Contribution of miRNAs, tRNAs and tRFs to Aberrant Signaling and Translation Deregulation in Lung Cancer

 , ,
, ,  , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
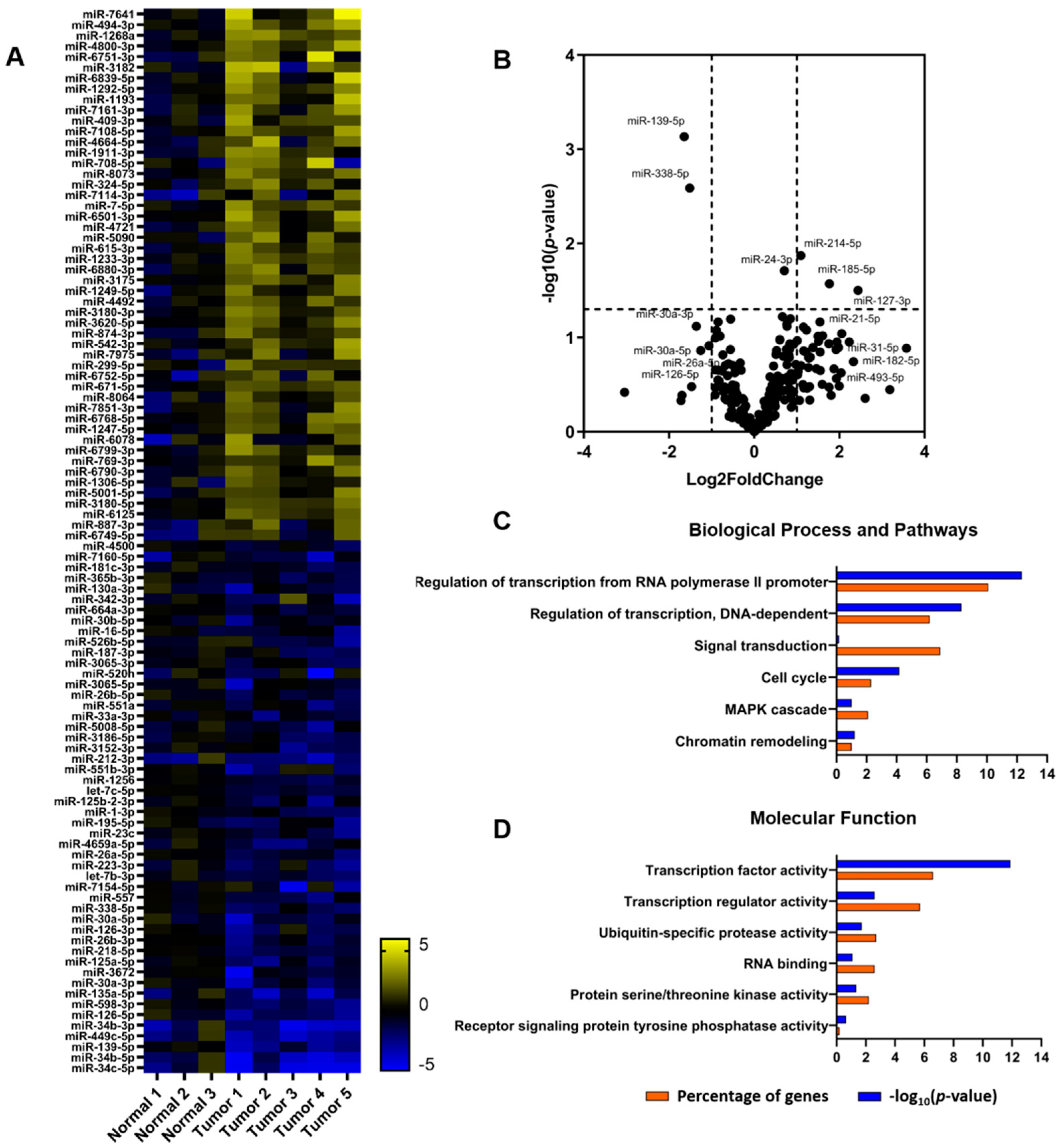
2.1. Patterns of miRNAs and tRFs Implicated in Transcription and Translation
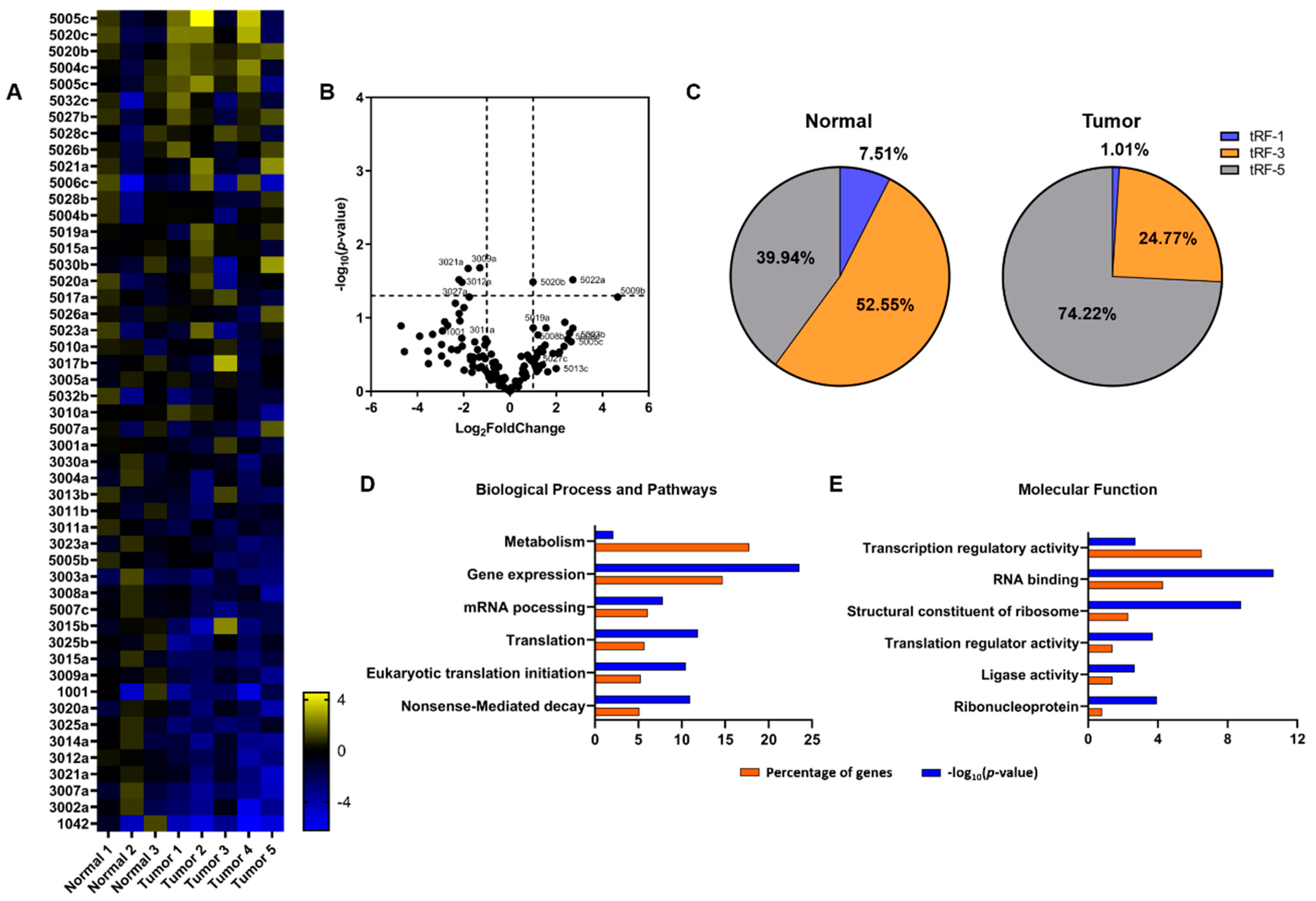
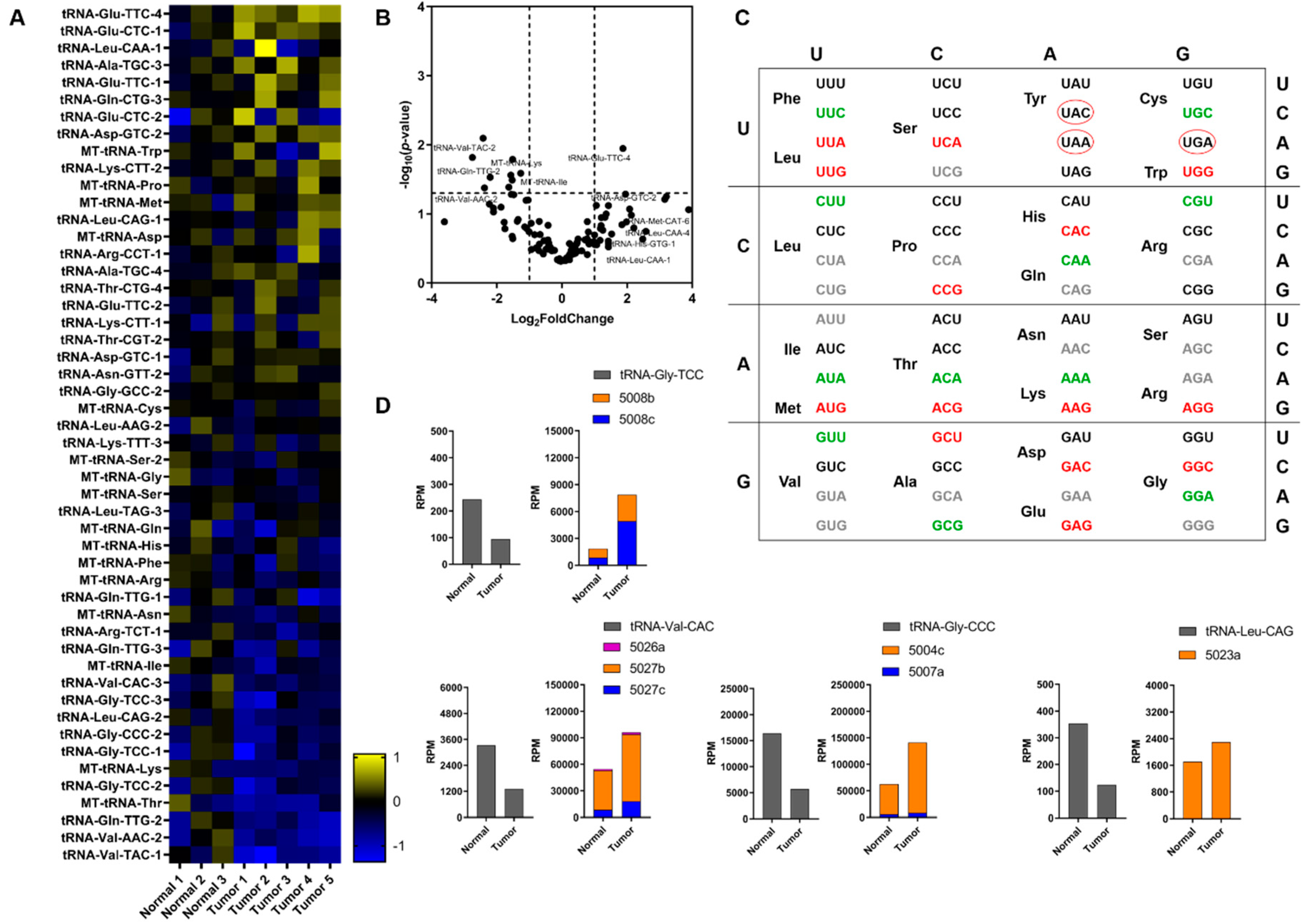
2.2. tRNA Profiling and Expression Levels of Related Genes
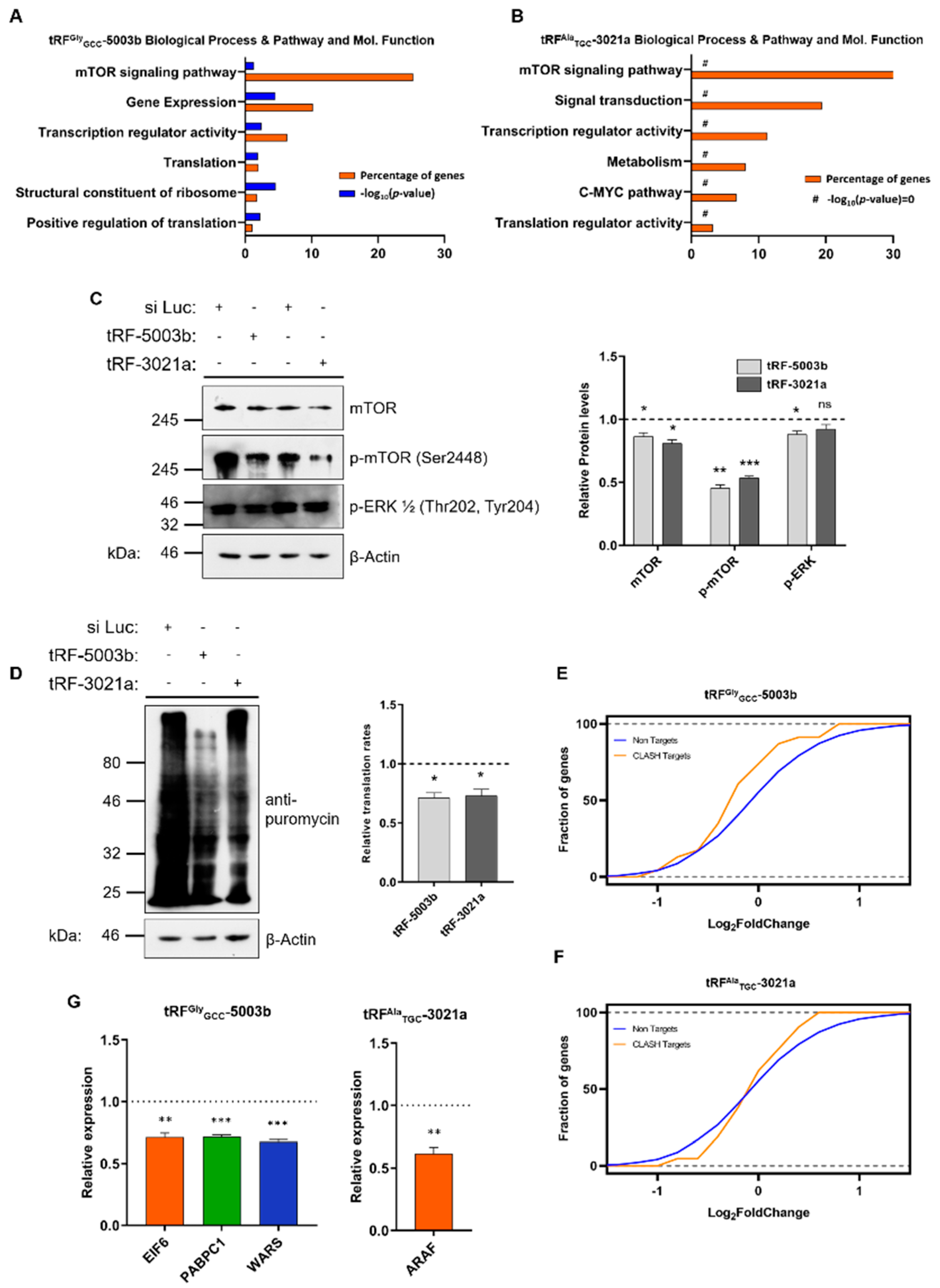
2.3. Specific tRFs Can Target Translation-Related Factors
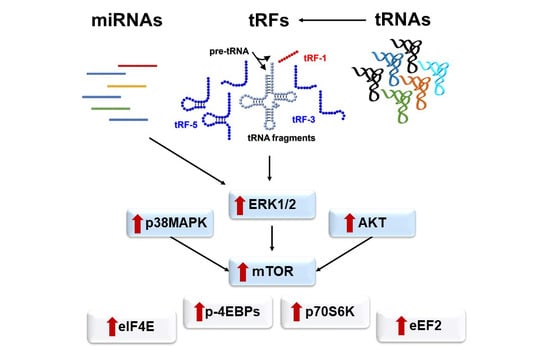
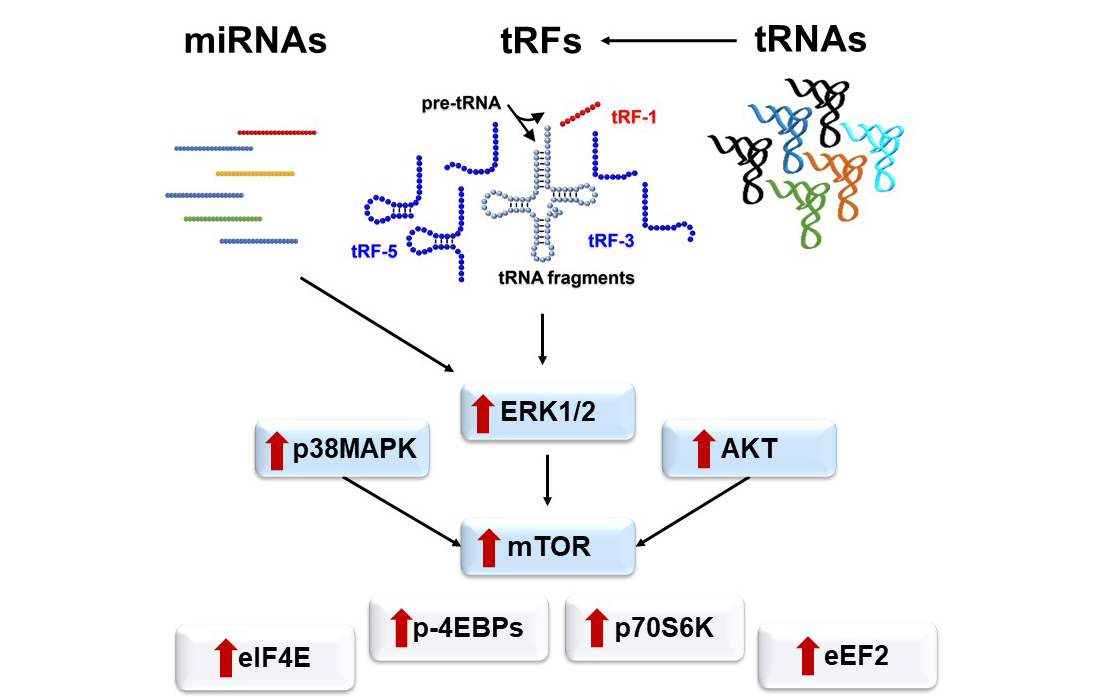
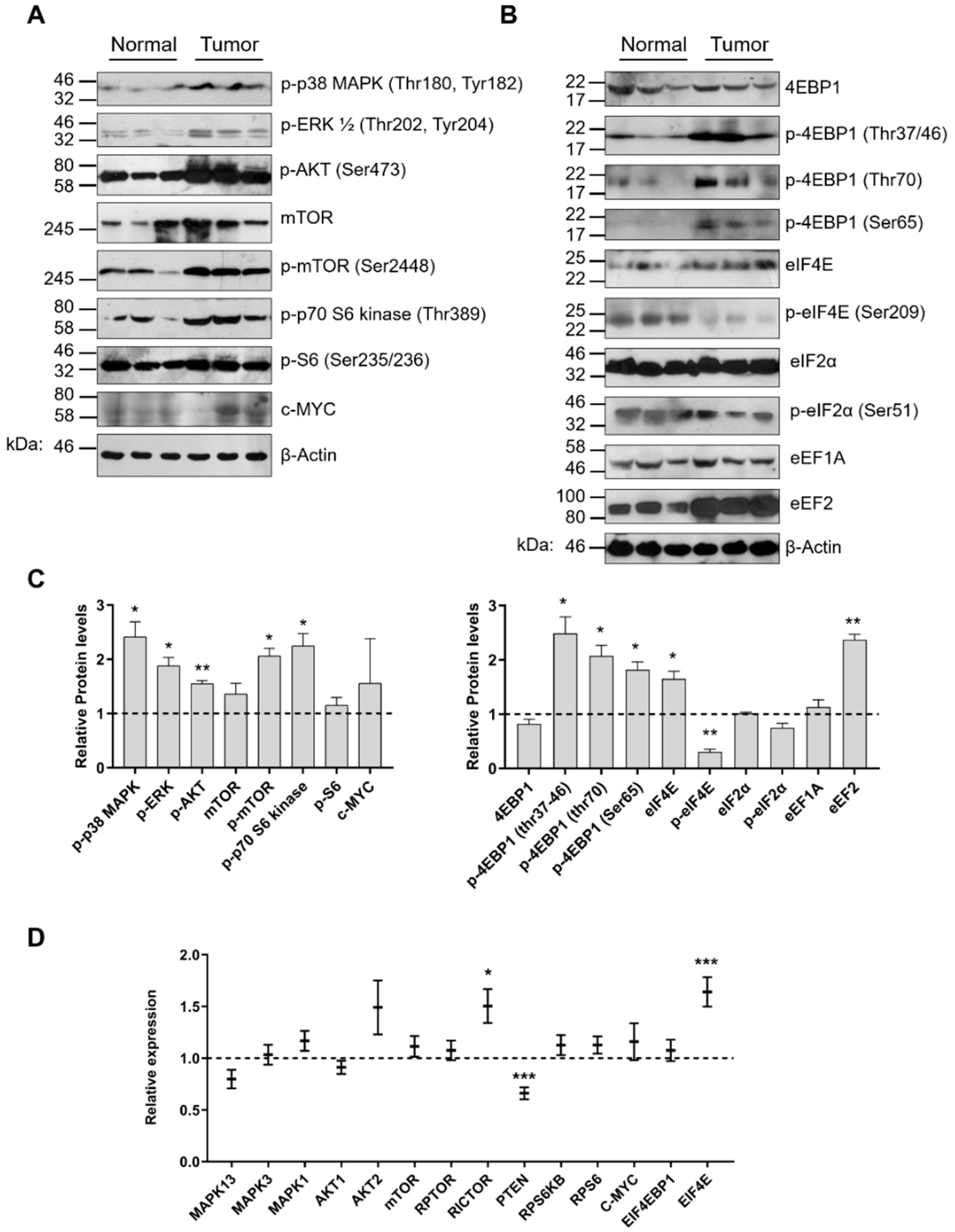
2.4. Aberrant Signaling Targets Important Translation Factors
2.5. Lung Cancer Specimens Exhibit Elevated Global Translation Rates
3. Discussion
4. Materials and Methods
4.1. Tumor Specimens and Cell Lines
4.2. Real-Time PCR and Western Blot Analyses
4.3. Next Generation Sequencing and Bioinformatics Analysis
4.4. Non-Radioactive Measurement of Translation Rates
4.5. Sedimentation Profile of Ribosomal Particles and Translation Efficiency Measurements
4.6. Supplementary Methods
Ribosomal Complex and Peptide-Bond Formation Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The role of non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P. The emerging complexity of the tRNA world: Mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 2017, 19, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Wilson, B.; Kumar, P.; Dutta, A. Noncanonical roles of tRNAs: tRNA fragments and beyond. Annu. Rev. Genet. 2020, 54. [Google Scholar] [CrossRef]
- Xie, Y.; Yao, L.; Yu, X.; Ruan, Y.; Li, Z.; Guo, J. Action mechanisms and research methods of tRNA-derived small RNAs. Signal Transduct. Target. Ther. 2020, 5, 109. [Google Scholar] [CrossRef]
