébé, C.; et al. Does bevacizumab impact anti-EGFR therapy efficacy in metastatic colorectal cancer? Oncotarget 2016, 7, 9309–9321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattingen, E.; Jurcoane, A.; Bähr, O.; Rieger, J.; Magerkurth, J.; Anti, S.; Steinbach, J.P.; Pilatus, U. Bevacizumab impairs oxidative energy metabolism and shows antitumoral effects in recurrent glioblastomas: A 31P/1H MRSI and quantitative magnetic resonance imaging study. Neuro-Oncology 2011, 13, 1349–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, S.; Kikuchi, H.; Iino, I.; Uehara, T.; Setoguchi, T.; Fujita, T.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Kitagawa, K.; et al. Anti-VEGF antibody therapy induces tumor hypoxia and stanniocalcin 2 expression and potentiates growth of human colon cancer xenografts. Int. J. Cancer 2014, 135, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Thiepold, A.L.; Lorenz, N.I.; Foltyn, M.; Engel, A.L.; Divé, I.; Urban, H.; Heller, S.; Bruns, I.; Hofmann, U.; Dröse, S.; et al. Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain 2017, 140, 2623–2638. [Google Scholar] [CrossRef]
- Lu, H.; Li, X.; Luo, Z.; Liu, J.; Fan, Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol. Cancer Ther. 2013, 12, 2187–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainer, J.L.; Sheehan, J.P.; Larner, J.M.; Jones, D.R. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J. Neurosurg. 2017, 126, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Thews, O.; Hoeckel, M. Treatment resistance of solid tumors: Role of hypoxia and anemia. Med. Oncol. 2001, 18, 243–259. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Tumor Hypoxia: Causative Mechanisms, Microregional Heterogeneities, and the Role of Tissue-Based Hypoxia Markers. Adv. Exp. Med. Biol. 2016, 923, 77–86. [Google Scholar] [CrossRef]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Mayer, A. Hypoxia in tumors: Pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv. Exp. Med. Biol. 2014, 812, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Saeki, T.; Osaki, A.; Yamane, T.; Kuji, I. Bevacizumab Induces Acute Hypoxia and Cancer Progression in Patients with Refractory Breast Cancer: Multimodal Functional Imaging and Multiplex Cytokine Analysis. Clin. Cancer Res. 2017, 23, 5769–5778. [Google Scholar] [CrossRef] [Green Version]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Chien, A.J.; Franco, S.X.; Stopeck, A.T.; Glencer, A.; Lahiri, S.; Arbushites, M.C.; Scott, J.; Park, J.W.; Hudis, C.; et al. A phase II study of lapatinib and bevacizumab as treatment for HER2-overexpressing metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 13–20. [Google Scholar] [CrossRef] [Green Version]
- COSMIC Database. Secondary COSMIC Database. Available online: https://www.cancer.sanger.ac.uk (accessed on 19 October 2020).
- Oden-Gangloff, A.; Di Fiore, F.; Bibeau, F.; Lamy, A.; Bougeard, G.; Charbonnier, F.; Blanchard, F.; Tougeron, D.; Ychou, M.; Boissière, F.; et al. TP53 mutations predict disease control in metastatic colorectal cancer treated with cetuximab-based chemotherapy. Br. J. Cancer 2009, 100, 1330–1335. [Google Scholar] [CrossRef] [Green Version]
- Sclafani, F.; Gonzalez, D.; Cunningham, D.; Hulkki Wilson, S.; Peckitt, C.; Tabernero, J.; Glimelius, B.; Cervantes, A.; Dewdney, A.; Wotherspoon, A.; et al. TP53 mutational status and cetuximab benefit in rectal cancer: 5-year results of the EXPERT-C trial. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.-L.; Liu, Y.; Lapke, N.; Hsu, H.-C.; Chen, H.-C.; Chen, S.-J.; Tan, K.T. TP53 GOF mutations predict good response and prolonged survival to bevacizumab treatment in metastatic colorectal cancer. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Hsu, H.C.; You, J.F.; Chen, S.J.; Chen, H.C.; Yeh, C.Y.; Tsai, W.S.; Hung, H.Y.; Yang, T.S.; Lapke, N.; Tan, K.T. TP53 DNA Binding Domain Mutations Predict Progression-Free Survival of Bevacizumab Therapy in Metastatic Colorectal Cancer. Cancers 2019, 11, 1079. [Google Scholar] [CrossRef] [Green Version]
