An Integrated Genomic Strategy to Identify CHRNB4 as a Diagnostic/Prognostic Biomarker for Targeted Therapy in Head and Neck Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
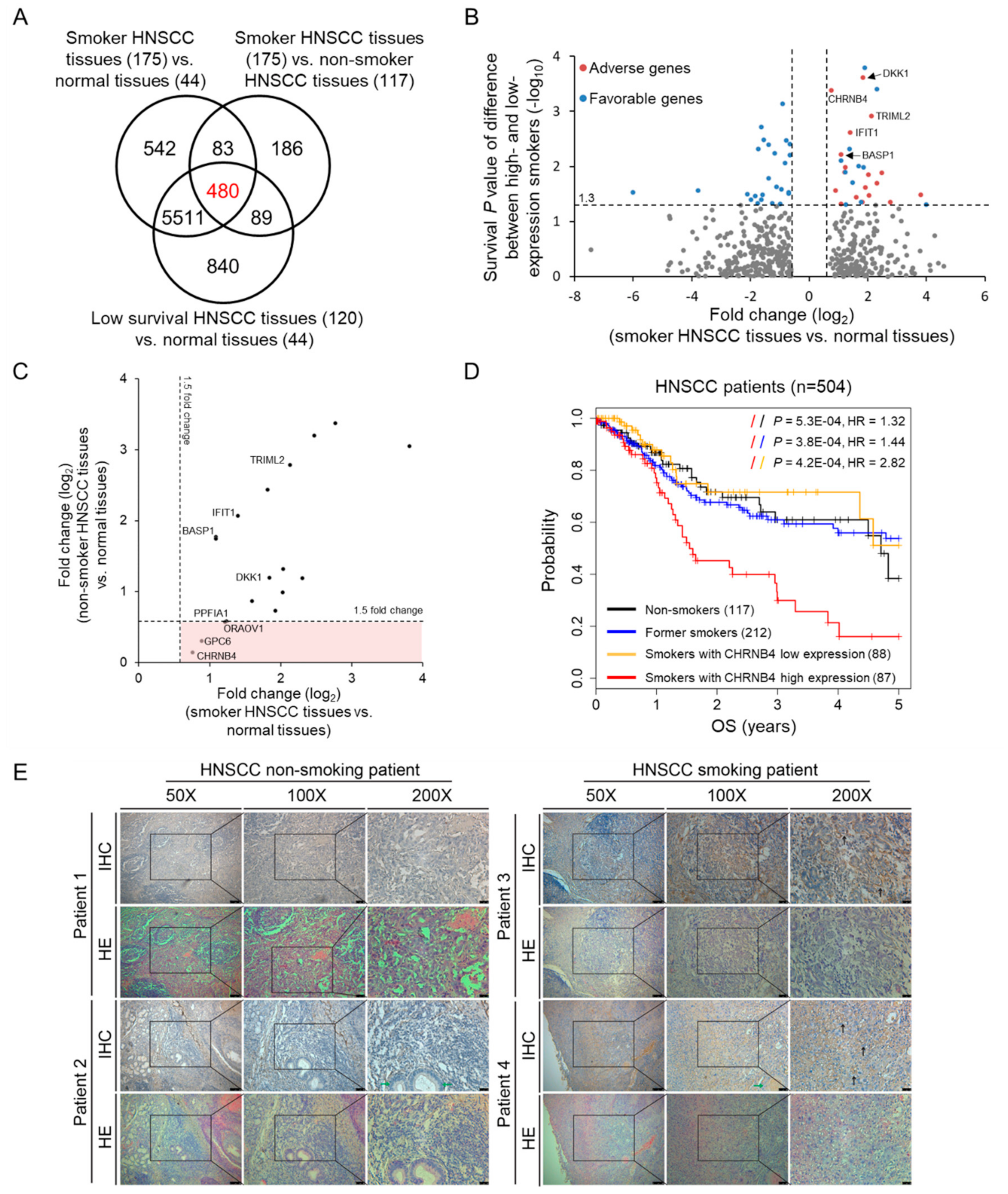
2.1. Identification of Smoking-Related and Prognostic Genes in Head and Neck Cancer
2.2. Association Cancer-Related Genes and CHRNB4
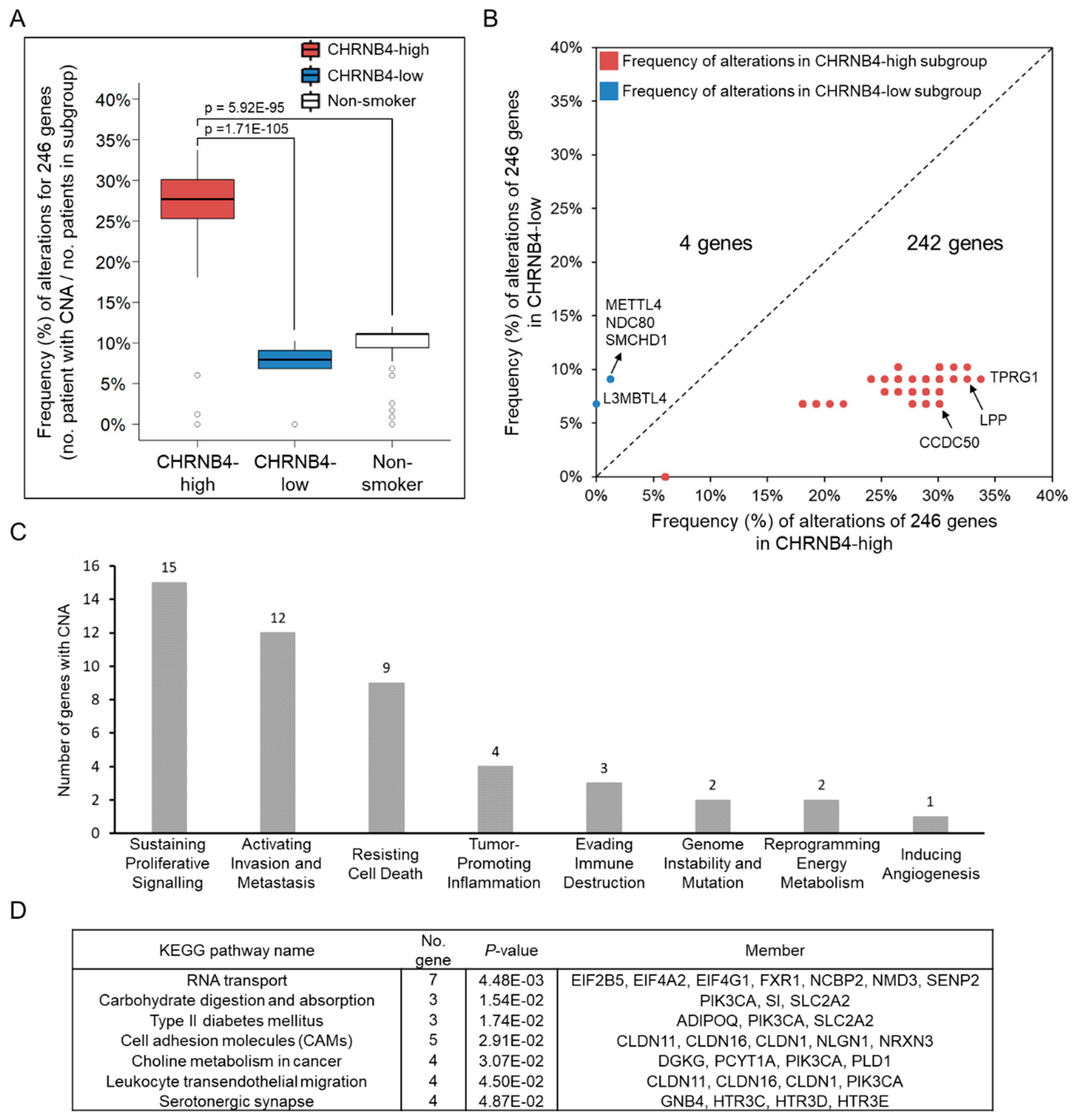
2.3. Relationship between Genetic Alterations and CHRNB4 Signature
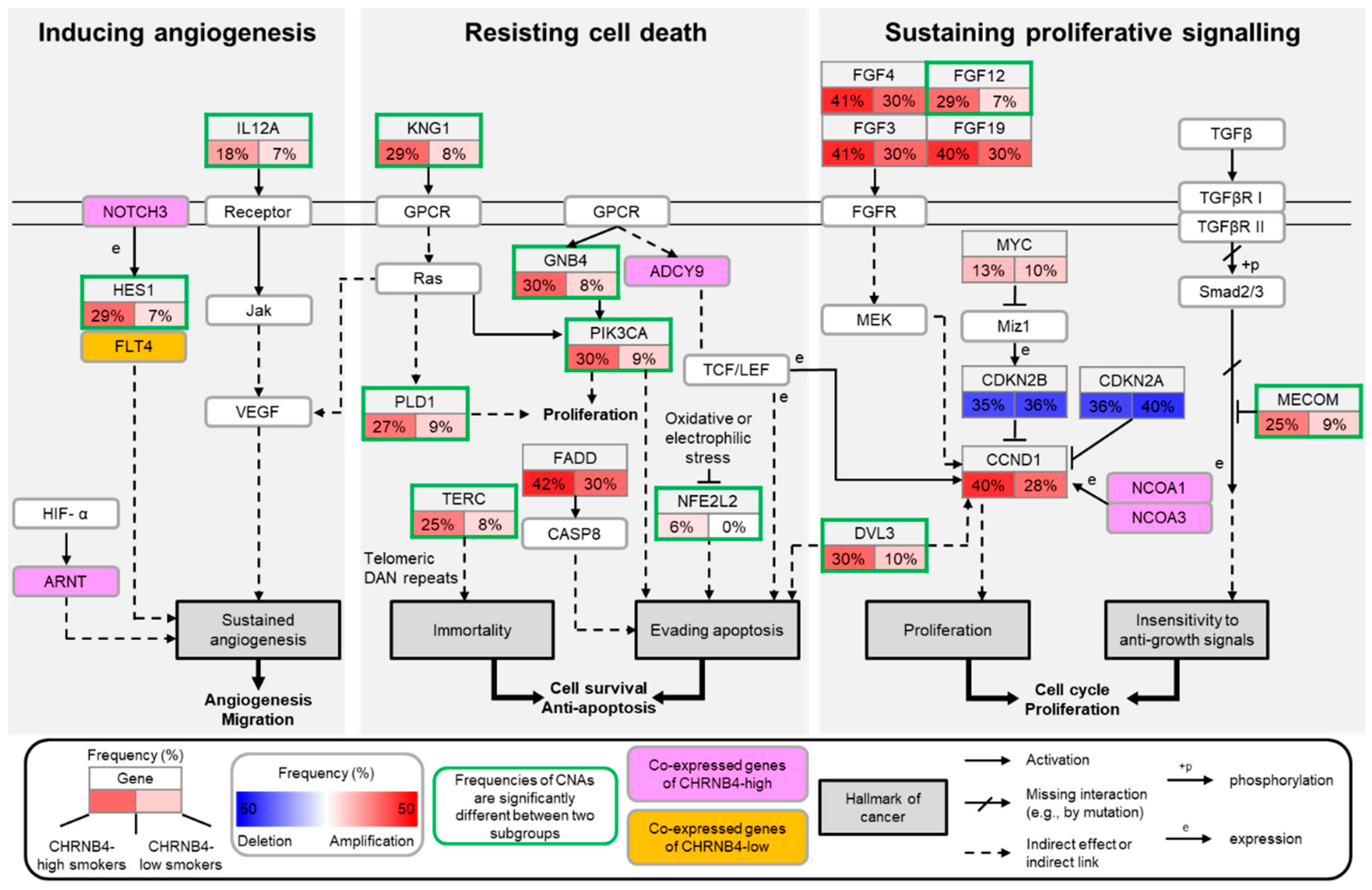
2.4. The Pathways and Downstream Targets of CHRNB4-High and -Low Subgroups
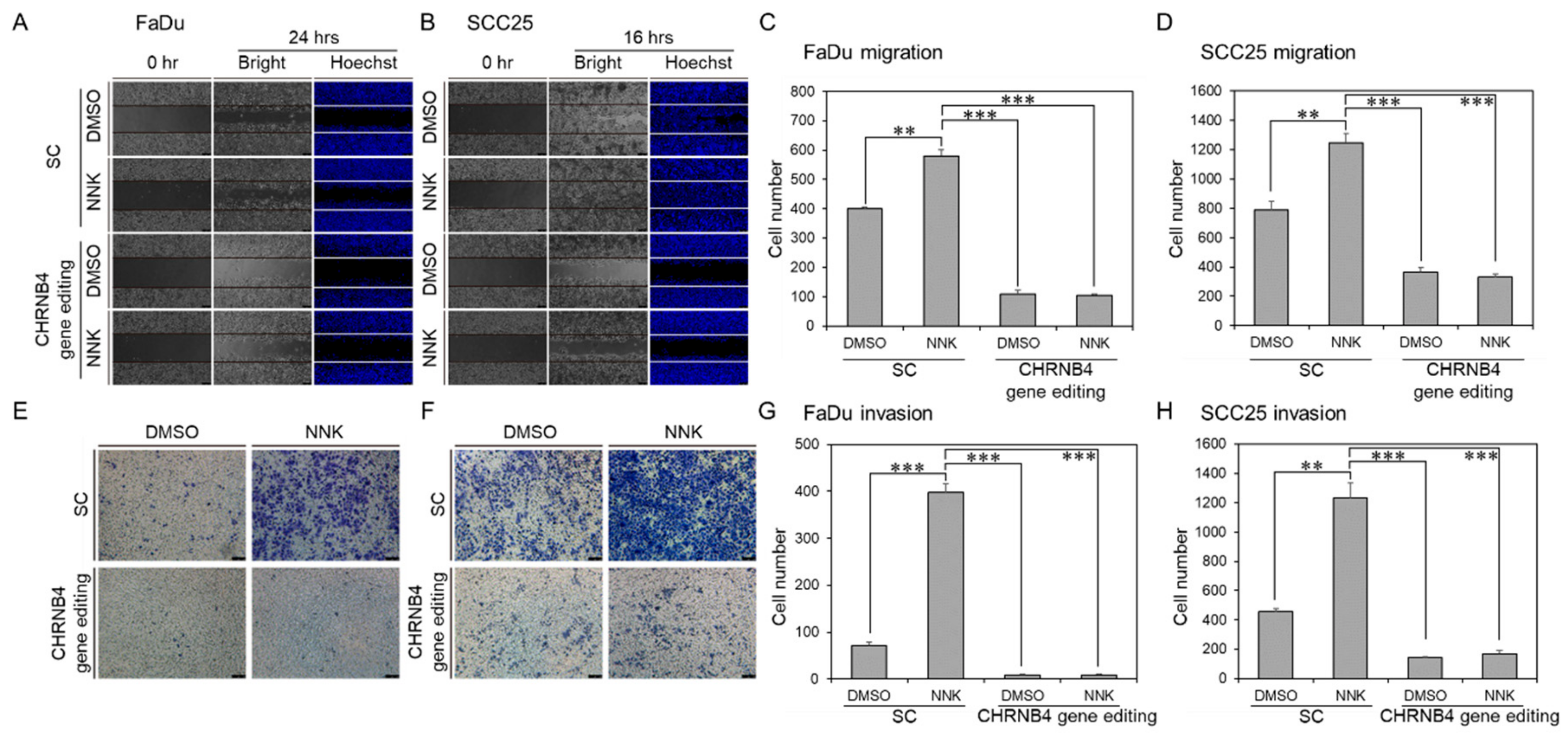
2.5. Validation of CHRNB4 Gene Editing in HNSCC Cells
2.6. CHRNB4 Promoted Cell Migration and Invasion in NNK-Treated HNSCC Cells
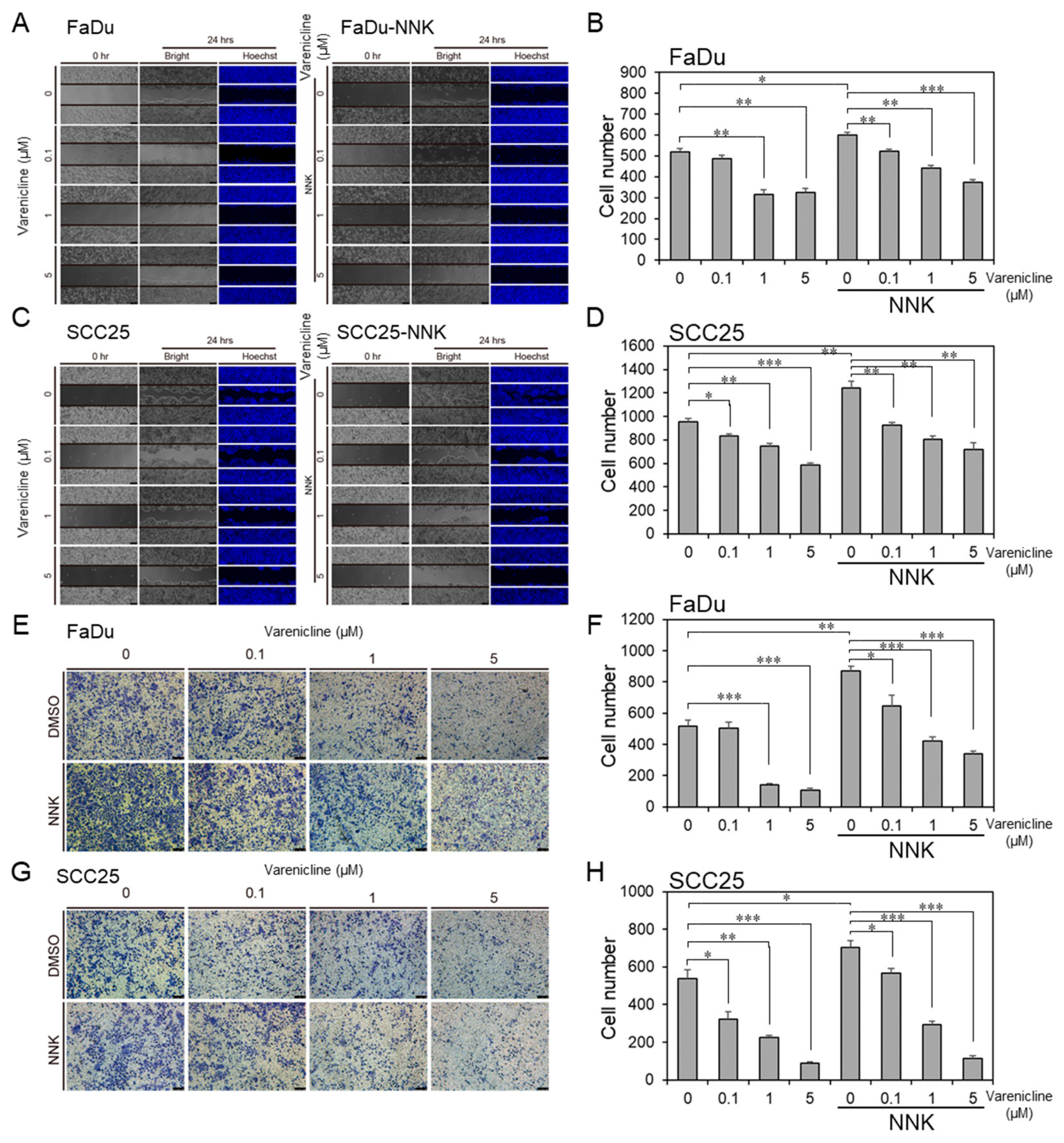
2.7. Varenicline, as a Repurposing Drug of CHRNB4, Inhibited Migration and Invasion in HNSCC Cell Lines
3. Discussion
4. Materials and Methods
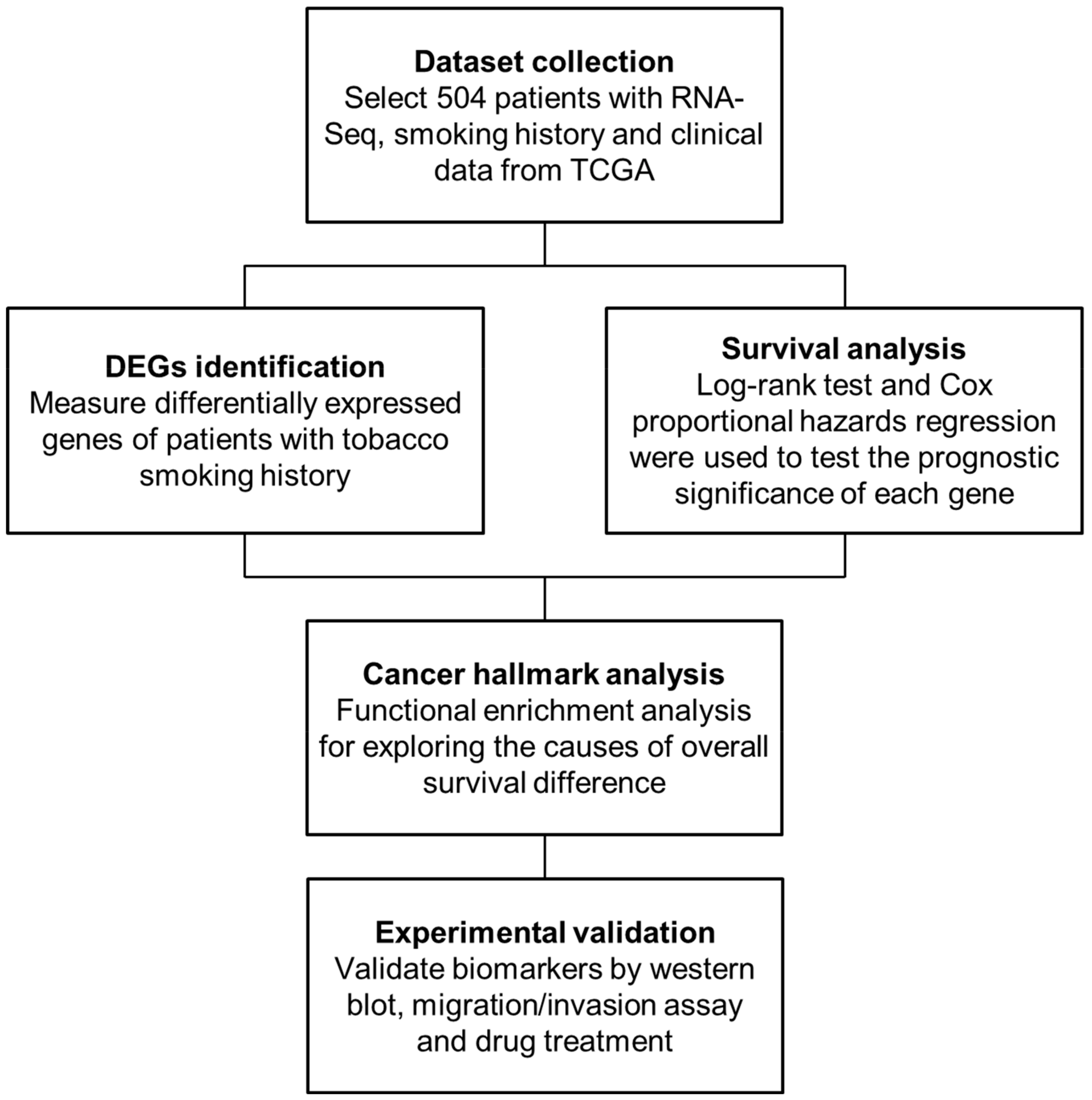
4.1. Patient Stratification and RNA-Seq Data Collection
4.2. Methods Overview
4.3. Identification of DEGs
4.4. Survival Analysis
4.5. Cancer Hallmark and Pathway Enrichment Analysis
4.6. Copy Number Alteration (CNA) Analysis
4.7. Cell Lines
4.8. CRISPR/Cas9 Gene Editing on CHRNB4 and Lentivirus Production
4.9. Western Blot Analysis
4.10. Migration and Invasion Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Lohavanichbutr, P.; Houck, J.; Fan, W.; Yueh, B.; Mendez, E.; Futran, N.; Doody, D.R.; Upton, M.P.; Farwell, D.G.; Schwartz, S.M.; et al. Genomewide Gene Expression Profiles of Hpv-Positive and Hpv-Negative Oropharyngeal Cancer: Potential Implications for Treatment Choices. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 180–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The Molecular Biology of Head and Neck Cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Suh, Y.; Amelio, I.; Urbano, T.G.; Tavassoli, M. Clinical Update on Cancer: Molecular Oncology of Head and Neck Cancer. Cell Death Dis. 2014, 5, e1018. [Google Scholar] [CrossRef] [Green Version]
- Spence, T.; Bruce, J.; Yip, K.W.; Liu, F.F. Hpv Associated Head and Neck Cancer. Cancers 2016, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Osazuwa-Peters, N.; Boakye, E.A.; Chen, B.Y.; Tobo, B.B.; Varvares, M.A. Association between Head and Neck Squamous Cell Carcinoma Survival, Smoking at Diagnosis, and Marital Status. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 43–50. [Google Scholar] [CrossRef]
- Hecht, S.S. Progress and Challenges in Selected Areas of Tobacco Carcinogenesis. Chem. Res. Toxicol. 2008, 21, 160–171. [Google Scholar] [CrossRef] [Green Version]
- Khariwala, S.S.; Hatsukami, D.; Hecht, S.S. Tobacco Carcinogen Metabolites and DNA Adducts as Biomarkers in Head and Neck Cancer: Potential Screening Tools and Prognostic Indicators. Head Neck 2012, 34, 441–447. [Google Scholar] [CrossRef]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. Egfr, P16, Hpv Titer, Bcl-Xl and P53, Sex, and Smoking as Indicators of Response to Therapy and Survival in Oropharyngeal Cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef] [Green Version]
