KLF5 Is Crucial for Androgen-AR Signaling to Transactivate Genes and Promote Cell Proliferation in Prostate Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
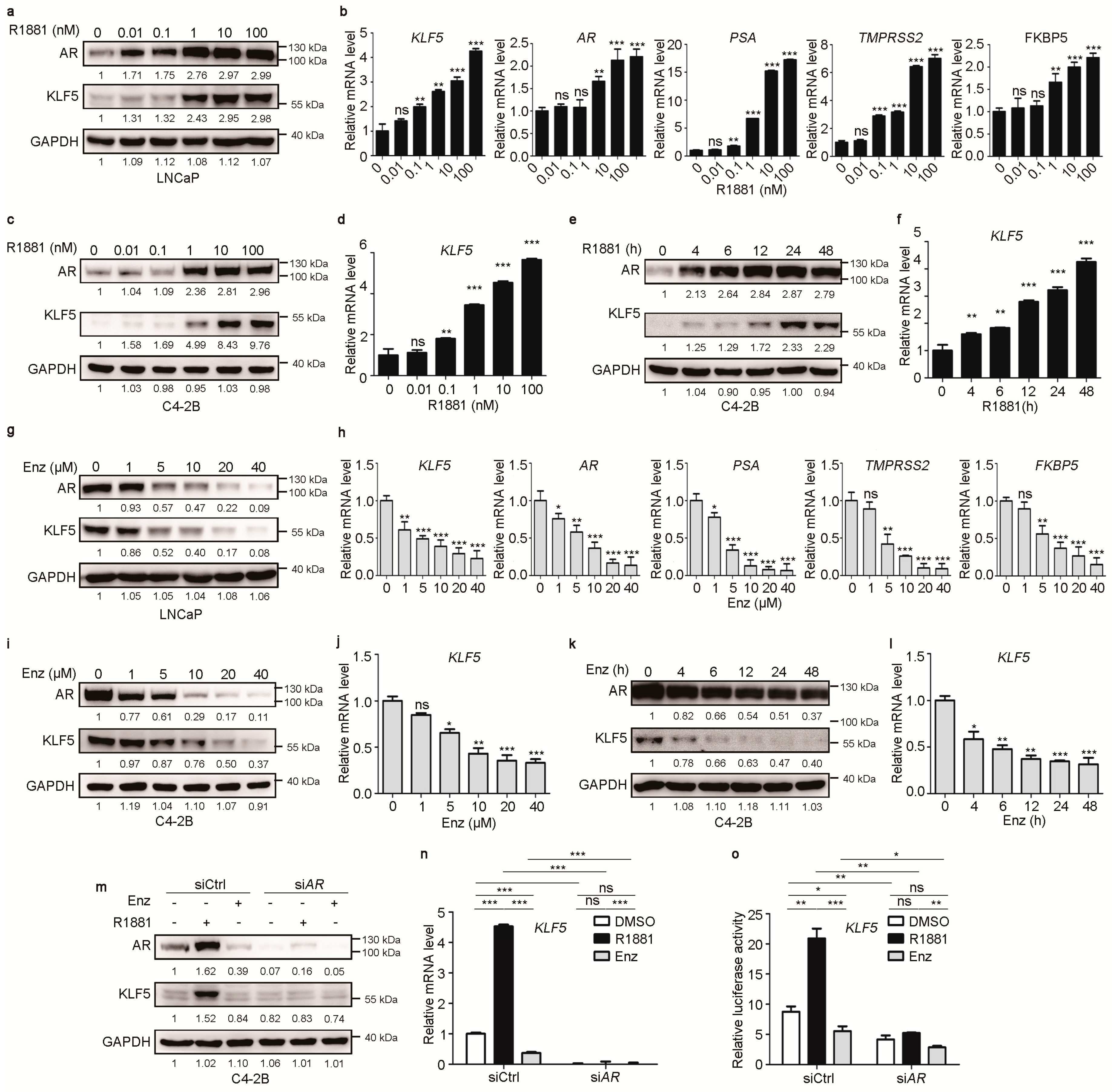
2.1. Androgen/AR Signaling Upregulates KLF5 Transcription in PCa cells
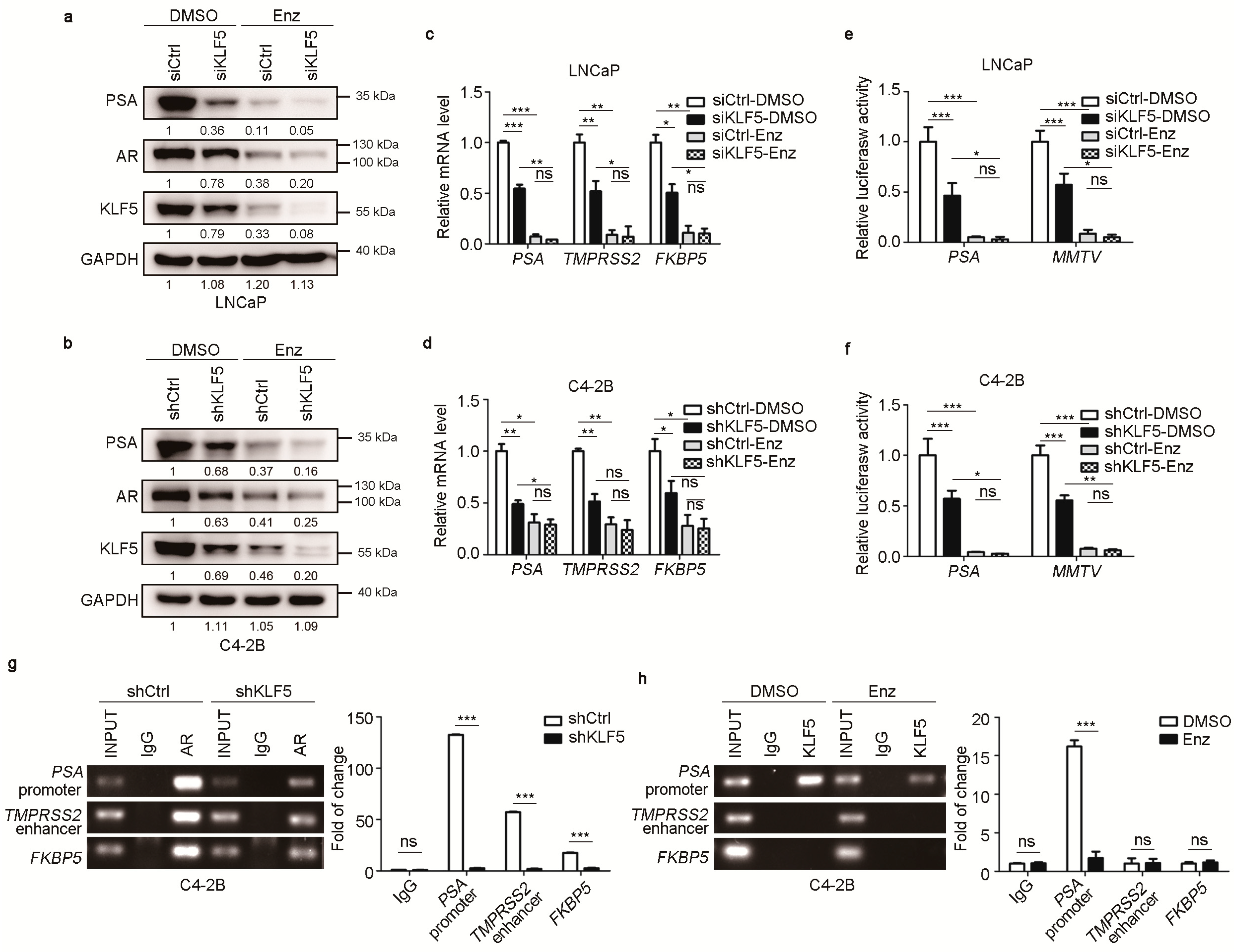
2.2. KLF5 is Crucial for Maintaining the Transcriptional Activity of AR in PCa Cells
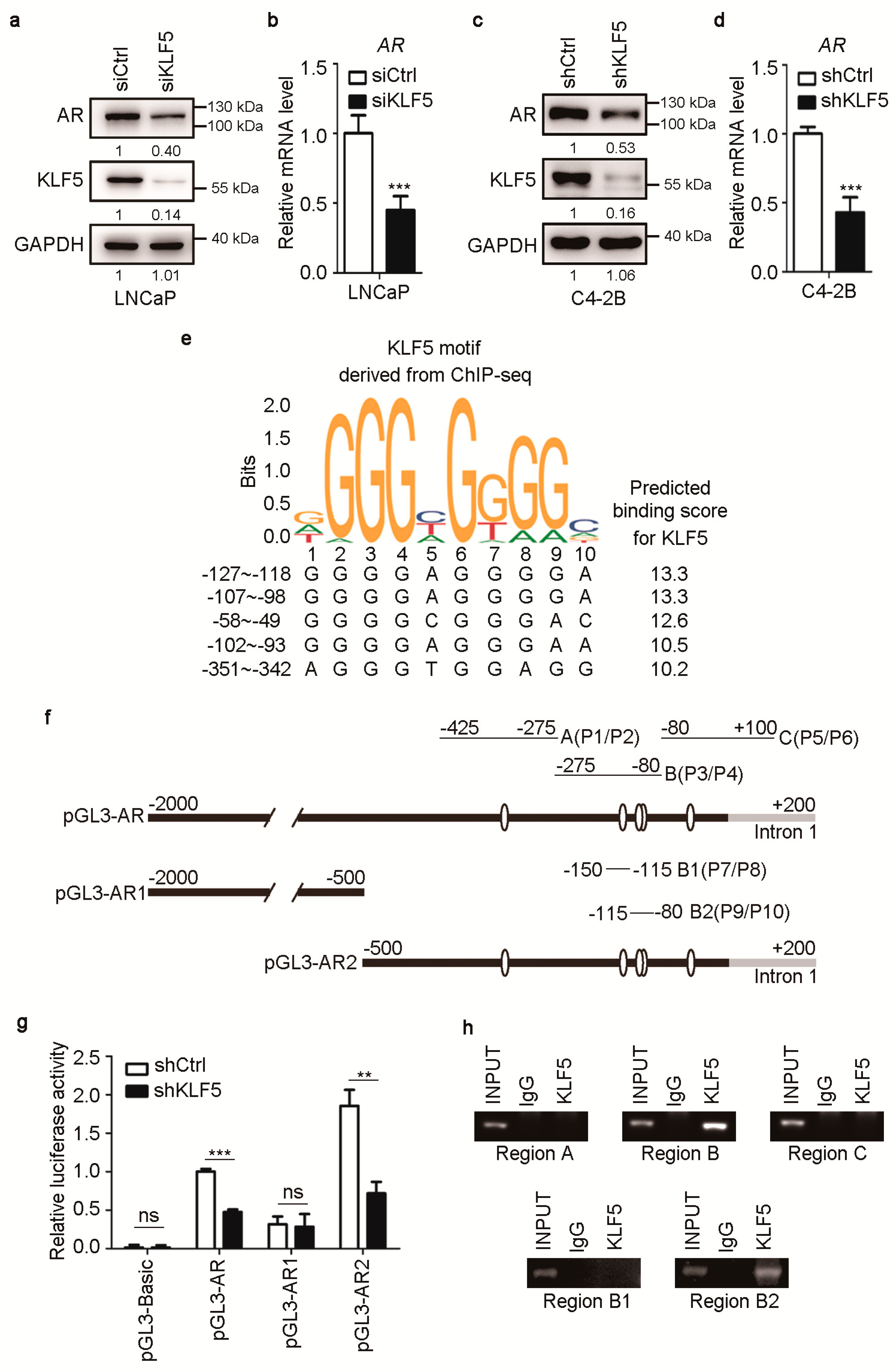
2.3. KLF5 also Promotes Transcription of the AR Gene in PCa Cells
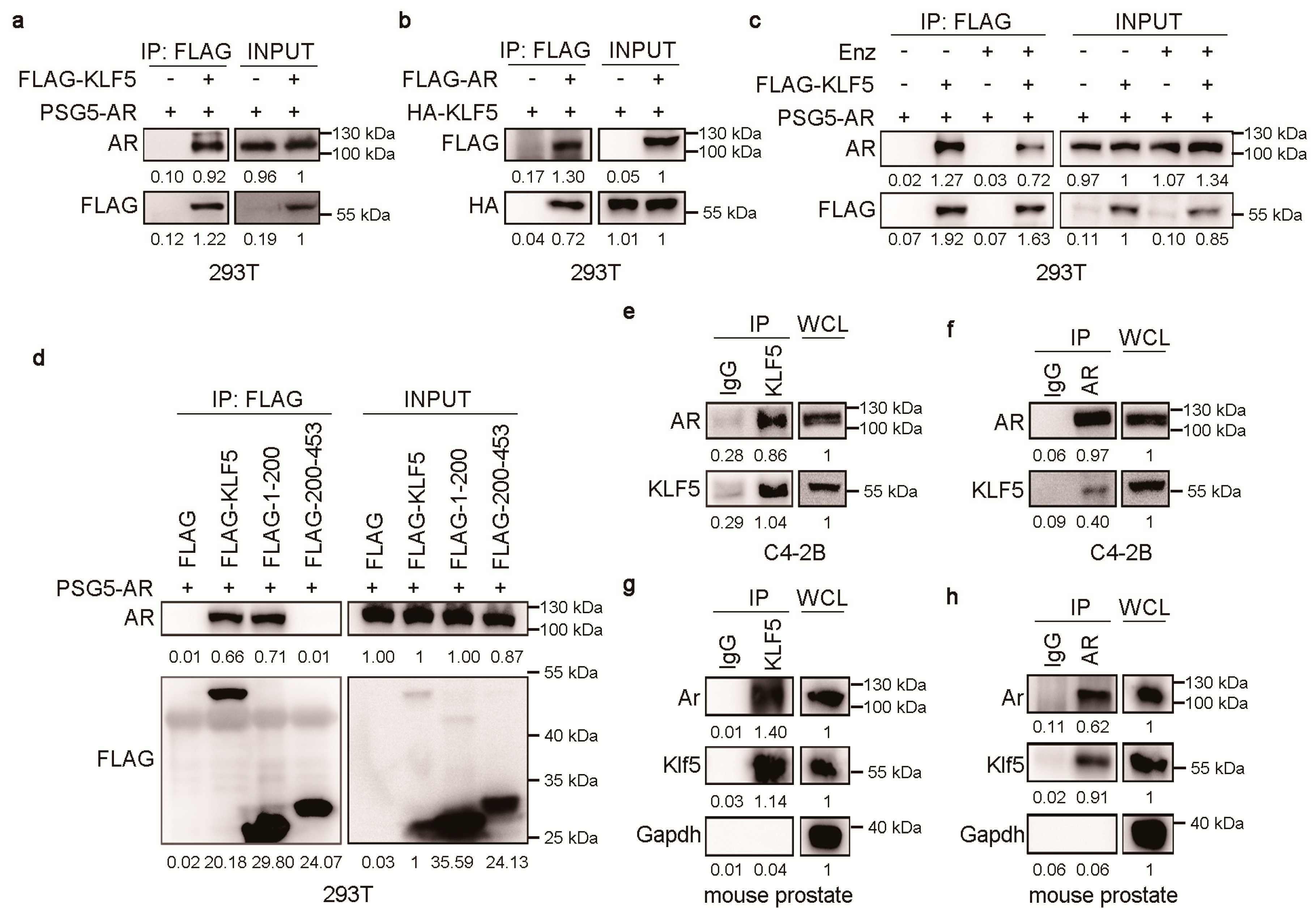
2.4. KLF5 Physically Associates with AR in Prostate Epithelial Cells
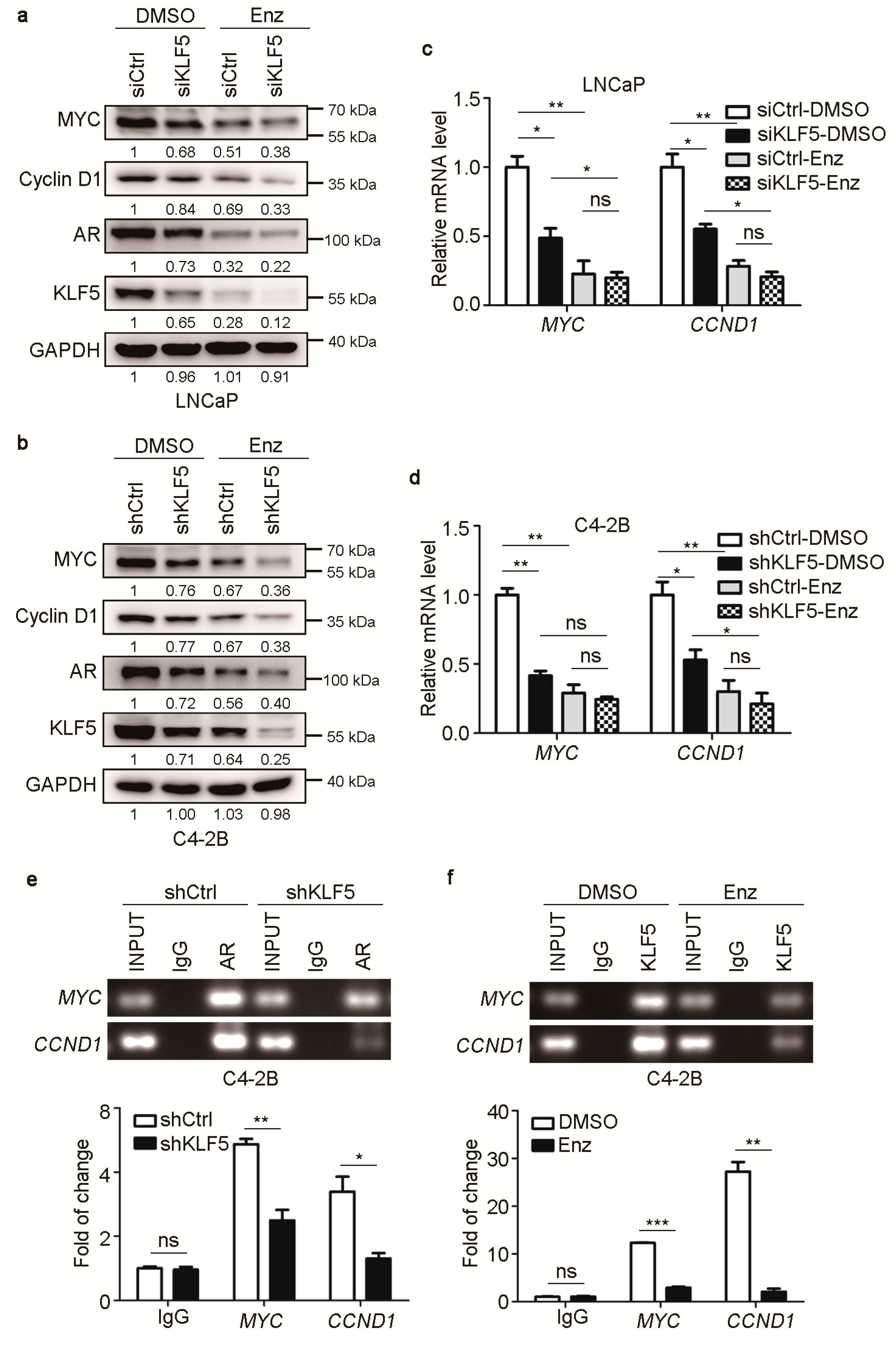
2.5. KLF5 Is also Crucial for AR-Mediated MYC and Cyclin D1 Expression in PCa Cells
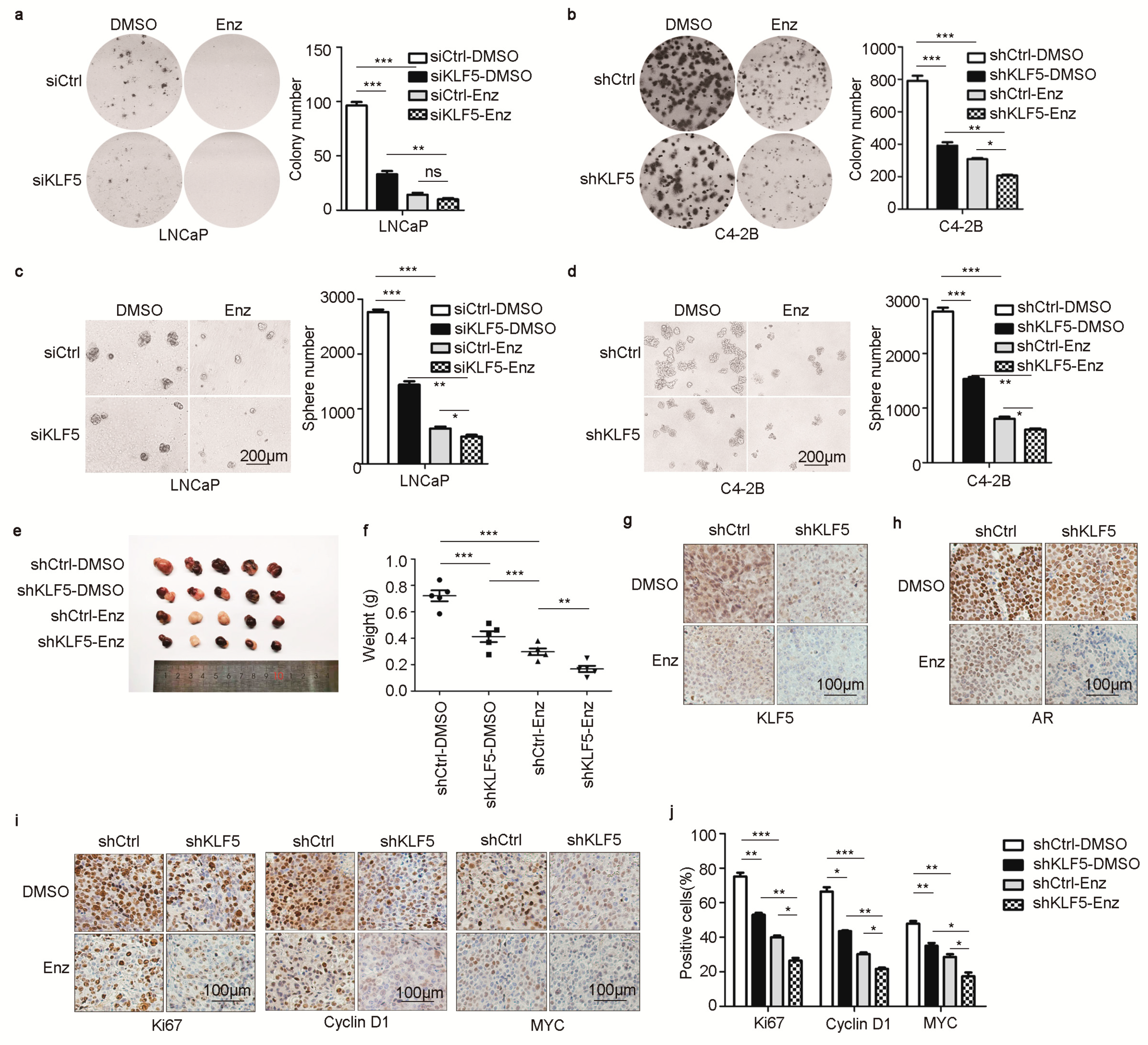
2.6. KLF5 Is Crucial for Androgen/AR to Promote Cell Proliferation and Tumor Growth in PCa Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture, and RNA Interference
4.2. Western Blot Analysis
4.3. RNA Extraction and Real-time qPCR
4.4. Construction of Expression and Luciferase Report Plasmids
4.5. Luciferase Reporter Assay
4.6. Colony Formation Assay
4.7. 3D Matrigel Assay
4.8. Immunoprecipitation
4.9. Mouse Xenograft Studies
4.10. Immunohistochemistry (IHC)
4.11. Chromatin Immunoprecipitation (ChIP)
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- El Sheikh, S.S.; Romanska, H.M.; Abel, P.; Domin, J.; Lalani el, N. Predictive value of PTEN and AR coexpression of sustained responsiveness to hormonal therapy in prostate cancer—A pilot study. Neoplasia 2008, 10, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.H.; Attard, G.