Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability

 , ,
, ,
Abstract
:1. Introduction
2. Results
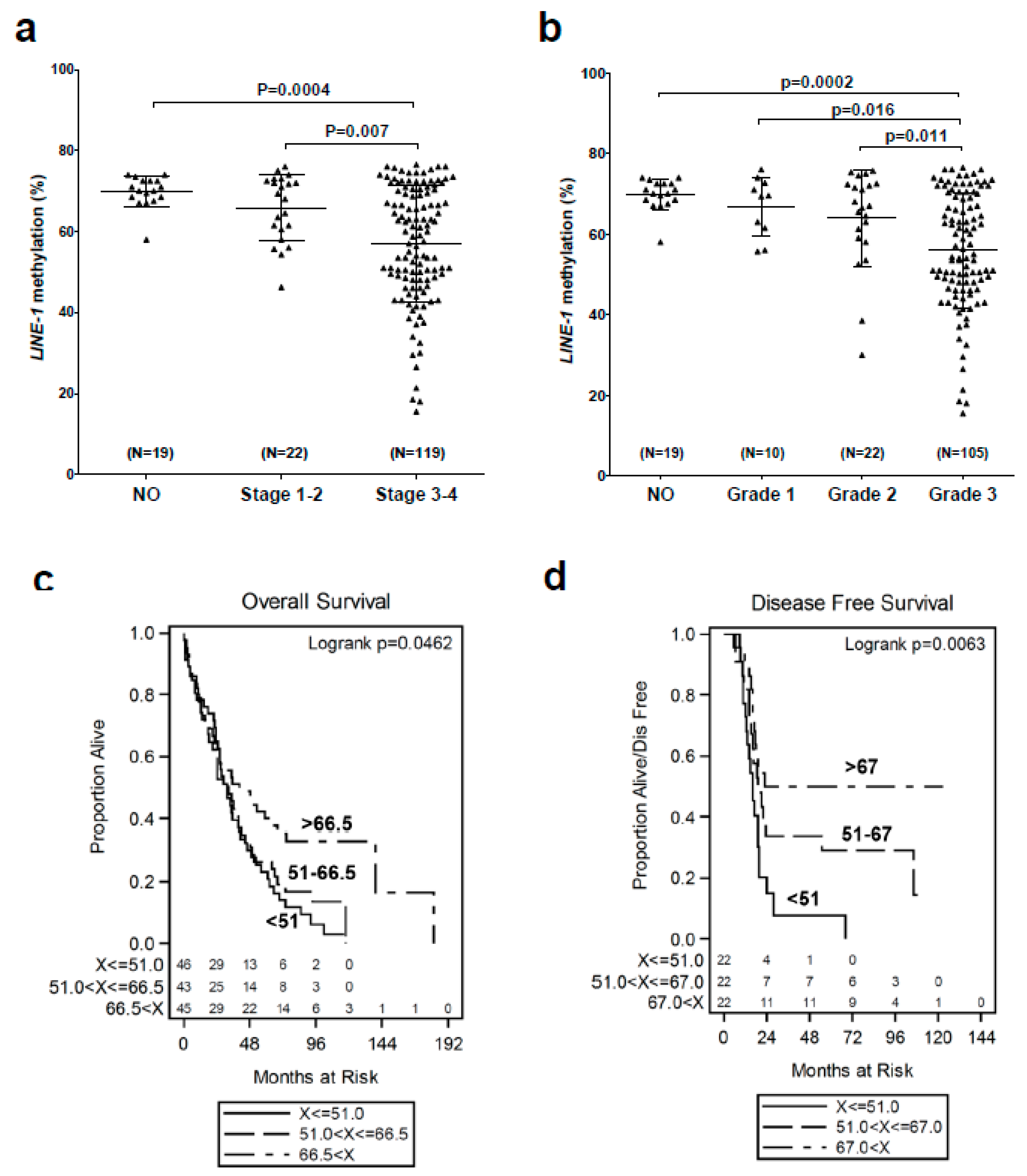
2.1. LINE-1 Hypomethylation is Associated with Disease Progression and Reduced Survival in EOC
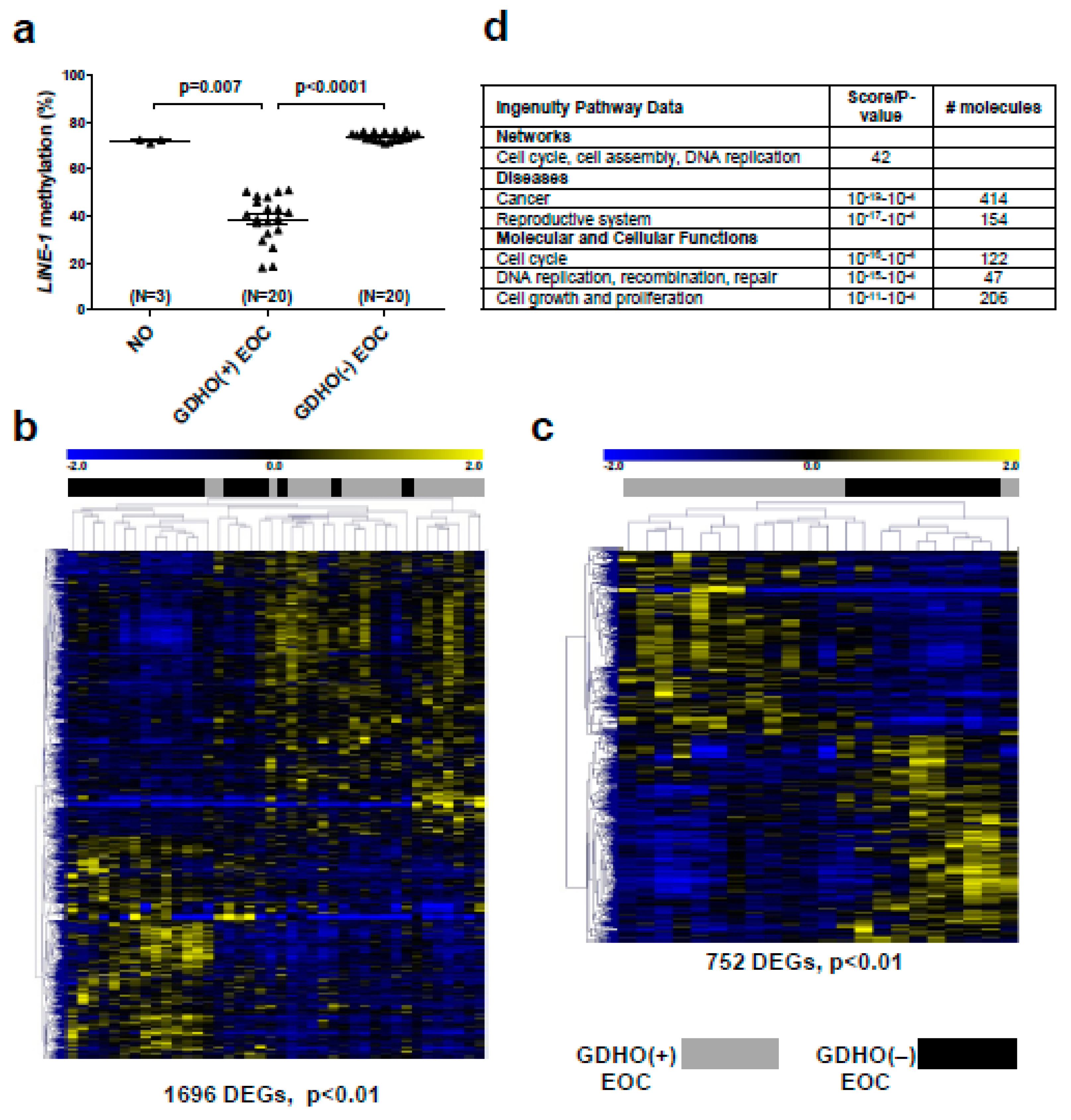
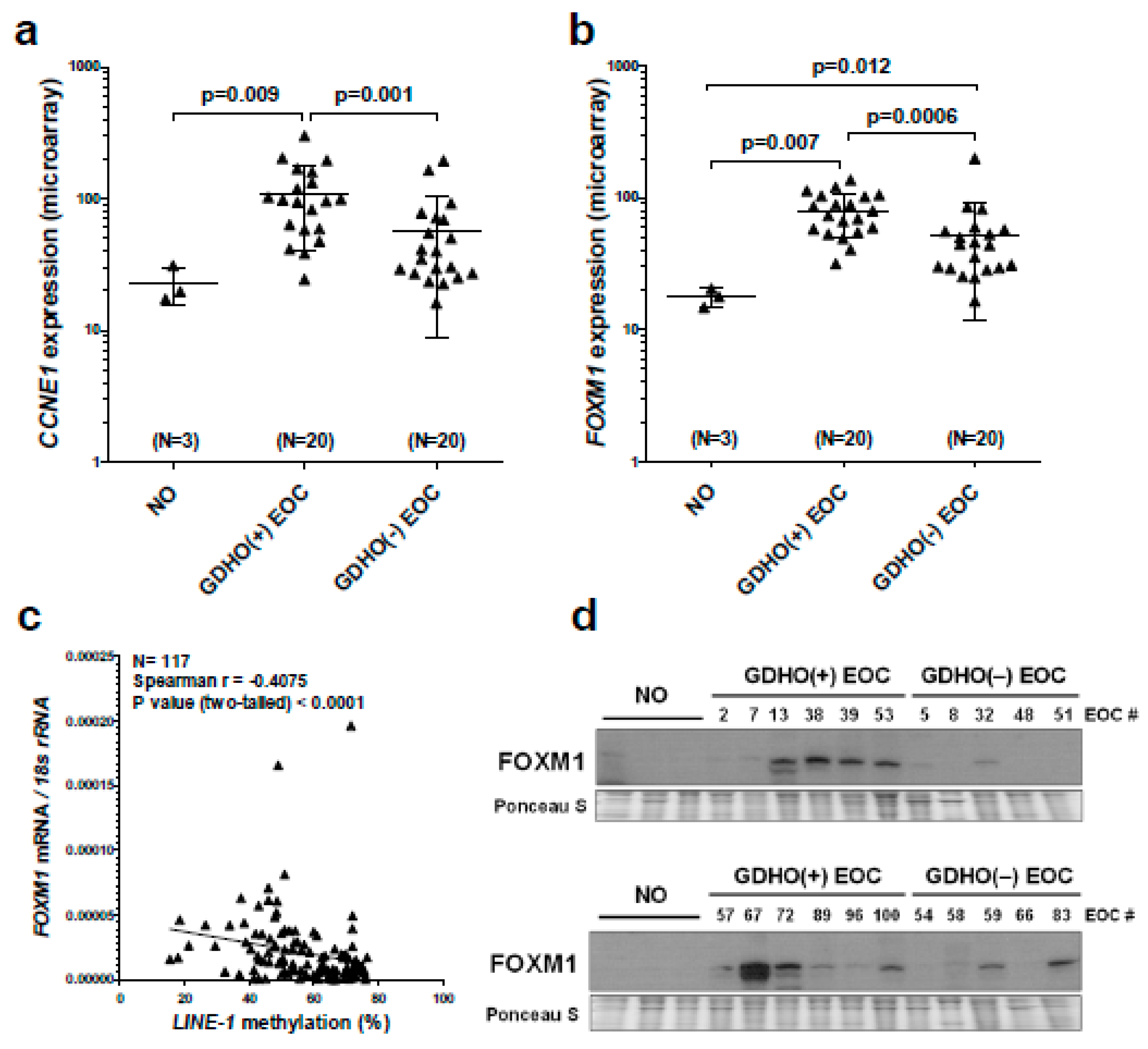
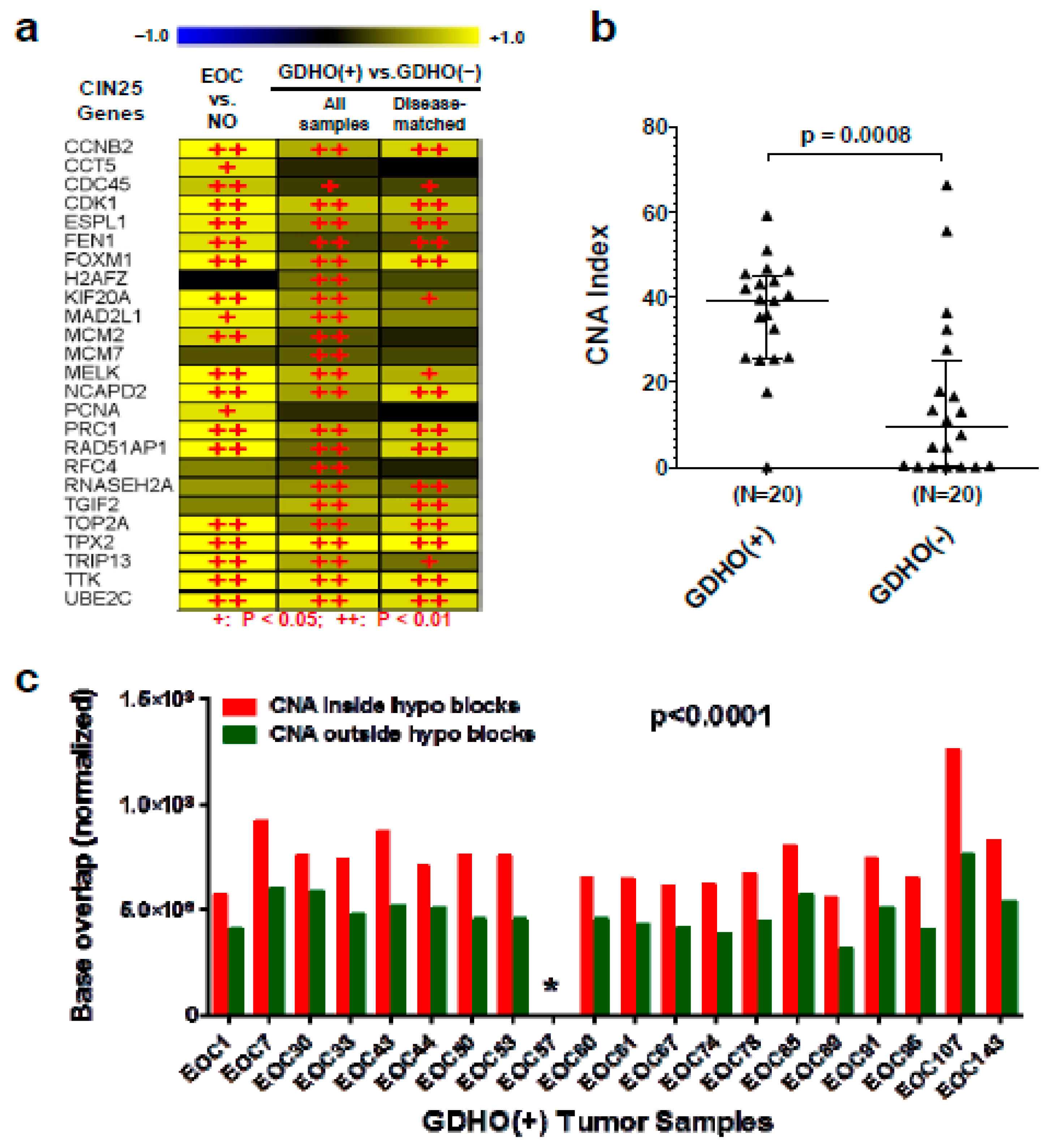
2.2. GDHO (+) EOC Tumors have Distinct Patterns of Gene Expression, including Enriched Signatures for Cell Proliferation
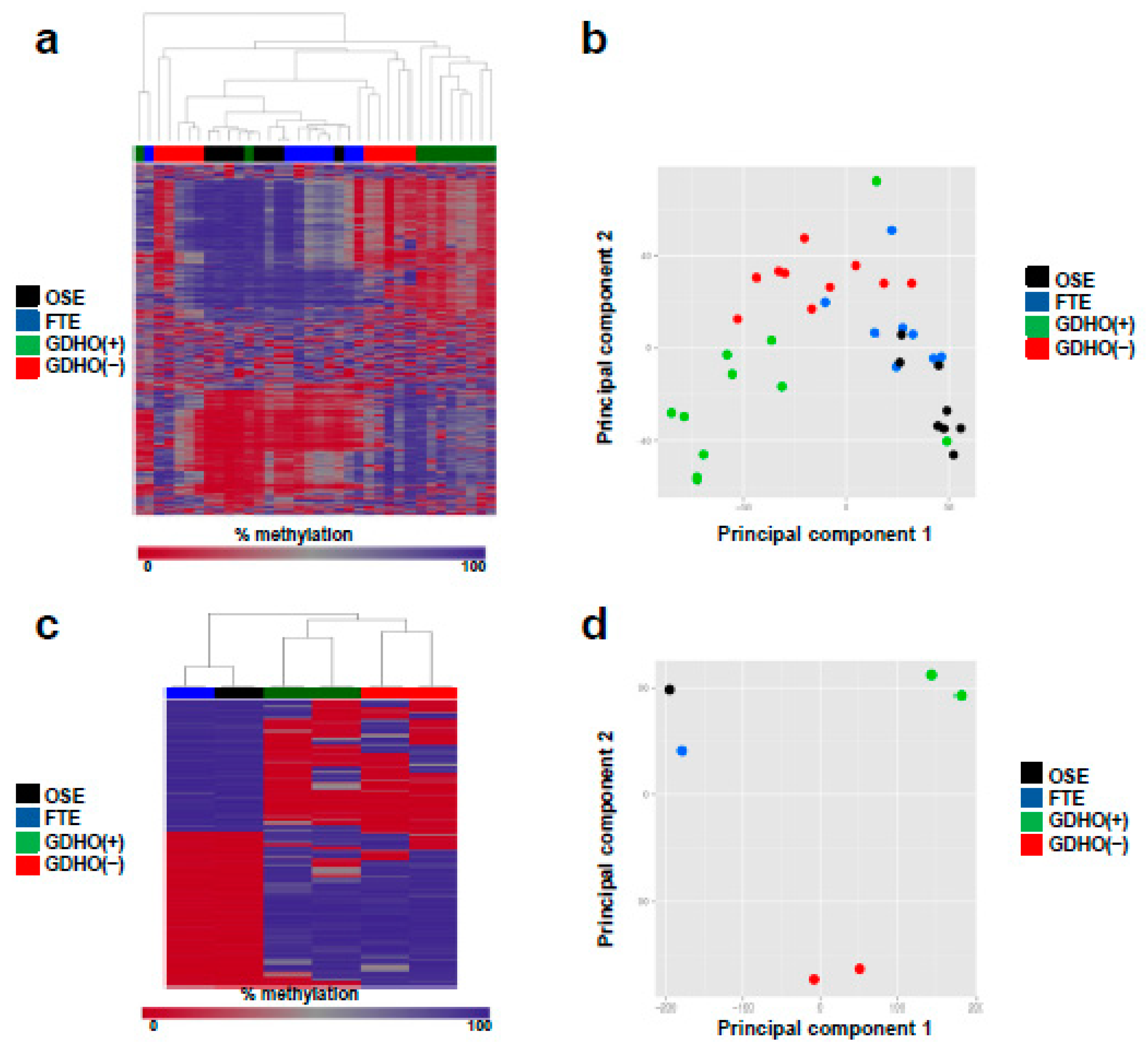
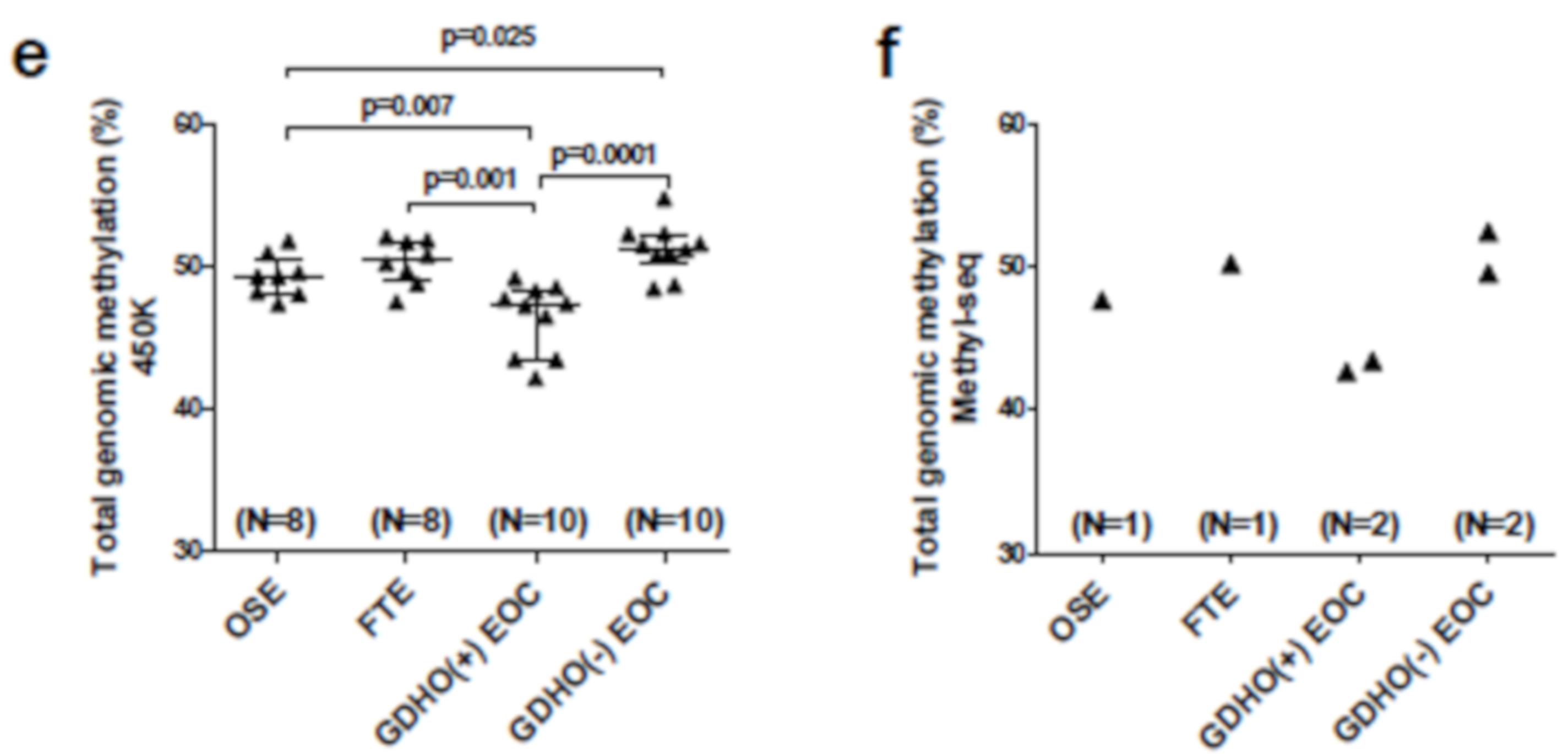
2.3. DNA Methylome Characteristics of GDHO (+) EOC
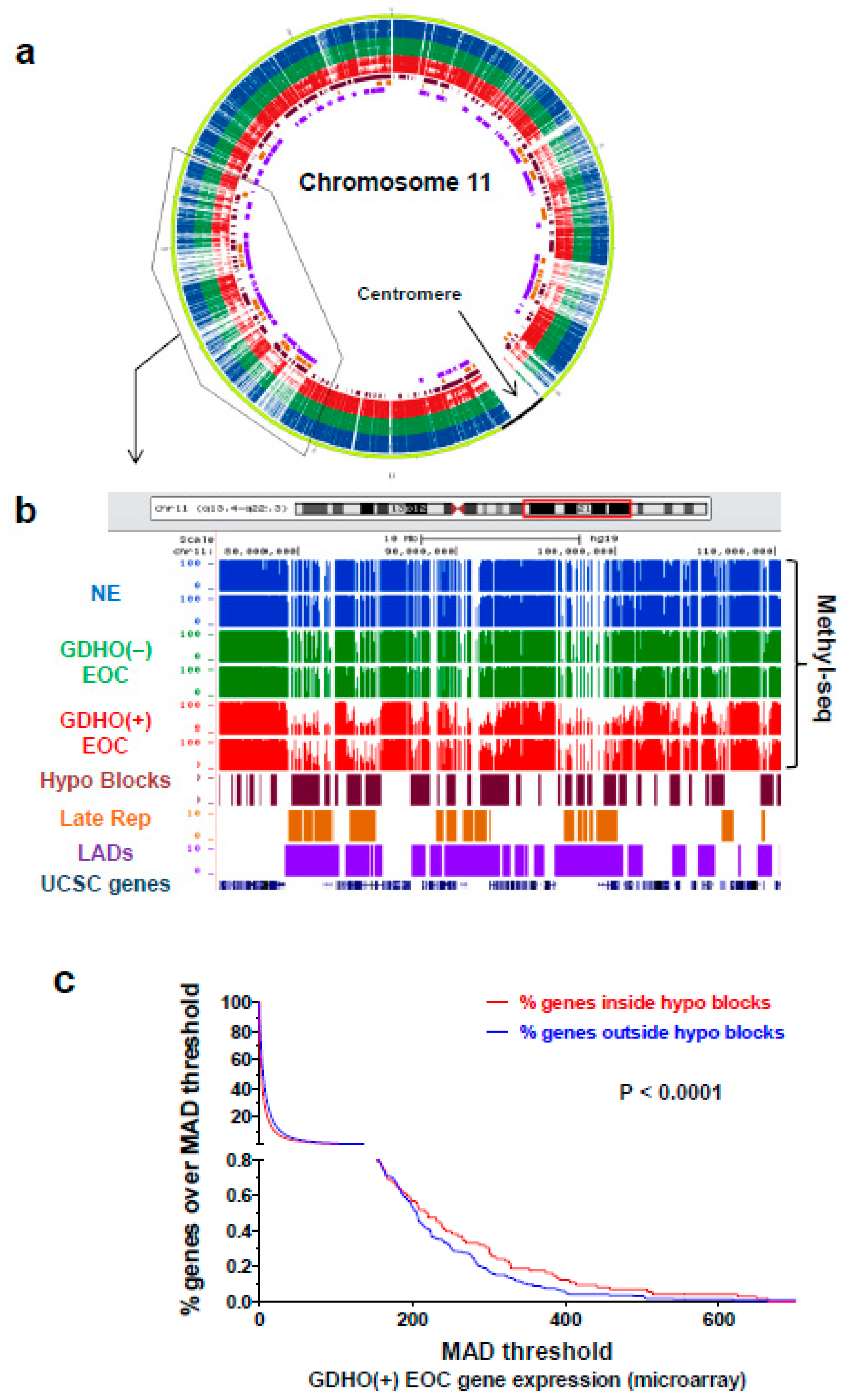
2.4. GDHO (+) EOC is Characterized by Hypomethylated Genomic Blocks
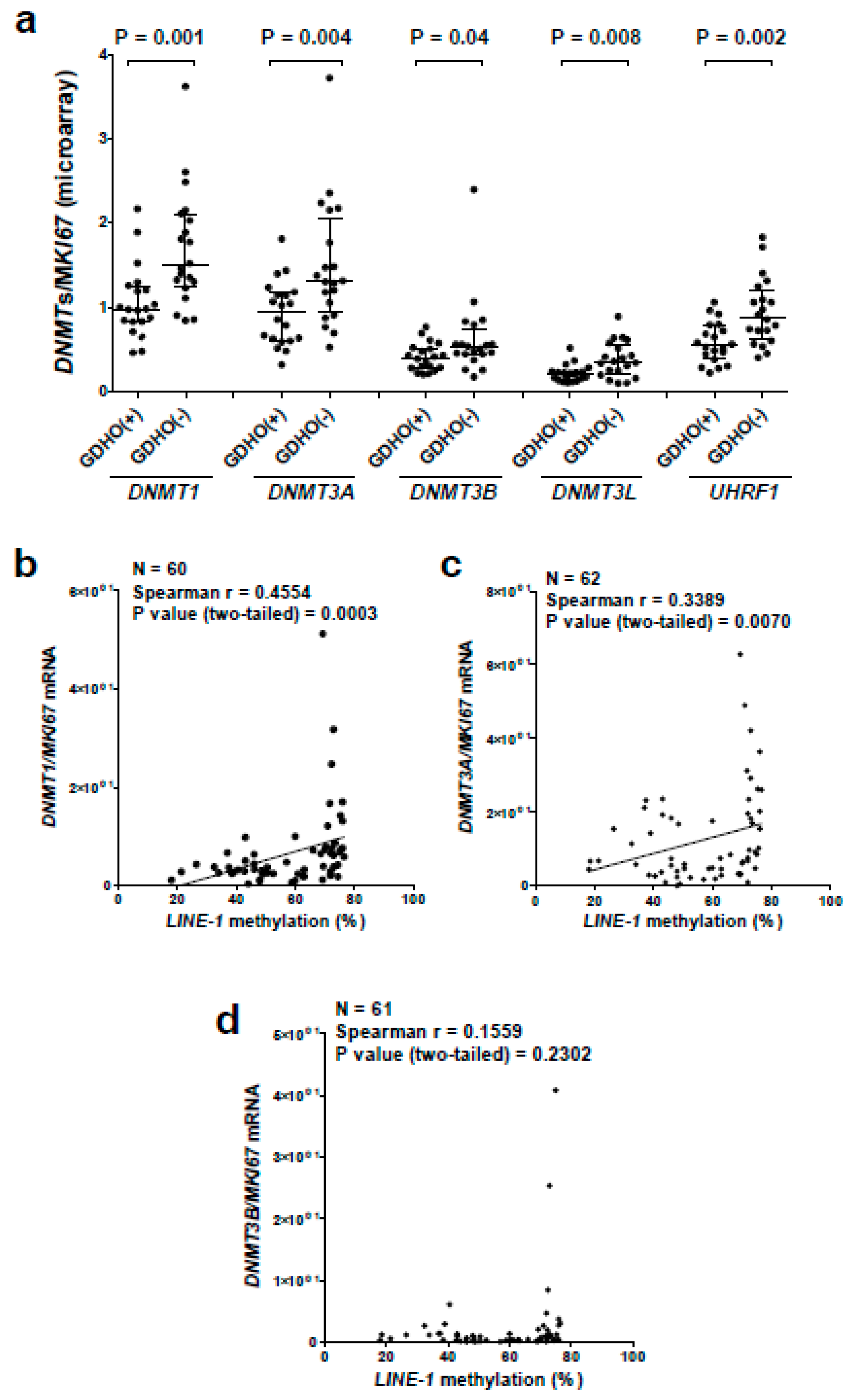
2.5. GDHO (+) EOC Tumors have Reduced Expression of Maintenance Methylation Components, when Normalized to Proliferation Markers
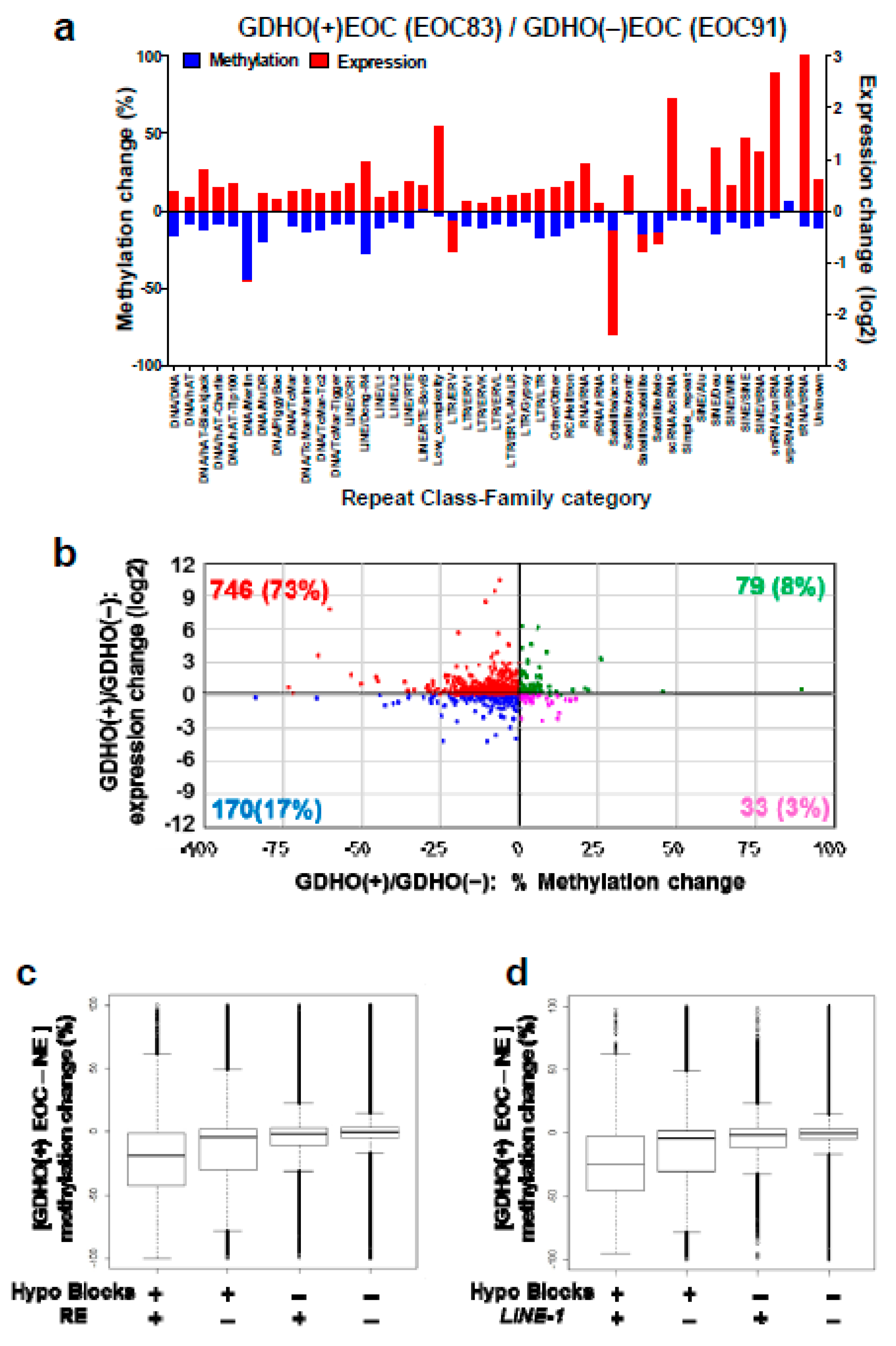
2.6. GDHO (+) EOC Shows Increased Expression of Repetitive Elements (RE)
3. Discussion
4. Methods
4.1. Human Tissues
4.2. DNA, RNA, and Protein Extractions
4.3. Cell lines and Drug Treatments
4.4. Gene Expression Microarrays
4.5. Reverse Transcriptase-Quantitative PCR (RT-qPCR)
4.6. Western Blot Analysis
4.7. Bisulfite Clonal Sequencing and Pyrosequencing
4.8. DNA Methylome Analyses
4.9. Determination of EOC Hypomethylated Blocks
4.10. Correlation of EOC Hypomethylated Blocks with Genomic Features
4.11. Total RNA Sequencing (RNA-seq)
4.12. Genomic Copy Number Analysis
4.13. Correlation of LINE-1 Methylation with EOC Patient Survival
4.14. Genomic Data Deposit and Public Access
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CG gene | cancer-germline or cancer-testis gene |
| CIN | chromosomal instability |
| CAN | copy number alteration |
| DAC | decitabine, 5-aza-2′-deoxycytidine |
| DEG | differentially expressed gene |
| DE-CG gene | differentially expressed CG gene |
| DMC | differentially methylated CpG site |
| DMR | differentially methylated region |
| DNMT | cytosine DNA methyltransferase |
| EOC | epithelial ovarian cancer |
| FDR | false discovery rate |
| FTE | fallopian tube epithelia |
| GDHO | global DNA hypomethylation |
| GSEA | gene set enrichment analysis |
| HGSOC | high-grade serous ovarian cancer |
| IPA | Ingenuity Pathway Analysis |
| LAD | lamina-associated domain |
| NE | normal epithelia (OSE + FTE) |
| NO | bulk normal ovary |
| OSE | ovarian surface epithelia |
| PCA | principal component analysis |
| RE | repetitive DNA elements |
| RMA | robust multichip average |