- Kuscu, C.; Kumar, P.; Kiran, M.; Su, Z.; Malik, A.; Dutta, A. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA 2018, 24, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Hasler, D.; Lehmann, G.; Murakawa, Y.; Klironomos, F.; Jakob, L.; Grässer, F.A.; Rajewsky, N.; Landthaler, M.; Meister, G. The lupus autoantigen la prevents mis-channeling of tRNA fragments into the human microRNA pathway. Mol. Cell 2016, 63, 110–124. [Google Scholar] [CrossRef]
- Kumar, P.; Kuscu, C.; Dutta, A. Biogenesis and function of transfer RNA-related fragments (tRFs). Trends Biochem. Sci. 2016, 41, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Telonis, A.G.; Loher, P.; Honda, S.; Jing, Y.; Palazzo, J.; Kirino, Y.; Rigoutsos, I. Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget 2015, 6, 24797–24822. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Kim, T.-H. Fine-Tuning of gene expression by tRNA-derived fragments during abiotic stress signal transduction. Int. J. Mol. Sci. 2018, 19, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.B.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.; Ivanov, P. tRNA fragments in human health and disease. FEBS Lett. 2014, 588, 4297–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Anaya, J.; Mudunuri, S.B.; Dutta, A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014, 12, 78. [Google Scholar] [CrossRef]
- Keam, S.; Hutvagner, G. tRNA-Derived Fragments (tRFs): Emerging new roles for an ancient RNA in the regulation of gene expression. Life 2015, 5, 1638–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, T.; Suresh, P.S.; Tsutsumi, R. tRFs: miRNAs in disguise. Gene 2016, 579, 133–138. [Google Scholar] [CrossRef]
- Balatti, V.; Nigita, G.; Veneziano, D.; Drusco, A.; Stein, G.S.; Messier, T.L.; Farina, N.H.; Lian, J.B.; Tomasello, L.; Liu, C.; et al. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 8071–8076. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Fuchs, G.; Wang, S.; Wei, W.; Zhang, Y.; Park, H.; Roy-Chaudhuri, B.; Li, P.; Xu, J.; Chu, K.; et al. A transfer-RNA-derived small RNA regulates ribosome biogenesis. Nature 2017, 552, 57–62. [Google Scholar] [CrossRef]