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Brewer, D.; Clark, J.; Flohr, P.; Edwards, S.; Berney, D.M.; et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br. J. Cancer 2010, 102, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Tu, J.J.; Rohan, S.; Kao, J.; Kitabayashi, N.; Mathew, S.; Chen, Y.T. Gene fusions between TMPRSS2 and ETS family genes in prostate cancer: Frequency and transcript variant analysis by RT-PCR and FISH on paraffin-embedded tissues. Mod. Pathol. 2007, 20, 921–928. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Bethel, C.R.; Chaudhary, J.; Anway, M.D.; Brown, T.R. Gene expression changes are age-dependent and lobe-specific in the brown Norway rat model of prostatic hyperplasia. Prostate 2009, 69, 838–850. [Google Scholar] [CrossRef] [Green Version]
- Begley, L.; Monteleon, C.; Shah, R.B.; Macdonald, J.W.; Macoska, J.A. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell 2005, 4, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Hammerer, P.; Madersbacher, S. Landmarks in hormonal therapy for prostate cancer. BJU Int. 2012, 110 (Suppl. 1), 23–29. [Google Scholar] [CrossRef]
- Pagliarulo, V.; Bracarda, S.; Eisenberger, M.A.; Mottet, N.; Schroder, F.H.; Sternberg, C.N.; Studer, U.E. Contemporary role of androgen deprivation therapy for prostate cancer. Eur. Urol. 2012, 61, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Sarwar, M.; Semenas, J.; Miftakhova, R.; Simoulis, A.; Robinson, B.; Gjorloff Wingren, A.; Mongan, N.P.; Heery, D.M.; Johnsson, H.; Abrahamsson, P.A.; et al. Targeted suppression of AR-V7 using PIP5K1alpha inhibitor overcomes enzalutamide resistance in prostate cancer cells. Oncotarget 2016, 7, 63065–63081. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef]