- Du, E.; Mazul, A.L.; Farquhar, D.; Brennan, P.; Anantharaman, D.; Abedi-Ardekani, B.; Weissler, M.C.; Hayes, D.N.; Olshan, A.F.; Zevallos, J.P. Long-Term Survival in Head and Neck Cancer: Impact of Site, Stage, Smoking, and Human Papillomavirus Status. Laryngoscope 2019, 129, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, T.; Altieri, A.; Chatenoud, L.; Gallus, S.; Bosetti, C.; Negri, E.; Franceschi, S.; Levi, F.; Talamini, R.; la Vecchia, C. Risk Factors for Oral and Pharyngeal Cancer in Young Adults. Oral. Oncol. 2004, 40, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.D.; Parkinson, E.K.; Harrison, P.R. Profiling Early Head and Neck Cancer. Nat. Rev. Cancer 2005, 5, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, J.; Straif, K.; Agudo, A.; Ahrens, W.; Santos, A.B.D.; Boccia, S.; Cadoni, G.; Canova, C.; Castellsague, X.; Chen, C.; et al. Low Frequency of Cigarette Smoking and the Risk of Head and Neck Cancer in the Inhance Consortium Pooled Analysis. Int. J. Epidemiol. 2016, 45, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Karam-Hage, M.; Cinciripini, P.M.; Gritz, E.R. Tobacco Use and Cessation for Cancer Survivors: An Overview for Clinicians. Ca A Cancer J. Clin. 2014, 64, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Shen, L.J.; Li, B.F.; Gao, J.; Xia, Y.F. Smoking Is a Poor Prognostic Factor for Male Nasopharyngeal Carcinoma Treated with Radiotherapy. Radiother. Oncol. 2014, 110, 409–415. [Google Scholar] [CrossRef]
- Gillison, M.L.; Zhang, Q.; Jordan, R.; Xiao, W.H.; Westra, W.H.; Trotti, A.; Spencer, S.; Harris, J.; Chung, C.H.; Ang, K.K. Tobacco Smoking and Increased Risk of Death and Progression for Patients with P16-Positive and P16-Negative Oropharyngeal Cancer. J. Clin. Oncol. 2012, 30, 2102–2111. [Google Scholar] [CrossRef]
- Fortin, A.; Wang, C.S.; Vigneault, E. Influence of Smoking and Alcohol Drinking Behaviors on Treatment Outcomes of Patients with Squamous Cell Carcinomas of the Head and Neck. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1062–1069. [Google Scholar] [CrossRef]
- Pytynia, K.B.; Grant, J.R.; Etzel, C.J.; Roberts, D.B.; Wei, Q.; Sturgis, E.M. Matched-Pair Analysis of Survival of Never Smokers and Ever Smokers with Squamous Cell Carcinoma of the Head and Neck. J. Clin. Oncol. 2004, 22, 3981–3988. [Google Scholar] [CrossRef]