| TET | Ten-eleven translocation methylcytosine dioxygenase |
| TF | transcription factor |
References
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Gehrke, C.W.; Kuo, K.C.; Ehrlich, M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988, 48, 1159–1161. [Google Scholar] [PubMed]
- Bedford, M.T.; van Helden, P.D. Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res. 1987, 47, 5274–5276. [Google Scholar] [PubMed]
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Garcia-Perez, J.L.; Badge, R.M.; Moran, J.V. LINE-1 elements in structural variation and disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- De Smet, C.; De Backer, O.; Faraoni, I.; Lurquin, C.; Brasseur, F.; Boon, T. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc. Natl. Acad. Sci. USA 1996, 93, 7149–7153. [Google Scholar] [CrossRef] [Green Version]
- Woloszynska-Read, A.; Zhang, W.; Yu, J.; Link, P.A.; Mhawech-Fauceglia, P.; Collamat, G.; Akers, S.N.; Ostler, K.R.; Godley, L.A.; Odunsi, K.; et al. Coordinated cancer germline antigen promoter and global DNA hypomethylation in ovarian cancer: Association with the BORIS/CTCF expression ratio and advanced stage. Clin. Cancer Res. 2011, 17, 2170–2180. [Google Scholar] [CrossRef] [Green Version]
- Akers, S.N.; Odunsi, K.; Karpf, A.R. Regulation of cancer germline antigen gene expression: Implications for cancer immunotherapy. Future Oncol. 2010, 6, 717–732. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Albahrani, M.; Zhang, W.; Kufel, C.N.; James, S.R.; Odunsi, K.; Klinkebiel, D.; Karpf, A.R. Epigenetic activation of POTE genes in ovarian cancer. Epigenetics 2019, 14, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Barger, C.J.; Eng, K.H.; Klinkebiel, D.; Link, P.A.; Omilian, A.; Bshara, W.; Odunsi, K.; Karpf, A.R. PRAME expression and promoter hypomethylation in epithelial ovarian cancer. Oncotarget 2016, 7, 45352–45369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Barger, C.J.; Link, P.A.; Mhawech-Fauceglia, P.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. DNA hypomethylation-mediated activation of Cancer/Testis Antigen 45 (CT45) genes is associated with disease progression and reduced survival in epithelial ovarian cancer. Epigenetics 2015, 10, 736–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, B.P.; Weisenberger, D.J.; Aman, J.F.; Hinoue, T.; Ramjan, Z.; Liu, Y.; Noushmehr, H.; Lange, C.P.; van Dijk, C.M.; Tollenaar, R.A.; et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2012, 44, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef]
