Tumor Cell Associated Hyaluronan-CD44 Signaling Promotes Pro-Tumor Inflammation in Breast Cancer

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
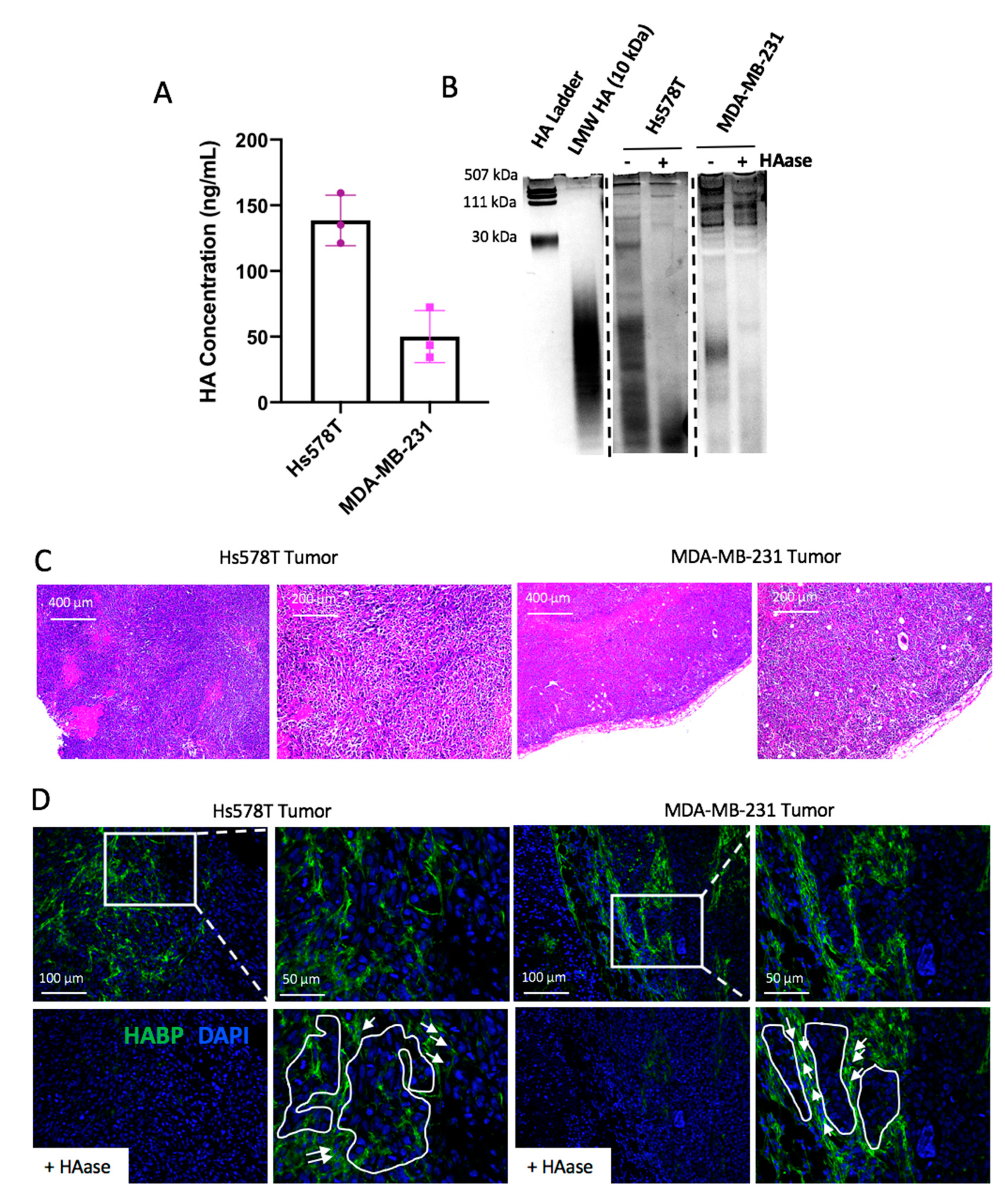
2.1. Hyaluronan Synthase 2 Expression in Tumor