The Role of PTEN Loss in Immune Escape, Melanoma Prognosis and Therapy Response

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
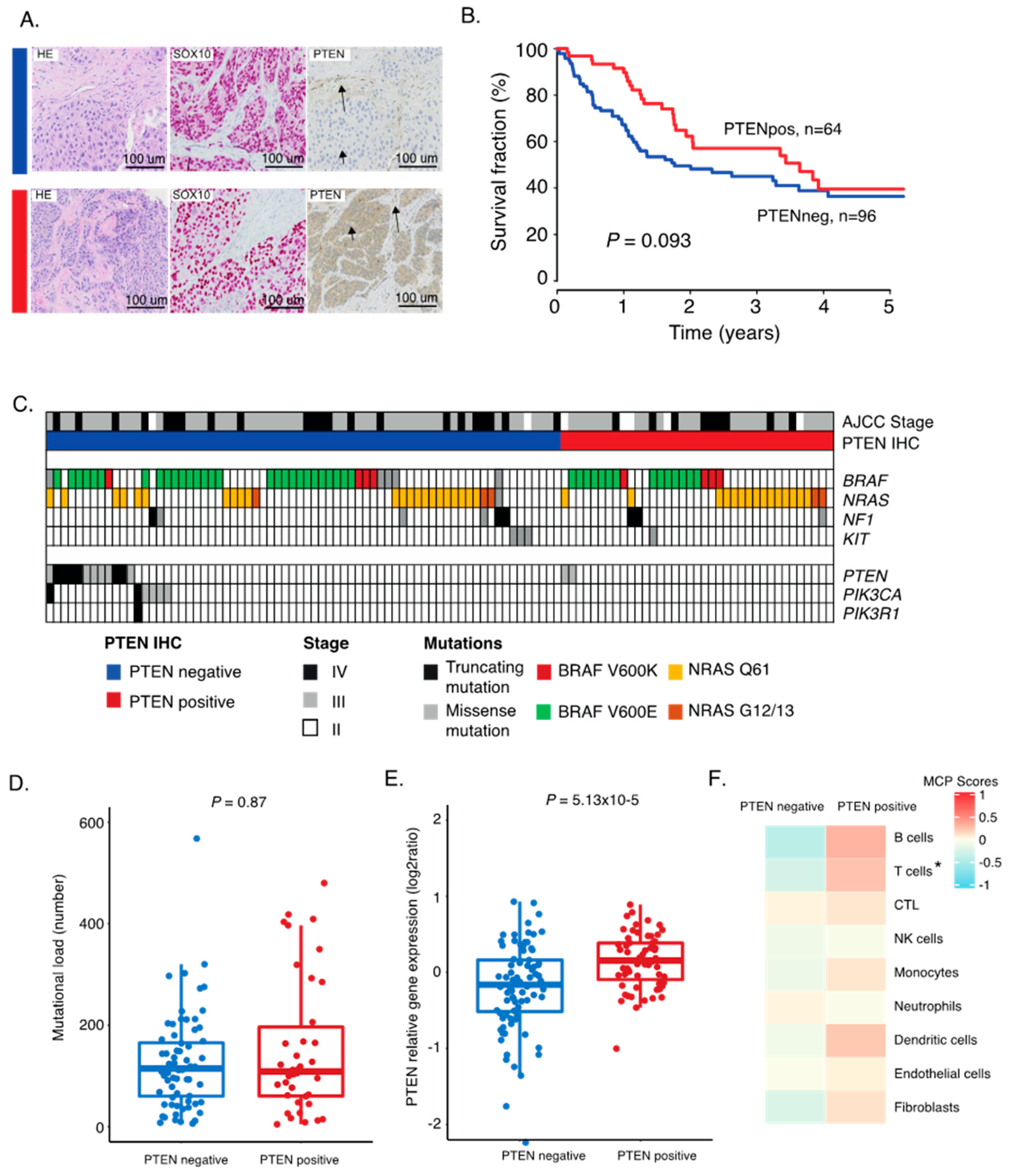
2.1. PTEN Protein Expression in Metastatic Melanoma
2.2. Correlation between PTEN Alteration and Tumor Immune Microenvironment
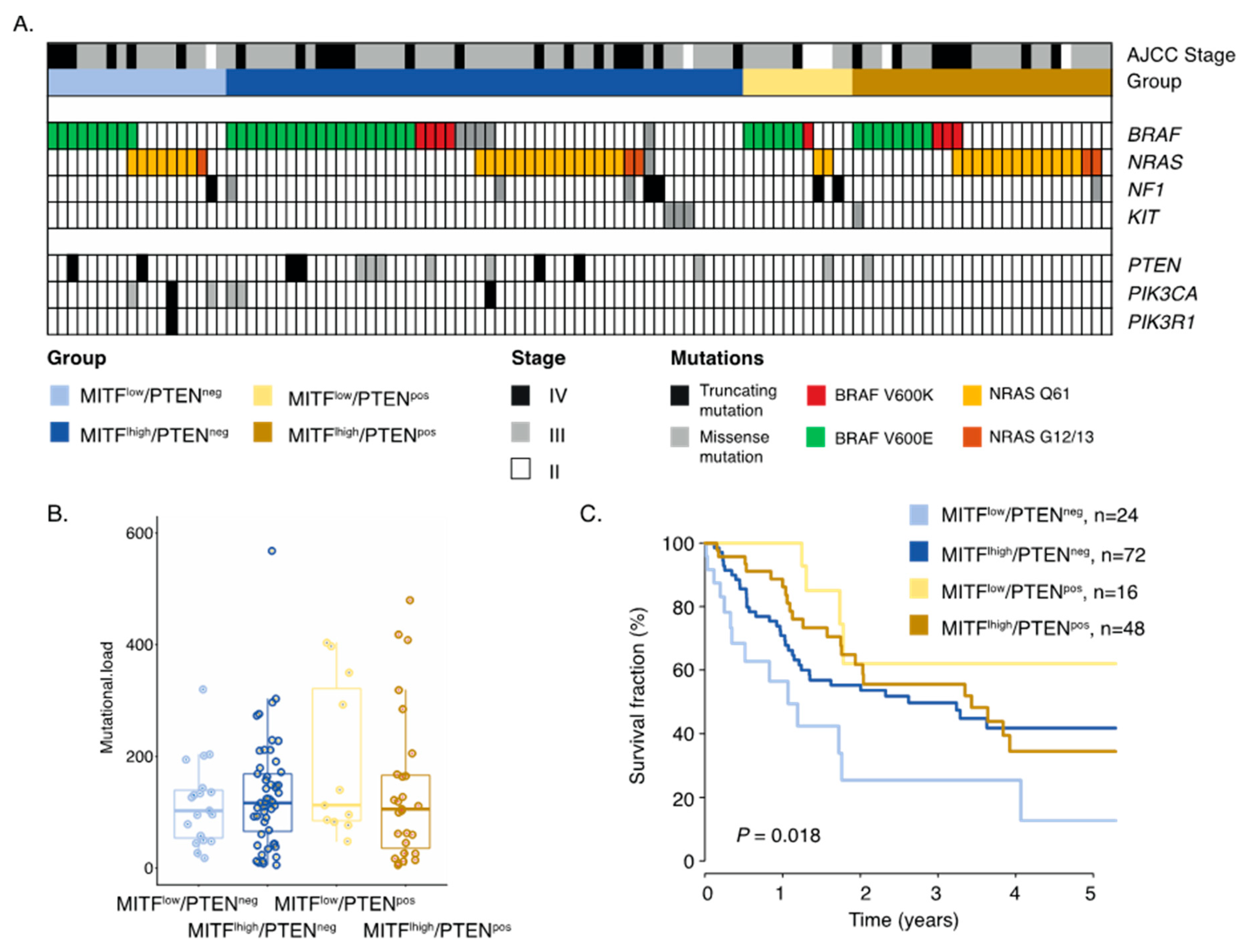
2.3. Inactivation of PTEN and Melanoma Cell Differentiation State Predicts Melanoma Survival
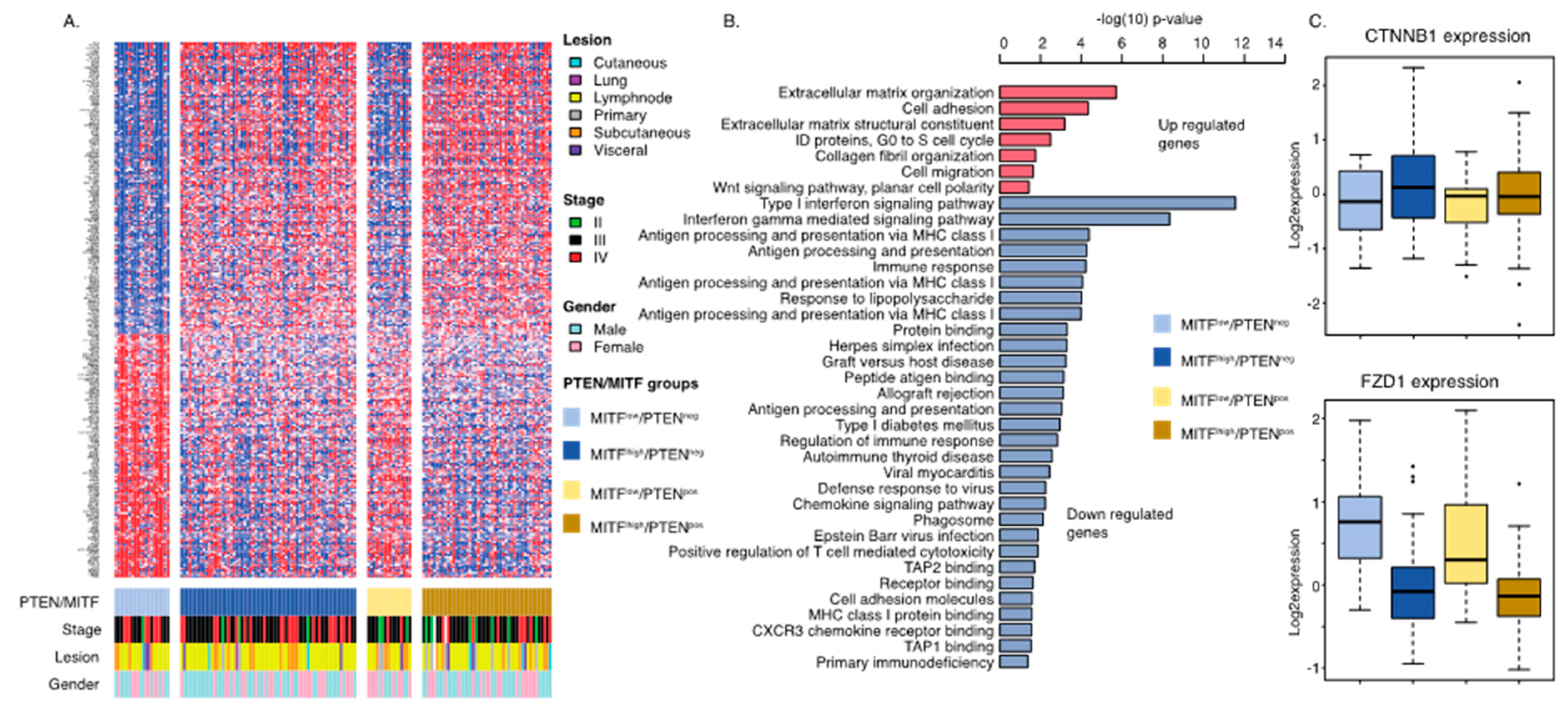
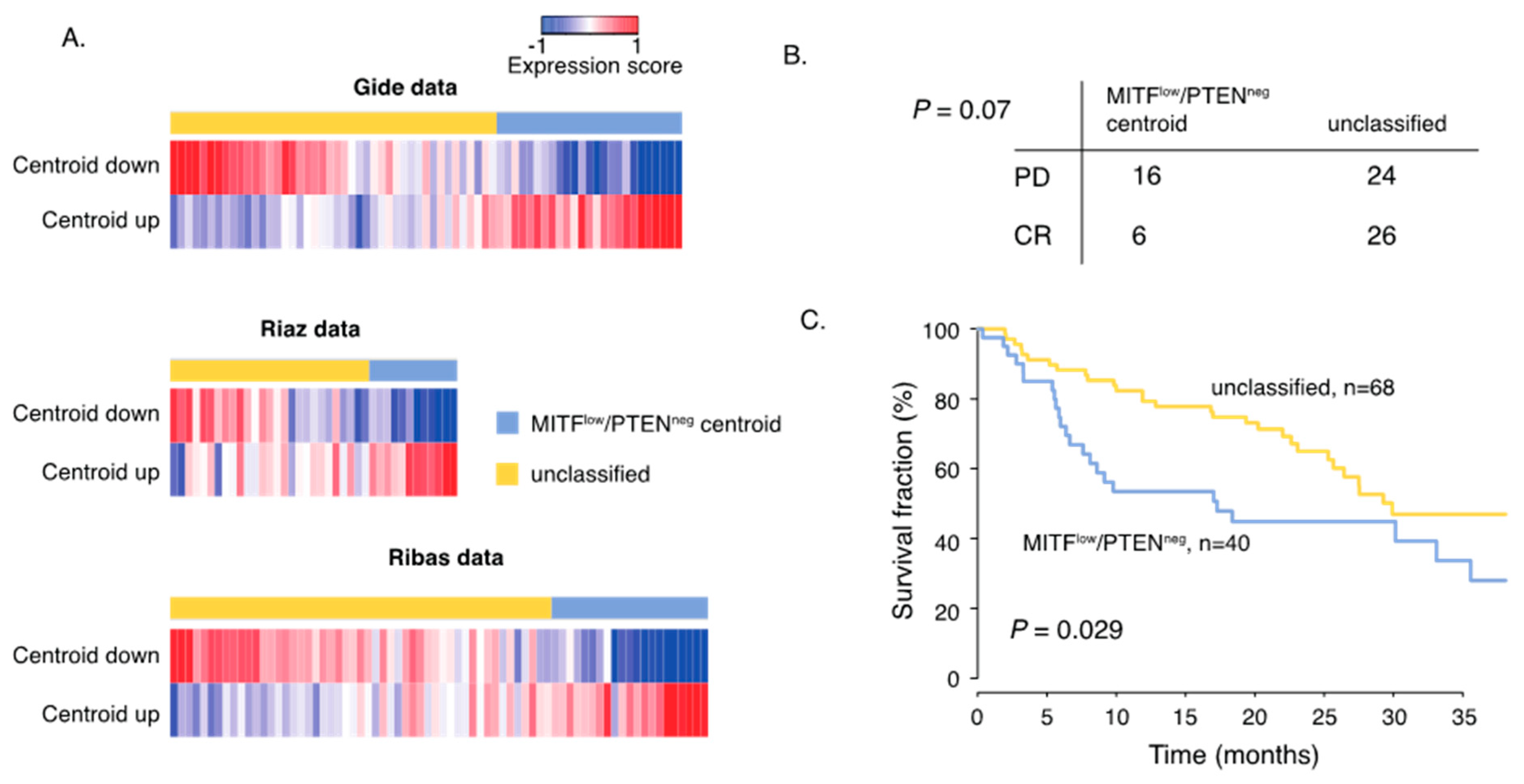
2.4. Gene Signature of Dedifferentiated PTEN Inactivated Melanoma Predicts Response to MAPK Inhibition and Immune Checkpoint Therapy
3. Discussion
4. Materials and Methods
4.1. Patient Material
4.2. Immunohistochemistry
4.3. Bioinformatic and Statistical Analyses
- T-cells: CD28, CD3D, CD5, TRAT1
- CD8 T-cells: CD8B, CD8A
- Cytotoxic lymphocytes: EOMES, GNLY, KLRC4-KLRK1
- NK cells: KIR2DL3, KIR2DL4, KIR3DS1, NCR1
- B lineage: CD19, CD79A, CD79B, MS4A1
- Monocytic lineage: ADAP2, CSF1R, RASSF4, TFEC
- Myeloid dendritic cells: CD1A, CD1B, CD1E, CLEC10A
- Neutrophils: CEACAM3, CXCR1, CXCR2, FCGR3B
- Endothelial cells: CDH5, MMRN1, MMRN2, VWF