- Lassen, P.; Lacas, B.; Pignon, J.P.; Trotti, A.; Zackrisson, B.; Zhang, Q.; Overgaard, J.; Blanchard, P.; March Collaborative Group. Prognostic Impact of Hpv-Associated P16-Expression and Smoking Status on Outcomes Following Radiotherapy for Oropharyngeal Cancer: The March-Hpv Project. Radiother. Oncol. 2018, 126, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Hafkamp, H.C.; Manni, J.J.; Haesvoets, A.; Voogd, A.C.; Schepers, M.; Bot, F.J.; Hopman, A.H.N.; Ramaekers, F.C.S.; Speel, E.J.M. Marked Differences in Survival Rate between Smokers and Nonsmokers with Hpv 16-Associated Tonsillar Carcinomas. Int. J. Cancer 2008, 122, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Kenny, P.J. Molecular Mechanisms Underlying Behaviors Related to Nicotine Addiction. Cold Spring Harb. Perspect. Med. 2013, 3, a012112. [Google Scholar] [CrossRef] [PubMed]
- Pistillo, F.; Fasoli, F.; Moretti, M.; McClure-Begley, T.; Zoli, M.; Marks, M.J.; Gotti, C. Chronic Nicotine and Withdrawal Affect Glutamatergic but Not Nicotinic Receptor Expression in the Mesocorticolimbic Pathway in a Region-Specific Manner. Pharmacol. Res. 2016, 103, 167–176. [Google Scholar] [CrossRef]
- Rollema, H.; Shrikhande, A.; Ward, K.M.; Tingley, F.D., 3rd; Coe, J.W.; O’Neill, B.T.; Tseng, E.; Wang, E.Q.; Mather, R.J.; Hurst, R.S.; et al. Pre-Clinical Properties of the Alpha4beta2 Nicotinic Acetylcholine Receptor Partial Agonists Varenicline, Cytisine and Dianicline Translate to Clinical Efficacy for Nicotine Dependence. Br. J. Pharmacol. 2010, 160, 334–345. [Google Scholar] [CrossRef]
- Rollema, H.; Hurst, R.S. The Contribution of Agonist and Antagonist Activities of Alpha4beta2* Nachr Ligands to Smoking Cessation Efficacy: A Quantitative Analysis of Literature Data. Psychopharmacology 2018, 235, 2479–2505. [Google Scholar] [CrossRef]
- Xi, Z.X. Preclinical Pharmacology, Efficacy and Safety of Varenicline in Smoking Cessation and Clinical Utility in High Risk Patients. Drug Healthc. Patient Saf. 2010, 2010, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Smulders, C.J.; Zwart, R.; Bermudez, I.; van Kleef, R.G.; Groot-Kormelink, P.J.; Vijverberg, H.P. Cholinergic Drugs Potentiate Human Nicotinic Alpha4beta2 Acetylcholine Receptors by a Competitive Mechanism. Eur. J. Pharmacol. 2005, 509, 97–108. [Google Scholar] [CrossRef]
- Lancaster, T.; Stead, L.F. Mecamylamine (a Nicotine Antagonist) for Smoking Cessation. Cochrane Database Syst. Rev. 2000, 2, CD001009. [Google Scholar] [CrossRef]
- Platek, A.J.; Jayaprakash, V.; Merzianu, M.; Platek, M.E.; Cohan, D.M.; Hicks, W.L.; Marimuthu, S.P.; Winslow, T.B.; Gupta, V.; Arshad, H.; et al. Smoking Cessation Is Associated with Improved Survival in Oropharynx Cancer Treated by Chemoradiation. Laryngoscope 2016, 126, 2733–2738. [Google Scholar] [CrossRef]
- Van Imhoff, L.C.R.; Kranenburg, G.G.J.; Macco, S.; Nijman, N.L.; van Overbeeke, E.J.; Wegner, I.; Grolman, W.; Pothen, A.J. Prognostic Value of Continued Smoking on Survival and Recurrence Rates in Patients with Head and Neck Cancer: A Systematic Review. Head Neck J. Sci. Spec. Head Neck 2016, 38, E2214–E2220. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Lee, H.H.; Kuei, C.H.; Lin, H.Y.; Lu, L.S.; Lee, F.P.; Chang, J.; Wang, J.Y.; Hsu, K.C.; Lin, Y.F. Nicotinic Acetylcholine Receptor Subunit Alpha-5 Promotes Radioresistance Via Recruiting E2f Activity in Oral Squamous Cell Carcinoma. J. Clin. Med. 2019, 8, 1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherl, C.; Schafer, R.; Schlabrakowski, A.; Tziridis, K.; Iro, H.; Wendler, O. Nicotinic Acetylcholine Receptors in Head and Neck Cancer and Their Correlation to Tumor Site and Progression. ORL J. Otorhinolaryngol. Relat. Spec. 2016, 78, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Nieh, S.; Jao, S.W.; Yang, C.Y.; Lin, Y.S.; Tseng, Y.H.; Liu, C.L.; Lee, T.Y.; Liu, T.Y.; Chu, Y.H.; Chen, S.F. Regulation of Tumor Progression Via the Snail-Rkip Signaling Pathway by Nicotine Exposure in Head and Neck Squamous Cell Carcinoma. Head Neck 2015, 37, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, T.; Tada, H.; Ibaragi, S.; Chen, C.; Sasano, T. Nicotine Exposure Induces the Proliferation of Oral Cancer Cells through the Alpha7 Subunit of the Nicotinic Acetylcholine Receptor. Biochem. Biophys. Res. Commun. 2019, 509, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, D.G.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Epithelial Cell Nicotinic Acetylcholine Receptor Expression in Head and Neck Squamous Cell Carcinoma Pathogenesis. Anticancer Res. 2007, 27, 835–839. [Google Scholar]
- Nakayama, H.; Numakawa, T.; Ikeuchi, T. Nicotine-Induced Phosphorylation of Akt through Epidermal Growth Factor Receptor and Src in Pc12h Cells. J. Neurochem. 2002, 83, 1372–1379. [Google Scholar] [CrossRef]
- Minna, J.D. Nicotine Exposure and Bronchial Epithelial Cell Nicotinic Acetylcholine Receptor Expression in the Pathogenesis of Lung Cancer. J. Clin. Investig. 2003, 111, 31–33. [Google Scholar] [CrossRef]
- Schuller, H.M.; Plummer, H.K., 3rd; Jull, B.A. Receptor-Mediated Effects of Nicotine and Its Nitrosated Derivative Nnk on Pulmonary Neuroendocrine Cells. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2003, 270, 51–58. [Google Scholar] [CrossRef]
- Schuller, H.M.; Jull, B.A.; Sheppard, B.J.; Plummer, H.K. Interaction of Tobacco-Specific Toxicants with the Neuronal Alpha(7) Nicotinic Acetylcholine Receptor and Its Associated Mitogenic Signal Transduction Pathway: Potential Role in Lung Carcinogenesis and Pediatric Lung Disorders. Eur. J. Pharmacol. 2000, 393, 265–277. [Google Scholar] [CrossRef]
- Schuller, H.M.; Castonguay, A.; Orloff, M.; Rossignol, G. Modulation of the Uptake and Metabolism of 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone by Nicotine in Hamster Lung. Cancer Res. 1991, 51, 2009–2014. [Google Scholar] [PubMed]
- Hecht, S.S. Tobacco Carcinogens, Their Biomarkers and Tobacco-Induced Cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Akopyan, G.; Bonavida, B. Understanding Tobacco Smoke Carcinogen Nnk and Lung Tumorigenesis. Int. J. Oncol. 2006, 29, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S.M.; Bornstein, S.; Li, Y.; Malkoski, S.P.; Wang, D.; Rustgi, A.K.; Kulesz-Martin, M.F.; Wang, X.J.; Lu, S.L. Tobacco-Specific Carcinogen Nitrosamine 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone Induces Akt Activation in Head and Neck Epithelia. Int. J. Oncol. 2011, 39, 1193–1198. [Google Scholar]