- Krishna, S.; Yim, D.G.; Lakshmanan, V.; Tirumalai, V.; Koh, J.L.; Park, J.E.; Cheong, J.K.; Low, J.L.; Lim, M.J.; Sze, S.K.; et al. Dynamic expression of tRNA-derived small RNAs define cellular states. EMBO Rep. 2019, 20, e47789. [Google Scholar] [CrossRef]
- Sobala, A.; Hutvagner, G. Small RNAs derived from the 5’ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013, 10, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Gebetsberger, J.; Wyss, L.; Mleczko, A.M.; Reuther, J.; Polacek, N. A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol. 2017, 14, 1364–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telonis, A.G.; Loher, P.; Magee, R.; Pliatsika, V.; Londin, E.; Kirino, Y.; Rigoutsos, I. tRNA fragments show intertwining with mRNAs of specific repeat content and have links to disparities. Cancer Res. 2019, 79, 3034–3049. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Sun, Q.; Liu, X.; Wang, P.; Wu, R.; Ma, Z. tRF-Leu-CAG promotes cell proliferation and cell cycle in non-small cell lung cancer. Chem. Biol. Drug Des. 2017, 90, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Fidalgo, A.; Varanda, A.S.; Oliveira, C.; Santos, M.A.S. tRNA Deregulation and its consequences in cancer. Trends Mol. Med. 2019, 25, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E. Controlling translation via modulation of tRNA levels. Wiley Interdiscip. Rev. RNA 2015, 6, 453–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavon-Eternod, M.; Gomes, S.; Rosner, M.R.; Pan, T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 2013, 19, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, S.; Ignatova, Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015, 16, 98–112. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Khoutorsky, A.; Mathews, M.B.; Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 791–807. [Google Scholar] [CrossRef]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sørensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.; Pereira, P.M.; Varanda, A.S.; Carvalho, J.; Azevedo, M.; Mateus, D.D.; Mendes, N.; Oliveira, P.; Trindade, F.; Pinto, M.T.; et