- Hon, G.C.; Hawkins, R.D.; Caballero, O.L.; Lo, C.; Lister, R.; Pelizzola, M.; Valsesia, A.; Ye, Z.; Kuan, S.; Edsall, L.E.; et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012, 22, 246–258. [Google Scholar] [CrossRef] [Green Version]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.A.; Steinhoff, C.; Florl, A.R. Methylation of endogenous human retroelements in health and disease. Curr. Top. Microbiol. Immunol. 2006, 310, 211–250. [Google Scholar]
- Karpf, A.R.; Matsui, S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [Green Version]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capella, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Suzuki, I.; Leodolter, A.; Alonso, S.; Horiuchi, S.; Yamashita, K.; Perucho, M. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 2006, 9, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Daskalos, A.; Nikolaidis, G.; Xinarianos, G.; Savvari, P.; Cassidy, A.; Zakopoulou, R.; Kotsinas, A.; Gorgoulis, V.; Field, J.K.; Liloglou, T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer. 2009, 124, 81–87. [Google Scholar] [CrossRef]
- Richards, K.L.; Zhang, B.; Baggerly, K.A.; Colella, S.; Lang, J.C.; Schuller, D.E.; Krahe, R. Genome-wide hypomethylation in head and neck cancer is more pronounced in HPV-negative tumors and is associated with genomic instability. PLOS ONE 2009, 4, e4941. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Bert, S.A.; Armstrong, N.J.; Caldon, C.E.; Song, J.Z.; Nair, S.S.; Gould, C.M.; Luu, P.L.; Peters, T.; Khoury, A.; et al. Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nat. Commun. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Dinh, H.Q.; Ramjan, Z.; Weisenberger, D.J.; Nicolet, C.M.; Shen, H.; Laird, P.W.; Berman, B.P. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 2018, 50, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Epping, M.T.; Wang, L.; Edel, M.J.; Carlee, L.; Hernandez, M.; Bernards, R. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell 2005, 122, 835–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpf, A.R.; Bai, S.; James, S.R.; Mohler, J.L.; Wilson, E.M. Increased expression of androgen receptor coregulator MAGE-11 in prostate cancer by DNA hypomethylation and cyclic AMP. Mol. Cancer Res. 2009, 7, 523–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oricchio, E.; Sciamanna, I.; Beraldi, R.; Tolstonog, G.V.; Schumann, G.G.; Spadafora, C. Distinct roles for LINE-1 and HERV-K retroelements in cell proliferation, differentiation and tumor progression. Oncogene 2007, 26, 