- Gankhuyag, N.; Lee, K.H.; Cho, J.Y. The Role of Nitrosamine (Nnk) in Breast Cancer Carcinogenesis. J. Mammary Gland Biol. Neoplasia 2017, 22, 159–170. [Google Scholar] [CrossRef]
- Jin, F.; Thaiparambil, J.; Donepudi, S.R.; Vantaku, V.; Piyarathna, D.W.B.; Maity, S.; Krishnapuram, R.; Putluri, V.; Gu, F.; Purwaha, P.; et al. Tobacco-Specific Carcinogens Induce Hypermethylation, DNA Adducts, DNA Damage in Bladder Cancer. Cancer Prev. Res. 2017, 10, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Lee, T.Y.; Ho, C.A.; Yang, C.Y.; Huang, W.Y.; Lin, Y.C.; Nieh, S.; Lin, Y.S.; Chen, S.F.; Lin, F.H. Exposure to Nicotine-Derived Nitrosamine Ketone and Arecoline Synergistically Facilitates Tumor Aggressiveness Via Overexpression of Epidermal Growth Factor Receptor and Its Downstream Signaling in Head and Neck Squamous Cell Carcinoma. PLoS ONE 2018, 13, e0201267. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Parker, J.S.; Karaca, G.; Wu, J.; Funkhouser, W.K.; Moore, D.; Butterfoss, D.; Xiang, D.; Zanation, A.; Yin, X.; et al. Molecular Classification of Head and Neck Squamous Cell Carcinomas Using Patterns of Gene Expression. Cancer Cell 2004, 5, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The Molecular Landscape of Head and Neck Cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Bragelmann, J.; et al. Integrative Analysis of Head and Neck Cancer Identifies Two Biologically Distinct Hpv and Three Non-Hpv Subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.H.; Jung, H.R.; Jung, A.R.; Lee, Y.C.; Kong, M.; Lee, J.S.; Eun, Y.G. Sox2 Activation Predicts Prognosis in Patients with Head and Neck Squamous Cell Carcinoma. Sci. Rep. 2018, 8, 1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprea, T.I.; Mestres, J. Drug Repurposing: Far Beyond New Targets for Old Drugs. AAPS J. 2012, 14, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug Repurposing in Oncology--Patient and Health Systems Opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer Drug Discovery by Repurposing: Teaching New Tricks to Old Dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy Plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.E.; Lingen, M.W.; Vokes, E.E. The Expanding Role of Systemic Therapy in Head and Neck Cancer. J. Clin. Oncol. 2004, 22, 1743–1752. [Google Scholar] [CrossRef]
- Kies, M.S.; Holsinger, F.C.; Lee, J.J.; William, W.N., Jr.; Glisson, B.S.; Lin, H.Y.; Lewin, J.S.; Ginsberg, L.E.; Gillaspy, K.A.; Massarelli, E.; et al. Induction Chemotherapy and Cetuximab for Locally Advanced Squamous Cell Carcinoma of the Head and Neck: Results from a Phase Ii Prospective Trial. J. Clin. Oncol. 2010, 28, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.Y.; Tseng, J.H.; Liu, K.H.; Lin, C.T.; Hsu, K.C.; Yang, J.M. Homopharma: A New Concept for Exploring the Molecular Binding Mechanisms and Drug Repurposing. BMC Genom. 2014, 15 (Suppl. S9), S8. [Google Scholar] [CrossRef] [Green Version]