- Fibroblasts: COL1A1, COL6A2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T-cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.S.; Schlegel, N.C.; Eichhoff1, O.M.; Widmer, D.S.; Praetorius, C.; Einarsson, S.O.; Valgeirsdottir, S.; Bergsteinsdottir, K.; Schepsky, A.; Dummer, R.; et al. Novel MITF targets identified using a two-step DNA microarray strategy. Pigment. Cell Melanoma Res. 2008, 21, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 295–1318. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Fisher, D.E.; Flaherty, K.T. Cell-state dynamics and therapeutic resistance in melanoma from the perspective of MITF and IFNγ pathways. Nat. Rev. Clin. Oncol. 2019, 16, 549–562. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumor immune responses. Nat. Rev. 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signaling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.; Chan, T.A. Immunotherapy and Oncogenic Pathways: The PTEN Connection. Cancer Discov. 2016, 6, 128–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seliger, B. Strategies of tumor immune evasion. Biodrugs 2005, 19, 347–354. [Google Scholar] [CrossRef]
- Nicolini, A.; Ferrari, P.; Rossi, G.; Carpi, A. Tumor growth and immune evasion as targets for a new strategy in advanced cancer. Endocrine-Related Cancer 2018, 25, R577–R604. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Gajewski, T.F. Mechanisms of Tumor Cell-Intrinsic Immune Evasion. Annu. Rev. Cancer Biol. 2017, 2, 213–228. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.F.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Wongchenko, M.J.; Robert, C.; Larkin, J.; Ascierto, P.A.; Dreno, B.; Maio, M.; Garbe, C.; Chapman, P.B.; Sosman, J.A.; et al. Genomic features of exceptional response in vemurafenib ± cobimetinib-treated patients with BRAFV600-mutated metastatic melanoma. Clin. Cancer Res. 2019, 25, 3239–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, A.; Webster, M.R.; Weeraratn, A.T. In the Wnt-er of life: Wnt signalling in melanoma and ageing. Br. J. Cancer 2016, 115, 1273–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucheit, A.B.; Chen, G.; Siroy, A.; Tetzlaff, M.; Broaddus, R.; Milton, D.; Fox, P.; Bassett, R.; Hwu, P.; Gershenwald, J.E.; et al. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin. Cancer Res. 2014, 20, 5527–5536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalanotti, F.; Cheng, D.T.; Shoushtari, A.N.; Panageas, K.S.; Momtaz, P.; Won, H.H.; Harding, J.J.; Merghoub, T.; Rosen, N.; Berger, M.F.; et al. PTEN Loss-of-Function Alterations Are Associated with Intrinsic Resistance to BRAF Inhibitors in Metastatic Melanoma. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B-cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lövgren, K.; Jumppanen, M.; Staaf, J.; Jönsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Torngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Entire Cohort (n = 169) | PTEN+ (n = 69) | PTEN− (n = 100) | p-Value |
|---|---|---|---|---|
| Gender n (%) | 1 | |||
| Male | 98 (58) | 39 (57) | 59 (59) | |
| Female | 69 (41) | 28 (41) | 41 (41) | |
| NA | 2 (1) | 2 (3) | - | |
| Age at diagnosis median (range) | 65 (22–91) | 65 (22–91) | 65 (30–88) | 0.54 |
| Stage | 0.003 | |||
| II | 18 (11) | 13 (19) | 5 (5) | |
| III | 99 (59) | 41 (59) | 58 (58) | |
| IV | 50 (29) | 13 (19) | 37 (37) | |
| NA | 2 (1) | 2 (3) | 0 | |
| Lesion type | 0.004 | |||
| Lymph node | 108 | 36 | 72 | |
| Subcutaneous | 35 | 16 | 19 | |
| Visceral | 10 | 4 | 6 | |
| Primary tumor | 14 | 11 | 3 | |
| NA | 2 | 2 | - | |
| Histological subtype | 0.5 | |||