- Itkonen, H.; Mills, I.G. Chromatin binding by the androgen receptor in prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 44–51. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Sogawa, K.; Imataka, H.; Yamasaki, Y.; Kusume, H.; Abe, H.; Fujii-Kuriyama, Y. cDNA cloning and transcriptional properties of a novel GC box-binding protein, BTEB2. Nucleic Acids Res. 1993, 21, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Ci, X.; Xing, C.; Zhang, B.; Zhang, Z.; Ni, J.J.; Zhou, W.; Dong, J.T. KLF5 inhibits angiogenesis in PTEN-deficient prostate cancer by attenuating AKT activation and subsequent HIF1alpha accumulation. Mol. Cancer 2015, 14, 91. [Google Scholar] [CrossRef] [Green Version]
- Diakiw, S.M.; D’Andrea, R.J.; Brown, A.L. The double life of KLF5: Opposing roles in regulation of gene-expression, cellular function, and transformation. IUBMB Life 2013, 65, 999–1011. [Google Scholar] [CrossRef]
- Dong, J.T.; Chen, C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell. Mol. Life Sci. 2009, 66, 2691–2706. [Google Scholar] [CrossRef]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-beta tumor suppression through a lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Gu, L.; Findley, H.W.; Chen, C.; Dong, J.T.; Yang, L.; Zhou, M. KLF5 interacts with P53 in regulating survivin expression in acute lymphoblastic leukemia. J. Biol. Chem. 2006, 281, 14711–14718. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Dong, X.Y.; Zhao, K.W.; Sun, X.; Li, Q.; Dong, J.T. Estrogen-induced interaction between KLF5 and estrogen receptor (ER) suppresses the function of ER in ER-positive breast cancer cells. Int. J. Cancer 2010, 126, 81–89. [Google Scholar] [CrossRef]