4226–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillman, J.C.; Pugacheva, E.M.; Barger, C.J.; Sribenja, S.; Rosario, S.; Albahrani, M.; Truskinovsky, A.M.; Stablewski, A.; Liu, S.; Loukinov, D.I.; et al. BORIS Expression in Ovarian Cancer Precursor Cells Alters the CTCF Cistrome and Enhances Invasiveness through GALNT14. Mol. Cancer Res. 2019, 17, 2051–2062. [Google Scholar] [CrossRef] [Green Version]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer 2013, 13, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Wild, L.; Flanagan, J.M. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim. Biophys. Acta 2010, 1806, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.; Polyak, K.; et al. RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, D.T.; Lipson, D.; Paul, S.; Brannigan, B.W.; Akhavanfard, S.; Coffman, E.J.; Contino, G.; Deshpande, V.; Iafrate, A.J.; Letovsky, S.; et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011, 331, 593–596. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef]
- Nephew, K.P.; Balch, C.; Zhang, S.; Huang, T.H. Epigenetics and ovarian cancer. Cancer Treat. Res. 2009, 149, 131–146. [Google Scholar] [CrossRef]
- Woloszynska-Read, A.; Mhawech-Fauceglia, P.; Yu, J.; Odunsi, K.; Karpf, A.R. Intertumor and intratumor NY-ESO-1 expression heterogeneity is associated with promoter-specific and global DNA methylation status in ovarian cancer. Clin. Cancer Res. 2008, 14, 3283–3290. [Google Scholar] [CrossRef] [Green Version]
- Widschwendter, M.; Jiang, G.; Woods, C.; Muller, H.M.; Fiegl, H.; Goebel, G.; Marth, C.; Muller-Holzner, E.; Zeimet, A.G.; Laird, P.W.; et al. DNA hypomethylation and ovarian cancer biology. Cancer Res. 2004, 64, 4472–4480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisanic, T.R., 2nd; Asaka, S.; Lin, S.F.; Yen, T.T.; Sun, H.; Bahadirli-Talbott, A.; Wang, T.H.; Burns, K.H.; Wang, T.L.; Shih, I.M. Long Interspersed Nuclear Element 1 Retrotransposons Become Deregulated during the Development of Ovarian Cancer Precursor Lesions. Am. J. Pathol. 2019, 189, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, S.N.; Moysich, K.; Zhang, W.; Collamat Lai, G.; Miller, A.; Lele, S.; Odunsi, K.; Karpf, A.R. LINE1 and Alu repetitive element DNA methylation in tumors and white blood cells from epithelial ovarian cancer patients. Gynecol. Oncol. 2014, 132, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, M.L.; George, L.K.; Grant, G.D.; Perou, C.M. Common markers of proliferation. Nat. Rev. Cancer 2006, 6, 99–106. [Google Scholar] [CrossRef]