| Unknown primary n (%) | 26 (15) | 9 (13) | 17 (17) | |
| SSM | 35 (21) | 11 (16) | 24 (24) | |
| NM | 57 (34) | 24 (35) | 33 (33) | |
| Other | 14 (8) | 5 (6) | 9 (9) | |
| NA | 37 (22) | 20 (29) | 17 (17) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrita, R.; Mitra, S.; Sanna, A.; Ekedahl, H.; Lövgren, K.; Olsson, H.; Ingvar, C.; Isaksson, K.; Lauss, M.; Carneiro, A.; et al. The Role of PTEN Loss in Immune Escape, Melanoma Prognosis and Therapy Response. Cancers 2020, 12, 742. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030742
Cabrita R, Mitra S, Sanna A, Ekedahl H, Lövgren K, Olsson H, Ingvar C, Isaksson K, Lauss M, Carneiro A, et al. The Role of PTEN Loss in Immune Escape, Melanoma Prognosis and Therapy Response. Cancers. 2020; 12(3):742. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030742
Chicago/Turabian StyleCabrita, Rita, Shamik Mitra, Adriana Sanna, Henrik Ekedahl, Kristina Lövgren, Håkan Olsson, Christian Ingvar, Karolin Isaksson, Martin Lauss, Ana Carneiro, and et al. 2020. "The Role of PTEN Loss in Immune Escape, Melanoma Prognosis and Therapy Response" Cancers 12, no. 3: 742. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030742