- Sun, R.; Chen, X.; Yang, V.W. Intestinal-enriched kruppel-like factor (kruppel-like factor 5) is a positive regulator of cellular proliferation. J. Biol. Chem. 2001, 276, 6897–6900. [Google Scholar] [CrossRef] [Green Version]
- McConnell, B.B.; Bialkowska, A.B.; Nandan, M.O.; Ghaleb, A.M.; Gordon, F.J.; Yang, V.W. Haploinsufficiency of Kruppel-like factor 5 rescues the tumor-initiating effect of the Apc(Min) mutation in the intestine. Cancer Res. 2009, 69, 4125–4133. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Benjamin, M.S.; Sun, X.; Otto, K.B.; Guo, P.; Dong, X.Y.; Bao, Y.; Zhou, Z.; Cheng, X.; Simons, J.W.; et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation in the TSU-Pr1 human bladder cancer cell line. Int. J. Cancer 2006, 118, 1346–1355. [Google Scholar] [CrossRef]
- Guo, P.; Dong, X.Y.; Zhao, K.; Sun, X.; Li, Q.; Dong, J.T. Opposing effects of KLF5 on the transcription of MYC in epithelial proliferation in the context of transforming growth factor beta. J. Biol. Chem. 2009, 284, 28243–28252. [Google Scholar] [CrossRef] [Green Version]
- Xing, C.; Fu, X.; Sun, X.; Guo, P.; Li, M.; Dong, J.T. Different expression patterns and functions of acetylated and unacetylated Klf5 in the proliferation and differentiation of prostatic epithelial cells. PLoS ONE 2013, 8, e65538. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ci, X.; Tao, R.; Ni, J.J.; Xuan, X.; King, J.L.; Xia, S.; Li, Y.; Frierson, H.F.; Lee, D.K.; et al. Klf5 acetylation regulates luminal differentiation of basal progenitors in prostate development and regeneration. Nat. Commun. 2020, 11, 997. [Google Scholar] [CrossRef]