al. Codon misreading tRNAs promote tumor growth in mice. RNA Biol. 2018, 15, 773–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or tumor suppressor? The duplicity of MicroRNAs in cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Mahlab, S.; Tuller, T.; Linial, M. Conservation of the relative tRNA composition in healthy and cancerous tissues. RNA 2012, 18, 640–652. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.G.; Reina, O.; Stephan-Otto Attolini, C.; Ribas de Pouplana, L. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl. Acad. Sci. USA 2019, 116, 8451–8456. [Google Scholar] [CrossRef] [Green Version]
- Telonis, A.G.; Rigoutsos, I. Race Disparities in the contribution of miRNA isoforms and tRNA-derived fragments to triple-negative breast cancer. Cancer Res. 2018, 78, 1140–1154. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Ge, J.; Li, T.; Shen, Y.; Guo, J. tRNA-derived fragments and tRNA halves: The new players in cancers. Cancer Lett. 2019, 452, 31–37. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, Y.; Gong, J.; Ruan, H.; Liu, C.-J.; Xiang, Y.; Cai, C.; Guo, A.-Y.; Ling, J.; Diao, L.; et al. Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers. Commun. Biol. 2018, 1, 234. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Inamura, K.; Ishikawa, Y. MicroRNA in lung cancer: Novel biomarkers and potential tools for treatment. J. Clin. Med. 2016, 5, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Kong, R.; He, Z.; Lin, L.-Y.; Qin, S.-S.; Chen, C.-Y.; Xie, Z.-Q.; Yu, F.; Sun, G.-Q.; Li, C.-G.; et al. High expression of miR-493-5p positively correlates with clinical prognosis of non small cell lung cancer by targeting oncogene ITGB1. Oncotarget 2017, 8, 47389–47399. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Ma, J.; Wang, Y.; Zan, J.; Wang, Z.; Zhu, Y.; Zhu, Y.; Ling, L.; Cao, L.; Liu, X.; et al. miR-21-5p induces cell proliferation by targeting TGFBI in non-small cell lung cancer cells. Exp. Ther. Med. 2018, 16, 4655–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, M.; Berti, G.; Russo, F.; Fazio, S.; Evangelista, M.; D’Aurizio, R.; Pellegrini, M.; Rainaldi, G. Discovering the miR-26a-5p targetome in prostate cancer cells. J. Cancer 2017, 8, 2729–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, S.M.; Achorn, C.; Kedersha, N.L.; Anderson, P.J.; Ivanov, P. YB-1 regulates tiRNA-induced stress granule formation but not translational repression. Nucleic Acids Res. 2016, 44, 6949–6960. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; You, S.; Hwang, D. Aminoacyl-tRNA synthetases and tumorigenesis: More than housekeeping. Nat. Rev. Cancer 2011, 11, 708–718. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef]
- Greenberg, A.K.; Basu, S.; Hu, J.; Yie, T.; Tchou-Wong, K.M.; Rom, W.N.; Lee, T.C. Selective p38 activation in human non–small cell lung cancer. Am. J. Respir. Cell Mol. Biol. 2002, 26, 558–564. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [PubMed]
- McKendrick, L.; Morley, S.J.; Pain, V.M.; Jagus, R.; Joshi, B. Phosphorylation of eukaryotic initiation factor 4E (eIF4E) at Ser209 is not required for protein synthesis in vitro and in vivo. Eur. J. Biochem. 2001, 268, 5375–5385. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.D.; Wek, R.C. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muaddi, H.; Majumder, M.; Peidis, P.; Papadakis, A.I.; Holcik, M.; Scheuner, D.; Kaufman, R.J.; Hatzoglou, M.; Koromilas, A.E. Phosphorylation of eIF2α at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol. Biol. Cell 2010, 21, 3220–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White-Gilbertson, S.; Kurtz, D.T.; Voelkel-Johnson, C. The role of protein synthesis in cell cycling and cancer. Mol. Oncol. 2009, 3, 402–408. [Google Scholar] [CrossRef]
- Võsa, U.; Vooder, T.; Kolde, R.; Vilo, J.; Metspalu, A.; Annilo, T. Meta-analysis of microRNA expression in lung cancer. Int. J. Cancer 2013, 132, 2884–2893. [Google Scholar] [CrossRef]