- Barger, C.J.; Zhang, W.; Hillman, J.; Stablewski, A.B.; Higgins, M.J.; Vanderhyden, B.C.; Odunsi, K.; Karpf, A.R. Genetic determinants of FOXM1 overexpression in epithelial ovarian cancer and functional contribution to cell cycle progression. Oncotarget 2015, 6, 27613–27627. [Google Scholar] [CrossRef] [Green Version]
- Saghafinia, S.; Mina, M.; Riggi, N.; Hanahan, D.; Ciriello, G. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 2018, 25, 1066–1080 e1068. [Google Scholar] [CrossRef] [Green Version]
- Teh, M.T.; Gemenetzidis, E.; Patel, D.; Tariq, R.; Nadir, A.; Bahta, A.W.; Waseem, A.; Hutchison, I.L. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinoma. PLOS ONE 2012, 7, e34329. [Google Scholar] [CrossRef]
- Woloszynska-Read, A.; James, S.R.; Link, P.A.; Yu, J.; Odunsi, K.; Karpf, A.R. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun. 2007, 7, 21. [Google Scholar]
- Karpf, A.R.; Jones, D.A. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002, 21, 5496–5503. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.G.; Sakabe, N.J.; de Oliveira, A.R.; Silva, M.C.; Mundstein, A.S.; Cohen, T.; Chen, Y.T.; Chua, R.; Gurung, S.; Gnjatic, S.; et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Research 2009, 37, D816–D819. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.T.; Wang, M.Y.; Chen, M.W.; Wei, L.H.; Chen, C.K.; Ko, C.H.; Jeng, Y.M.; Sung, P.L.; Jan, Y.H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.N.; Zhang, Y.X.; Wang, X.M.; Liu, Y.J.; Zhang, Y.Q.; Que, Y.H.; Zhao, W.J. ATAD2 is highly expressed in ovarian carcinomas and indicates poor prognosis. Asian Pac. J. Cancer Prev. 2014, 15, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with down-regulation of E-cadherin. Int. J. Cancer. 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Chang, B.; Yang, F.; Guo, X.; Cai, K.Q.; Xiao, X.S.; Wang, H.; Sen, S.; Hung, M.C.; Mills, G.B.; et al. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin. Cancer Res. 2010, 16, 3171–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, A.; Li, J.; Messersmith, W.A.; Laheru, D.; Rudek, M.A.; Maniar, M.; Hidalgo, M.; Baker, S.D.; Donehower, R.C. Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J. Clin. Oncol. 2008, 26, 5504–5510. [Google Scholar] [CrossRef] [PubMed]
- Wrzeszczynski, K.O.; Varadan, V.; Byrnes, J.; Lum, E.; Kamalakaran, S.; Levine, D.A.; Dimitrova, N.; Zhang, M.Q.; Lucito, R. Identification of tumor suppressors and oncogenes from genomic and epigenetic features in ovarian cancer. PLOS ONE 2011, 6, e28503. [Google Scholar] [CrossRef]