- Xing, C.; Ci, X.; Sun, X.; Fu, X.; Zhang, Z.; Dong, E.N.; Hao, Z.Z.; Dong, J.T. Klf5 deletion promotes Pten deletion-initiated luminal-type mouse prostate tumors through multiple oncogenic signaling pathways. Neoplasia 2014, 16, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Frigo, D.E.; Sherk, A.B.; Wittmann, B.M.; Norris, J.D.; Wang, Q.; Joseph, J.D.; Toner, A.P.; Brown, M.; McDonnell, D.P. Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Mol. Endocrinol. 2009, 23, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Moon, J.S.; Park, S.W.; Koh, Y.K.; Ahn, Y.H.; Kim, K.S. KLF5 enhances SREBP-1 action in androgen-dependent induction of fatty acid synthase in prostate cancer cells. Biochem. J. 2009, 417, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Ai, J.; Wang, Y.; Dar, J.A.; Liu, J.; Liu, L.; Nelson, J.B.; Wang, Z. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol. Endocrinol. 2009, 23, 1963–1972. [Google Scholar] [CrossRef] [Green Version]
- Koochekpour, S. Androgen receptor signaling and mutations in prostate cancer. Asian J. Androl. 2010, 12, 639–657. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [Green Version]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef] [Green Version]
- Ferraldeschi, R.; Pezaro, C.; Karavasilis, V.; de Bono, J. Abiraterone and novel antiandrogens: Overcoming castration resistance in prostate cancer. Annu. Rev. Med. 2013, 64, 1–13. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Pattison, J.M.; Posternak, V.; Cole, M.D. Transcription Factor KLF5 Binds a Cyclin E1 Polymorphic Intronic Enhancer to Confer Increased Bladder Cancer Risk. Nat. Commun. 