- Gantenbein, N.; Bernhart, E.; Anders, I.; Golob-Schwarzl, N.; Krassnig, S.; Wodlej, C.; Brcic, L.; Lindenmann, J.; Fink-Neuboeck, N.; Gollowitsch, F.; et al. Influence of eukaryotic translation initiation factor 6 on non–small cell lung cancer development and progression. Eur. J. Cancer 2018, 101, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Comtesse, N.; Keller, A.; Diesinger, I.; Bauer, C.; Kayser, K.; Huwer, H.; Lenhof, H.-P.; Meese, E. Frequent overexpression of the genes FXR1, CLAPM1 and EIF4G located on amplicon 3q26-27 in squamous cell carcinoma of the lung. Int. J. Cancer 2007, 120, 2538–2544. [Google Scholar] [CrossRef]
- Imielinski, M.; Greulich, H.; Kaplan, B.; Araujo, L.; Amann, J.; Horn, L.; Schiller, J.; Villalona-Calero, M.A.; Meyerson, M.; Carbone, D.P. Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J. Clin. Investig. 2014, 124, 1582–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Sheng, C.; Yin, Y.; Wen, S.; Yang, G.; Cheng, Z.; Zhu, Q. PABPC1 interacts with AGO2 and is responsible for the microRNA mediated gene silencing in high grade hepatocellular carcinoma. Cancer Lett. 2015, 367, 49–57. [Google Scholar] [CrossRef]
- Kuang, M.; Zheng, D.; Tao, X.; Peng, Y.; Pan, Y.; Zheng, S.; Zhang, Y.; Li, H.; Yuan, C.; Zhang, Y.; et al. tRNA-based prognostic score in predicting survival outcomes of lung adenocarcinomas. Int. J. Cancer 2019, 145, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kim, Y.; Jin, K.S.; Kim, H.S.; Kim, J.H.; Wang, D.; Park, M.; Jo, C.H.; Kwon, N.H.; Kim, D.; et al. Structure of the ArgRS–GlnRS–AIMP1 complex and its implications for mammalian translation. Proc. Natl. Acad. Sci. USA 2014, 111, 15084–15089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, Y.-G.; Kim, E.-K.; Kim, T.; Park, H.; Park, H.-S.; Choi, E.-J.; Kim, S. Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001, 276, 6030–6036. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-S.; Kim, M.-S.; Huh, S.