- Liao, Y.P.; Chen, L.Y.; Huang, R.L.; Su, P.H.; Chan, M.W.; Chang, C.C.; Yu, M.H.; Wang, P.H.; Yen, M.S.; Nephew, K.P.; et al. Hypomethylation signature of tumor-initiating cells predicts poor prognosis of ovarian cancer patients. Hum. Mol. Genet. 2014, 23, 1894–1906. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef]
- Lee, E.J.; Pei, L.; Srivastava, G.; Joshi, T.; Kushwaha, G.; Choi, J.H.; Robertson, K.D.; Wang, X.; Colbourne, J.K.; Zhang, L.; et al. Targeted bisulfite sequencing by solution hybrid selection and massively parallel sequencing. Nucleic Acids Res. 2011, 39, e127. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: an information aesthetic for comparative genomics. Genome Research 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.D.; Keyomarsi, K.; Gonzales, F.A.; Velicescu, M.; Jones, P.A. Differential mRNA expression of the human DNA methyltransferases (DNMTs) 1, 3a and 3b during the G(0)/G(1) to S phase transition in normal and tumor cells. Nucleic Acids Res. 2000, 28, 2108–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, M.; Kim, J.K.; Esteve, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Xiong, W.; Hahn, M.A.; Jin, S.G. The role of 5-hydroxymethylcytosine in human cancer. Cell Tissue Res. 2014, 356, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Woloszynska-Read, A.; James, S.R.; Song, C.; Jin, B.; Odunsi, K.; Karpf, A.R. BORIS/CTCFL expression is insufficient for cancer-germline antigen gene expression and DNA hypomethylation in ovarian cell lines. Cancer Immun. 2010, 10, 6. [Google Scholar]
- Ferreira, H.J.; Heyn, H.; Garcia Del Muro, X.; Vidal, A.; Larriba, S.; Munoz, C.; Villanueva, A.; Esteller, M. Epigenetic loss of the PIWI/piRNA machinery in human testicular tumorigenesis. Epigenetics 2013, 9, 113–118. [Google Scholar] [CrossRef]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 Overexpression Drives DNA Hypomethylation and Hepatocellular Carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar] [CrossRef]
- Desjobert, C.; El Mai, M.; Gerard-Hirne, T.; Guianvarc’h, D.; Carrier, A.; Pottier, C.; Arimondo, P.B.; Riond, J. Combined analysis of DNA methylation and cell cycle in cancer cells. Epigenetics 2014. [Google Scholar] [CrossRef]
- Shipony, Z.; Mukamel, Z.; Cohen, N.M.; Landan, G.; Chomsky, E.; Zeliger, S.R.; Fried, Y.C.; Ainbinder, E.; Friedman, N.; Tanay, A. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 2014. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.L.; Hlady, R.A.; Opavska, J.; Klinkebiel, D.; Pirruccello, S.J.; Talmon, G.A.; Sharp, J.G.; Wu, L.; Jaenisch, R.; Simpson, M.A.; et al. Tumor suppressor functions of Dnmt3a and Dnmt3b in the prevention of malignant mouse lymphopoiesis. Leukemia 2014, 28, 1138–1142. [Google Scholar] [CrossRef] [PubMed]