2016, 14, 1078–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.M. Prostate-specific antigen in screening of prostate cancer. J. Clin. Lab. Anal. 1994, 8, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magee, J.A.; Chang, L.W.; Stormo, G.D.; Milbrandt, J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology 2006, 147, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Chen, C.; Bolton, E.C. Androgen Receptor-Mediated Growth Suppression of HPr-1AR and PC3-Lenti-AR Prostate Epithelial Cells. PLoS ONE 2015, 10, e0138286. [Google Scholar] [CrossRef]
- Petre-Draviam, C.E.; Cook, S.L.; Burd, C.J.; Marshall, T.W.; Wetherill, Y.B.; Knudsen, K.E. Specificity of cyclin D1 for androgen receptor regulation. Cancer Res. 2003, 63, 4903–4913. [Google Scholar]
- Maina, P.K.; Shao, P.; Liu, Q.; Fazli, L.; Tyler, S.; Nasir, M.; Dong, X.; Qi, H.H. c-MYC drives histone demethylase PHF8 during neuroendocrine differentiation and in castration-resistant prostate cancer. Oncotarget 2016, 7, 75585–75602. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Jiang, M.; Grabowska, M.M.; Li, J.; Connelly, Z.M.; Zhang, J.; Hayward, S.W.; Cates, J.M.; Han, G.; Yu, X. Androgen receptor differentially regulates the proliferation of prostatic epithelial cells in vitro and in vivo. Oncotarget 2016, 7, 70404–70419. [Google Scholar] [CrossRef]
- Chiu, C.M.; Yeh, S.H.; Chen, P.J.; Kuo, T.J.; Chang, C.J.; Chen, P.J.; Yang, W.J.; Chen, D.S. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc. Natl. Acad. Sci. USA 2007, 104, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Zhao, K.W.; Dong, X.Y.; Sun, X.; Dong, J.T. Acetylation of KLF5 alters the assembly of p15 transcription factors in transforming growth factor-beta-mediated induction in epithelial cells. J. Biol. Chem. 2009, 284, 18184–18193. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, B.; Wu, Q.; Ci, X.; Zhao, R.; Zhang, Z.; Xia, S.; Su, D.; Chen, J.; Ma, G.; et al. Interruption of KLF5 acetylation converts its function from tumor suppressor to tumor promoter in prostate cancer cells. Int. J. Cancer 2015, 136, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Kumekawa, M.; Fukuda, G.; Shimizu, S.; Konno, K.; Odawara, M. Inhibition of monocyte chemoattractant protein-1 by Kruppel-like factor 5 small interfering RNA in the tumor necrosis factor- alpha-activated human umbilical vein endothelial cells. Biol. Pharm. Bull. 2008, 31, 1609–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, I.; Unden, A.B.; Toftgard, R. Human Kruppel-like factor5/KLF5: Synergy with NF-kappaB/Rel factors and expression in human skin and hair follicles. Eur. J. Cell Biol. 2002, 81, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Zhang, R.N.; Wen, Y.; Yin, W.N.; Bai, D.; Zheng, G.Y.; Li, J.S.; Zheng, B.; Wen, J.K. 1, 25(OH)2D3-induced interaction of vitamin D receptor with p50 subunit of NF-kappaB suppresses the interaction between KLF5 and p50, contributing to inhibition of LPS-induced macrophage proliferation. Biochem. Biophys. Res. Commun. 2017, 482, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Chanchevalap, S.; Nandan, M.O.; McConnell, B.B.; Charrier, L.; Merlin, D.; Katz, J.P.; Yang, V.W. Kruppel-like factor 5 is an important mediator for lipopolysaccharide-induced proinflammatory response in intestinal epithelial cells. Nucleic Acids Res. 2006, 34, 1216–1223. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Hyytinen, E.R.; Sun, X.; Helin, H.J.; Koivisto, P.A.; Frierson, H.F.; Vessella, R.L.; Dong, J.T. Deletion, mutation, and loss of expression of KLF6 in human prostate cancer. Am. J. Pathol. 2003, 162, 1349–1354. [Google Scholar] [CrossRef] [Green Version]