- McMurray, H.R.; Sampson, E.R.; Compitello, G.; Kinsey, C.; Newman, L.; Smith, B.; Chen, S.R.; Klebanov, L.; Salzman, P.; Yakovlev, A.; et al. Synergistic response to oncogenic mutations defines gene class critical to cancer phenotype. Nature 2008, 453, 1112–1116. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Elledge, S.J. Cancer: Deconstructing oncogenesis. Nature 2008, 453, 995–996. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Baba, Y.; Sagiya, Y.; Gotou, M.; Nakamura, K.; Sawada, H.; Yamanaka, K.; Sakakibara, Y.; Mori, I.; Hikichi, Y.; et al. Biologic Response of Colorectal Cancer Xenograft Tumors to Sequential Treatment with Panitumumab and Bevacizumab. Neoplasia 2018, 20, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Mésange, P.; Bouygues, A.; Ferrand, N.; Sabbah, M.; Escargueil, A.E.; Savina, A.; Chibaudel, B.; Tournigand, C.; André, T.; de Gramont, A.; et al. Combinations of Bevacizumab and Erlotinib Show Activity in Colorectal Cancer Independent of RAS Status. Clin. Cancer Res. 2018, 24, 2548–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournigand, C.; Chibaudel, B.; Samson, B.; Scheithauer, W.; Vernerey, D.; Mésange, P.; Lledo, G.; Viret, F.; Ramée, J.F.; Tubiana-Mathieu, N.; et al. Bevacizumab with or without erlotinib as maintenance therapy in patients with metastatic colorectal cancer (GERCOR DREAM.; OPTIMOX3): A randomised, open-label, phase 3 trial. Lancet. Oncol. 2015, 16, 1493–1505. [Google Scholar] [CrossRef]
- Stefanini, M.O.; Wu, F.T.; Mac Gabhann, F.; Popel, A.S. Increase of plasma VEGF after intravenous administration of bevacizumab is predicted by a pharmacokinetic model. Cancer Res. 2010, 70, 9886–9894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibovitz, A.; Stinson, J.C.; McCombs, W.B.; McCoy, C.E.; Mazur, K.C.; Mabry, N.D. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976, 36, 4562–4569. [Google Scholar]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Wischhusen, J.; Naumann, U.; Ohgaki, H.; Rastinejad, F.; Weller, M. CP-31398, a novel p53-stabilizing agent, induces p53-dependent and p53-independent glioma cell death. Oncogene 2003, 22, 8233–8245. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.; Kim, S.; Wilson, C.; Lehár, J.; Kryukov, G.; Garraway, L.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Grady, J.E.; Lummis, W.L.; Smith, C.G. An improved tissue culture assay. III. Alternate methods for measuring cell growth. Cancer Res 1960, 20, 1114–1117. [Google Scholar]
- Wanka, C.; Brucker, D.P.; Bähr, O.; Ronellenfitsch, M.; Weller, M.; Steinbach, J.P.; Rieger, J. Synthesis of cytochrome C oxidase 2: A p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene 2012, 31, 3764–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE Guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]
- Charan, J.; Kantharia, N.D. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 2013, 4, 303–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sample Size Calculations (IACUC), from Boson University, Reviewed January 2014. Available online: https://www.bu.edu/researchsupport/compliance/animal-care/working-with-animals/research/sample-size-calculations-iacuc/ (accessed on 19 October 2020).
- Mignion, L.; Acciardo, S.; Gourgue FJoudiou, N.; Caignet, X.; Goebbels, R.; Corbet, C.; Feron, O.; Bouzin, C.; Cani, P.; Machiels, J.; et al. Metabolic Imaging Using Hyperpolarized Pyruvate-Lactate Exchange Assesses Response or Resistance to the EGFR Inhibitor Cetuximab in Patient-Derived HNSCC Xenografts. Clin. Cancer Res. 2020, 26, 1932–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, T.G.; Gaustad, J.V.; Rofstad, E.K. Bevacizumab treatment of meningeal melanoma metastases. J. Transl. Med. 2020, 18, 13. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KRAS | TP53 | PTEN | EGFR | RB | |
|---|---|---|---|---|---|
| LIM1215 | wildtype | wildtype | wildtype | wildtype | wildtype |
| SW948 | mutant | mutant | wildtype | wildtype | wildtype |
| LNT-229 | wildtype | wildtype | wildtype | wildtype | wildtype |
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| 18S | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
| SDHA | TGGGAACAAGAGGGCATCTG | CCACCACTGCATCAAATTCATG |
| EGFR | GCGTTCGGCACGGTGTATAA | GGCTTTCGGAGATGTTGCTTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urban, H.; Maurer, G.D.; Luger, A.-L.; Lorenz, N.I.; Sauer, B.; Stroh, C.; Trojan, J.; Mittelbronn, M.; Steinbach, J.P.; Harter, P.N.; et al. Cetuximab-Mediated Protection from Hypoxia- Induced Cell Death: Implications for Therapy Sequence in Colorectal Cancer. Cancers 2020, 12, 3050. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103050
Urban H, Maurer GD, Luger A-L, Lorenz NI, Sauer B, Stroh C, Trojan J, Mittelbronn M, Steinbach JP, Harter PN, et al. Cetuximab-Mediated Protection from Hypoxia- Induced Cell Death: Implications for Therapy Sequence in Colorectal Cancer. Cancers. 2020; 12(10):3050. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103050
Chicago/Turabian StyleUrban, Hans, Gabriele D. Maurer, Anna-Luisa Luger, Nadja I. Lorenz, Benedikt Sauer, Christopher Stroh, Jörg Trojan, Michel Mittelbronn, Joachim P. Steinbach, Patrick N. Harter, and et al. 2020. "Cetuximab-Mediated Protection from Hypoxia- Induced Cell Death: Implications for Therapy Sequence in Colorectal Cancer" Cancers 12, no. 10: 3050. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103050