- Kinney, S.R.; Moser, M.T.; Pascual, M.; Greally, J.M.; Foster, B.A.; Karpf, A.R. Opposing roles of Dnmt1 in early and late-stage murine prostate cancer. Mol. Cell. Biol. 2010, 30, 4159–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.; Liang, G.; Sharma, S.; Lin, J.C.; Choi, S.H.; Han, H.; Yoo, C.B.; Egger, G.; Yang, A.S.; Jones, P.A. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 2009, 29, 5366–5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Cui, X.; Jiang, J.; Cao, D.; He, Y.; Wang, H. Uncoordinated expression of DNA methylation-related enzymes in human cancer. Epigenetics Chromatin 2017, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Song, Z.; Fu, Y.; Yu, Z.; Zhao, L.; Zhao, H.; Yao, W.; Huang, D.; Mi, X.; Wang, E.; et al. Clinicopathological significance and prognostic value of DNA methyltransferase 1, 3a, and 3b expressions in sporadic epithelial ovarian cancer. PLOS ONE 2012, 7, e40024. [Google Scholar] [CrossRef] [Green Version]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef] [Green Version]
- Cadieux, B.; Ching, T.T.; VandenBerg, S.R.; Costello, J.F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006, 66, 8469–8476. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Odunsi, K.; Matsuzaki, J.; James, S.R.; Mhawech-Fauceglia, P.; Tsuji, T.; Miller, A.; Zhang, W.; Akers, S.N.; Griffiths, E.A.; Miliotto, A.; et al. Epigenetic potentiation of NY-ESO-1 vaccine therapy in human ovarian cancer. Cancer Immunol. Res. 2014, 2, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpf, A.R. A potential role for epigenetic modulatory drugs in the enhancement of cancer/germ-line antigen vaccine efficacy. Epigenetics 2006, 1, 116–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.H.; Varadan, V.; Kamalakaran, S.; Zhang, M.Q.; Dimitrova, N.; Hicks, J. Major chromosomal breakpoint intervals in breast cancer co-localize with differentially methylated regions. Front. Oncol. 2012, 2, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 microarray software suite. Methods Enzymol 2006, 411, 134–193. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- James, S.R.; Link, P.A.; Karpf, A.R. Epigenetic regulation of X-linked cancer/germline antigen genes by DNMT1 and DNMT3b. Oncogene 2006, 25, 6975–6985. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Assenov, Y.; Muller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [Green Version]