- Kuperstein, I.; Bonnet, E.; Nguyen, H.A.; Cohen, D.; Viara, E.; Grieco, L.; Fourquet, S.; Calzone, L.; Russo, C.; Kondratova, M.; et al. Atlas of Cancer Signalling Network: A Systems Biology Resource for Integrative Analysis of Cancer Data with Google Maps. Oncogenesis 2015, 4, e160. [Google Scholar] [CrossRef] [Green Version]
- Pinero, J.; Ramirez-Anguita, J.M.; Sauch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The Disgenet Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar]
- Yi, H.; Wang, K.; Jin, J.F.; Jin, H.; Yang, L.; Zou, Y.; Du, B.; Liu, X. Elevated Adenylyl Cyclase 9 Expression Is a Potential Prognostic Biomarker for Patients with Colon Cancer. Med. Sci. Monit. 2018, 24, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavon, M.A.; Parreno, M.; Tellez-Gabriel, M.; Leon, X.; Arroyo-Solera, I.; Lopez, M.; Cespedes, M.V.; Casanova, I.; Gallardo, A.; Lopez-Pousa, A.; et al. Ckmt1 and Ncoa1 Expression as a Predictor of Clinical Outcome in Patients with Advanced-Stage Head and Neck Squamous Cell Carcinoma. Head Neck 2016, 38 (Suppl. S1), E1392–E1403. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Koslowski, M.; Paret, C.; Schmidt, M.; Tureci, O.; Sahin, U. Ncoa3 Is a Selective Co-Activator of Estrogen Receptor Alpha-Mediated Transactivation of Plac1 in Mcf-7 Breast Cancer Cells. BMC Cancer 2013, 13, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayamori, K.; Katsube, K.; Sakamoto, K.; Ohyama, Y.; Hirai, H.; Yukimori, A.; Ohata, Y.; Akashi, T.; Saitoh, M.; Harada, K.; et al. Notch3 Is Induced in Cancer-Associated Fibroblasts and Promotes Angiogenesis in Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0154112. [Google Scholar] [CrossRef]
- Knechtel, G.; Szkandera, J.; Stotz, M.; Hofmann, G.; Langsenlehner, U.; Krippl, P.; Samonigg, H.; Renner, W.; Langner, C.; Dehchamani, D.; et al. Single Nucleotide Polymorphisms in the Hypoxia-Inducible Factor-1 Gene and Colorectal Cancer Risk. Mol. Carcinog. 2010, 49, 805–809. [Google Scholar] [CrossRef]
- Borglykke, A.; Grarup, N.; Sparso, T.; Linneberg, A.; Fenger, M.; Jeppesen, J.; Hansen, T.; Pedersen, O.; Jorgensen, T. Genetic Variant Scl2a2 Is Associated with Risk of Cardiovascular Disease—Assessing the Individual and Cumulative Effect of 46 Type 2 Diabetes Related Genetic Variants. PLoS ONE 2012, 7, e50418. [Google Scholar] [CrossRef]
- Shi, H.; Lu, Y.; Du, J.; Du, W.C.; Ye, X.H.; Yu, X.F.; Ma, J.H.; Cheng, J.L.; Gao, Y.Q.; Cao, Y.Y.; et al. Application of Back Propagation Artificial Neural Network on Genetic Variants in Adiponectin Adipoq, Peroxisome Proliferator-Activated Receptor-Gamma, Retinoid X Receptor-Alpha Genes and Type 2 Diabetes Risk in a Chinese Han Population. Diabetes Technol. Ther. 2012, 14, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin Promotes Tumor Growth in Human Hepatocellular Cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef]