- Thalmann, G.N.; Sikes, R.A.; Wu, T.T.; Degeorges, A.; Chang, S.M.; Ozen, M.; Pathak, S.; Chung, L.W. LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate 2000, 44, 91–103. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, Z.; Xia, S.; Xing, C.; Ci, X.; Li, X.; Zhao, R.; Tian, S.; Ma, G.; Zhu, Z.; et al. KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol. Cell. Biol. 2013, 33, 4919–4935. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Zhao, D.; Yan, L.; Jiang, W.; Kim, J.S.; Gu, B.; Liu, Q.; Wang, R.; Xia, B.; Zhao, J.C.; et al. BMI1 regulates androgen receptor in prostate cancer independently of the polycomb repressive complex 1. Nat. Commun. 2018, 9, 500. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, B.; Liu, M.; Fu, X.; Ci, X.; A, J.; Fu, C.; Dong, G.; Wu, R.; Zhang, Z.; et al. KLF5 Is Crucial for Androgen-AR Signaling to Transactivate Genes and Promote Cell Proliferation in Prostate Cancer Cells. Cancers 2020, 12, 748. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030748
Li J, Zhang B, Liu M, Fu X, Ci X, A J, Fu C, Dong G, Wu R, Zhang Z, et al. KLF5 Is Crucial for Androgen-AR Signaling to Transactivate Genes and Promote Cell Proliferation in Prostate Cancer Cells. Cancers. 2020; 12(3):748. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030748
Chicago/Turabian StyleLi, Juan, Baotong Zhang, Mingcheng Liu, Xing Fu, Xinpei Ci, Jun A, Changying Fu, Ge Dong, Rui Wu, Zhiqian Zhang, and et al. 2020. "KLF5 Is Crucial for Androgen-AR Signaling to Transactivate Genes and Promote Cell Proliferation in Prostate Cancer Cells" Cancers 12, no. 3: 748. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030748