-H.; Park, J.; Chung, J.; Kang, S.S.; Choi, E.-J. Akt (Protein Kinase B) negatively regulates SEK1 by means of protein phosphorylation. J. Biol. Chem. 2002, 277, 2573–2578. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-S.; Han, J.M.; Kang, T.; Park, Y.I.; Kim, H.M.; Kim, S. Antitumor activity of the novel human cytokine AIMP1 in an in vivo tumor model. Mol. Cells 2006, 21, 213–217. [Google Scholar]
- Gerber, A.; Ito, K.; Chu, C.-S.; Roeder, R.G. Gene-specific control of tRNA expression by RNA polymerase II. Mol. Cell 2020, 78, 765–778.e7. [Google Scholar] [CrossRef]
- Vicent, S.; López-Picazo, J.M.; Toledo, G.; Lozano, M.D.; Torre, W.; Garcia-Corchón, C.; Quero, C.; Soria, J.-C.; Martín-Algarra, S.; Manzano, R.G.; et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br. J. Cancer 2004, 90, 1047–1052. [Google Scholar] [CrossRef]
- Shahbazian, D.; Roux, P.P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.B.; Blenis, J.; Pende, M.; Sonenberg, N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef]
- Balsara, B.R. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis 2004, 25, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Grafanaki, K.; Anastasakis, D.; Kyriakopoulos, G.; Skeparnias, I.; Georgiou, S.; Stathopoulos, C. Translation regulation in skin cancer from a tRNA point of view. Epigenomics 2019, 11, 215–245. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zheng, M.; Lu, D.; Wang, J.; Jiang, W.; Sha, O. Anti-tumor activities and apoptotic mechanism of ribosome-inactivating proteins. Chin. J. Cancer 2015, 34, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.C.; Ruggero, D. Targeting eukaryotic translation initiation factor 4E (eIF4E) in cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Ramalingam, S.S.; Kauh, J.; Xu, Z.; Khuri, F.R.; Sun, S.-Y. Phosphorylated eukaryotic translation initiation factor 4 (eIF4E) is elevated in human cancer tissues. Cancer Biol. 2009, 8, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Mountain, C.F. Revisions in the international system for staging lung cancer. Chest 1997, 111, 1710–1717. [Google Scholar] [CrossRef] [Green Version]