- Hambek, M.; Werner, C.; Baghi, M.; Gstottner, W.; Knecht, R. Prestimulation of Head and Neck Cancer Cells with Growth Factors Enhances Treatment Efficacy. Anticancer Res. 2006, 26, 1091–1095. [Google Scholar] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy Quantitative Assessment of Genome Editing by Sequence Trace Decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.C.; Chen, Y.F.; Lin, S.R.; Yang, J.M. Igemdock: A Graphical Environment of Enhancing Gemdock Using Pharmacological Interactions and Post-Screening Analysis. BMC Bioinform. 2011, 12 (Suppl. S1), S33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.M.; Chen, C.C. Gemdock: A Generic Evolutionary Method for Molecular Docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Neuchrist, C.; Erovic, B.M.; Handisurya, A.; Fischer, M.B.; Steiner, G.E.; Hollemann, D.; Gedlicka, C.; Saaristo, A.; Burian, M. Vascular Endothelial Growth Factor C and Vascular Endothelial Growth Factor Receptor 3 Expression in Squamous Cell Carcinomas of the Head and Neck. Head Neck 2003, 25, 464–474. [Google Scholar] [CrossRef]
- Quesada-Calvo, F.; Massot, C.; Bertrand, V.; Longuespee, R.; Bletard, N.; Somja, J.; Mazzucchelli, G.; Smargiasso, N.; Baiwir, D.; de Pauw-Gillet, M.C.; et al. Olfm4, Kng1 and Sec24c Identified by Proteomics and Immunohistochemistry as Potential Markers of Early Colorectal Cancer Stages. Clin. Proteom. 2017, 14, 9. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.W.; Choi, K.Y.; Do, S.M. Phospholipase D Meets Wnt Signaling: A New Target for Cancer Therapy. Cancer Res. 2011, 71, 293–297. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.C.; Shahab, S.; Weed, S.A. Actin Cytoskeletal Mediators of Motility and Invasion Amplified and Overexpressed in Head and Neck Cancer. Clin. Exp. Metastasis 2008, 25, 289–304. [Google Scholar] [CrossRef]
- Jehan, Z.; Bavi, P.; Sultana, M.; Abubaker, J.; Bu, R.; Hussain, A.; Alsbeih, G.; Al-Sanea, N.; Abduljabbar, A.; Ashari, L.H.; et al. Frequent Pik3ca Gene Amplification and Its Clinical Significance in Colorectal Cancer. J. Pathol. 2009, 219, 337–346. [Google Scholar] [CrossRef]
- Cao, Y.; Bryan, T.M.; Reddel, R.R. Increased Copy Number of the Tert and Terc Telomerase Subunit Genes in Cancer Cells. Cancer Sci. 2008, 99, 1092–1099. [Google Scholar] [CrossRef]
- Leinonen, H.M.; Kansanen, E.; Polonen, P.; Heinaniemi, M.; Levonen, A.L. Dysregulation of the Keap1-Nrf2 Pathway in Cancer. Biochem. Soc. Trans. 