- Favorov, A.; Mularoni, L.; Cope, L.M.; Medvedeva, Y.; Mironov, A.A.; Makeev, V.J.; Wheelan, S.J. Exploring massive, genome scale datasets with the GenometriCorr package. PLoS Comput. Biol. 2012, 8, e1002529. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 2014, 47, 11–12. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Region | Number of Regions | Size of Regions (bp) | % of Genome | % Regions in Hypo Blocks | Correlation | Jaccard Test P-Value 3,4 |
| EOC Hypomethylated Blocks | 2208 | 8.81E + 08 | 29 | 100 | N/A | N/A |
| LADs | 1231 | 1.13E + 09 | 37 | 36 | Direct | <0.01 |
| Early Replicating | 910 | 7.10E + 08 | 23 | 25 | Indirect | <0.01 |
| Late-Replicating | 1155 | 5.81E + 08 | 19 | 36 | Direct | <0.01 |
| CpG islands | 27,537 | 2.10E + 07 | 1 | 32 | Direct | <0.01 |
| Genes | 28,489 | 1.41E + 09 | 46 | 32 | Direct | <0.01 |
| # binding sites | # binding sites in hypo blocks | % binding sites in hypo blocks | Fold Enrichment | Correlation | Projection Test P-value | |
| EZH2 Sites | 14,818 | 6103 | 41 | 1.41 | Direct | <0.01 |
| SUZ12 Sites | 5772 | 2497 | 43 | 1.48 | Direct | <0.01 |
| CTCF Sites | 162,209 | 52,216 | 32 | 1.10 | Direct | <0.01 |
| # peaks | # peaks in hypo blocks | % peaks in hypo blocks | Fold Enrichment | Correlation | Jaccard Test P-value | |
| H3K4me3 | 33,116 | 8655 | 26 | 0.90 | Indirect | <0.01 |
| H3K9me3 | 49,328 | 18,264 | 37 | 1.28 | Direct | <0.01 |
| H3K27ac | 67,989 | 17,935 | 26 | 0.91 | Indirect | <0.01 |
| H3K27me3 | 40,126 | 15,428 | 38 | 1.32 | Direct | <0.01 |
| H3K36me3 | 33,342 | 6303 | 19 | 0.65 | Indirect | <0.01 |
| # of repeats | # repeats in hypo blocks | % repeats in hypo blocks | Fold Change | Correlation | Jaccard Test P-value | |
| LINE-1 | 951,780 | 264,352 | 28 | 0.96 | Direct | <0.01 |
| Alu/SINE | 1,194,734 | 292,495 | 24 | 0.84 | Indirect | <0.01 |
| Satellites | 6775 | 1052 | 16 | 0.64 | Indirect | <0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers 2020, 12, 764. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030764
Zhang W, Klinkebiel D, Barger CJ, Pandey S, Guda C, Miller A, Akers SN, Odunsi K, Karpf AR. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers. 2020; 12(3):764. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030764
Chicago/Turabian StyleZhang, Wa, David Klinkebiel, Carter J. Barger, Sanjit Pandey, Chittibabu Guda, Austin Miller, Stacey N. Akers, Kunle Odunsi, and Adam R. Karpf. 2020. "Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability" Cancers 12, no. 3: 764. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030764