- Peltier, H.J.; Latham, G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008, 14, 844–852. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; da Silva, A.M.; Calin, G.; Pantel, K. Data normalization strategies for MicroRNA quantification. Clin. Chem. 2015, 61, 1333–1342. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.-S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinos, G.; Wilson, D.N.; Teraoka, Y.; Szaflarski, W.; Fucini, P.; Kalpaxis, D.; Nierhaus, K.H. Dissecting the ribosomal inhibition mechanisms of edeine and pactamycin. Mol. Cell 2004, 13, 113–124. [Google Scholar] [CrossRef]
- Budkevich, T.V.; El’skaya, A.V.; Nierhaus, K.H. Features of 80S mammalian ribosome and its subunits. Nucleic Acids Res. 2008, 36, 4736–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skeparnias, I.; Anastasakis, D.; Grafanaki, K.; Kyriakopoulos, G.; Alexopoulos, P.; Dougenis, D.; Scorilas, A.; Kontos, C.K.; Stathopoulos, C. Contribution of miRNAs, tRNAs and tRFs to Aberrant Signaling and Translation Deregulation in Lung Cancer. Cancers 2020, 12, 3056. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103056
Skeparnias I, Anastasakis D, Grafanaki K, Kyriakopoulos G, Alexopoulos P, Dougenis D, Scorilas A, Kontos CK, Stathopoulos C. Contribution of miRNAs, tRNAs and tRFs to Aberrant Signaling and Translation Deregulation in Lung Cancer. Cancers. 2020; 12(10):3056. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103056
Chicago/Turabian StyleSkeparnias, Ilias, Dimitrios Anastasakis, Katerina Grafanaki, George Kyriakopoulos, Panagiotis Alexopoulos, Dimitrios Dougenis, Andreas Scorilas, Christos K. Kontos, and Constantinos Stathopoulos. 2020. "Contribution of miRNAs, tRNAs and tRFs to Aberrant Signaling and Translation Deregulation in Lung Cancer" Cancers 12, no. 10: 3056. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103056