2015, 43, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.; Mak, C.S.; Yung, M.M.; Leung, T.H.; Wong, O.G.; Cheung, A.N.; Ngan, H.Y. Ampk Activators Suppress Cervical Cancer Cell Growth through Inhibition of Dvl3 Mediated Wnt/Beta-Catenin Signaling Activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, L.; Peiffer, D.A.; Zhou, L.; Chan, O.T.; Bibikova, M.; Wickham-Garcia, E.; Lu, S.H.; Zhan, Q.; Wang-Rodriguez, J.W.; et al. Genomic Profiling of 766 Cancer-Related Genes in Archived Esophageal Normal and Carcinoma Tissues. Int. J. Cancer 2008, 122, 2249–2254. [Google Scholar] [CrossRef]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic Variation at Mecom, Tert, Jak2 and Hbs1l-Myb Predisposes to Myeloproliferative Neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Lee, C.H.; Chuang, Y.H.; Lee, J.Y.; Chiu, Y.Y.; Lee, Y.H.W.; Jong, Y.J.; Hwang, J.K.; Huang, S.H.; Chen, L.C.; et al. Membrane Protein-Regulated Networks across Human Cancers. Nat. Commun. 2019, 10, 3131. [Google Scholar] [CrossRef] [Green Version]
- Gaude, E.; Frezza, C. Tissue-Specific and Convergent Metabolic Transformation of Cancer Correlates with Metastatic Potential and Patient Survival. Nat. Commun. 2016, 7, 13041. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. Kegg: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The Cbio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the Cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. Gistic2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale Crispr-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.C.; Tam, K.W.; Liu, W.N.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Lee, A.W.; Yang, J.M.; Lin, C.L.; et al. Crispr/Cas9 Genome Editing of Epidermal Growth Factor Receptor Sufficiently Abolished Oncogenicity in Anaplastic Thyroid Cancer. Dis. Markers 2018, 2018, 3835783. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chow, J.M.; Hsieh, Y.Y.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Shabangu, B.M.; Lee, C.H. Hdac1,2 Knock-out and Hdaci Induced Cell Apoptosis in Imatinib-Resistant K562 Cells. Int. J. Mol. Sci. 2019, 20, 2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.L.; Tsai, M.L.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Huang, L.C.; Lee, C.H. Hdac1 and Hdac2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, Y.-H.; Lee, C.-H.; Lin, C.-Y.; Liu, C.-L.; Huang, S.-H.; Lee, J.-Y.; Chiu, Y.-Y.; Lee, J.-C.; Yang, J.-M. An Integrated Genomic Strategy to Identify CHRNB4 as a Diagnostic/Prognostic Biomarker for Targeted Therapy in Head and Neck Cancer. Cancers 2020, 12, 1324. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051324
Chuang Y-H, Lee C-H, Lin C-Y, Liu C-L, Huang S-H, Lee J-Y, Chiu Y-Y, Lee J-C, Yang J-M. An Integrated Genomic Strategy to Identify CHRNB4 as a Diagnostic/Prognostic Biomarker for Targeted Therapy in Head and Neck Cancer. Cancers. 2020; 12(5):1324. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051324
Chicago/Turabian StyleChuang, Yi-Hsuan, Chia-Hwa Lee, Chun-Yu Lin, Chia-Lin Liu, Sing-Han Huang, Jung-Yu Lee, Yi-Yuan Chiu, Jih-Chin Lee, and Jinn-Moon Yang. 2020. "An Integrated Genomic Strategy to Identify CHRNB4 as a Diagnostic/Prognostic Biomarker for Targeted Therapy in Head and Neck Cancer" Cancers 12, no. 5: 1324. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051324