Cells is Associated with the Triple Negative Breast Cancer Subtype
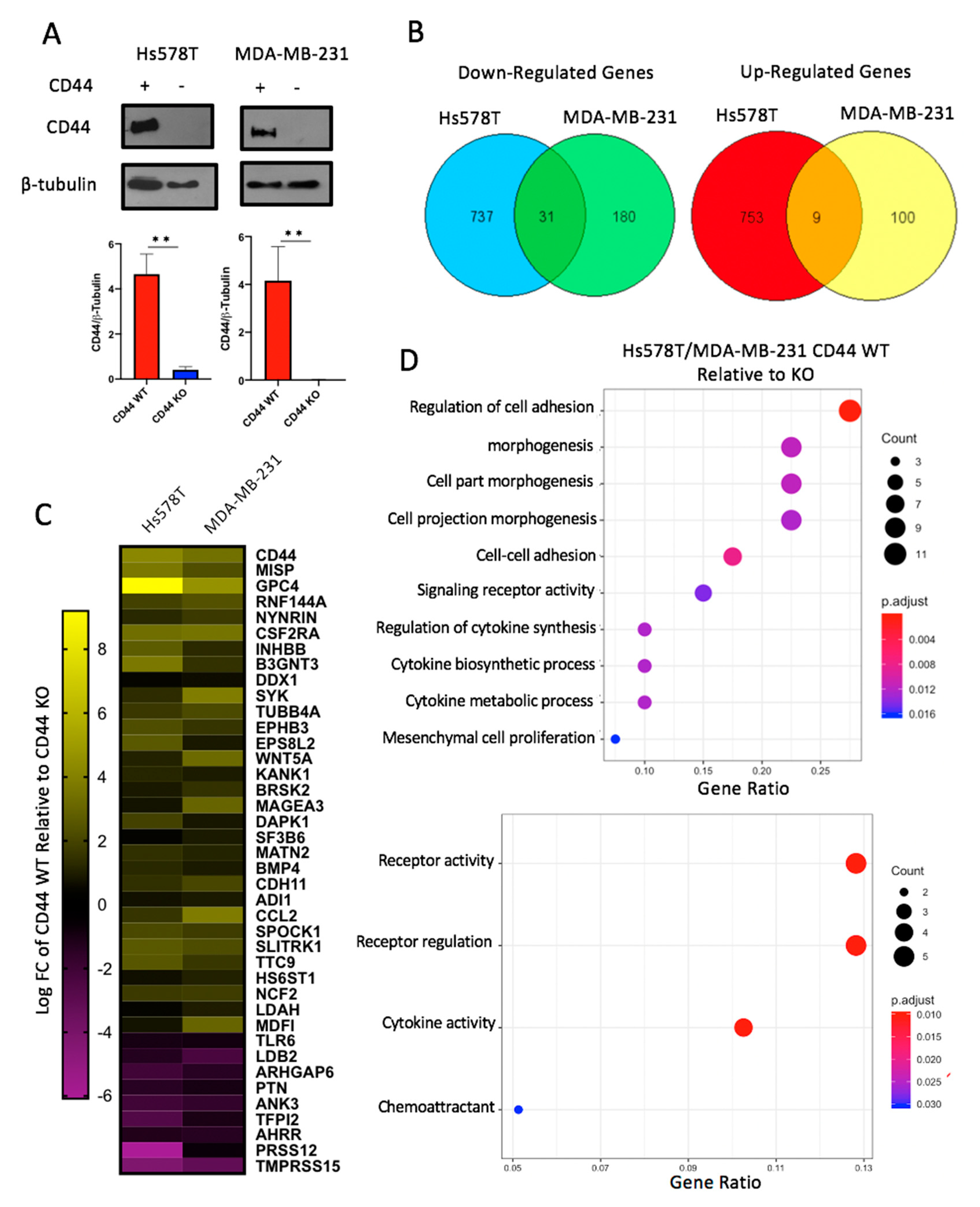
2.2. CD44 Deletion in Breast Cancer Cell Lines Decreases Expression of Genes Involved in Cellular Adhesion and Cytokine Activity
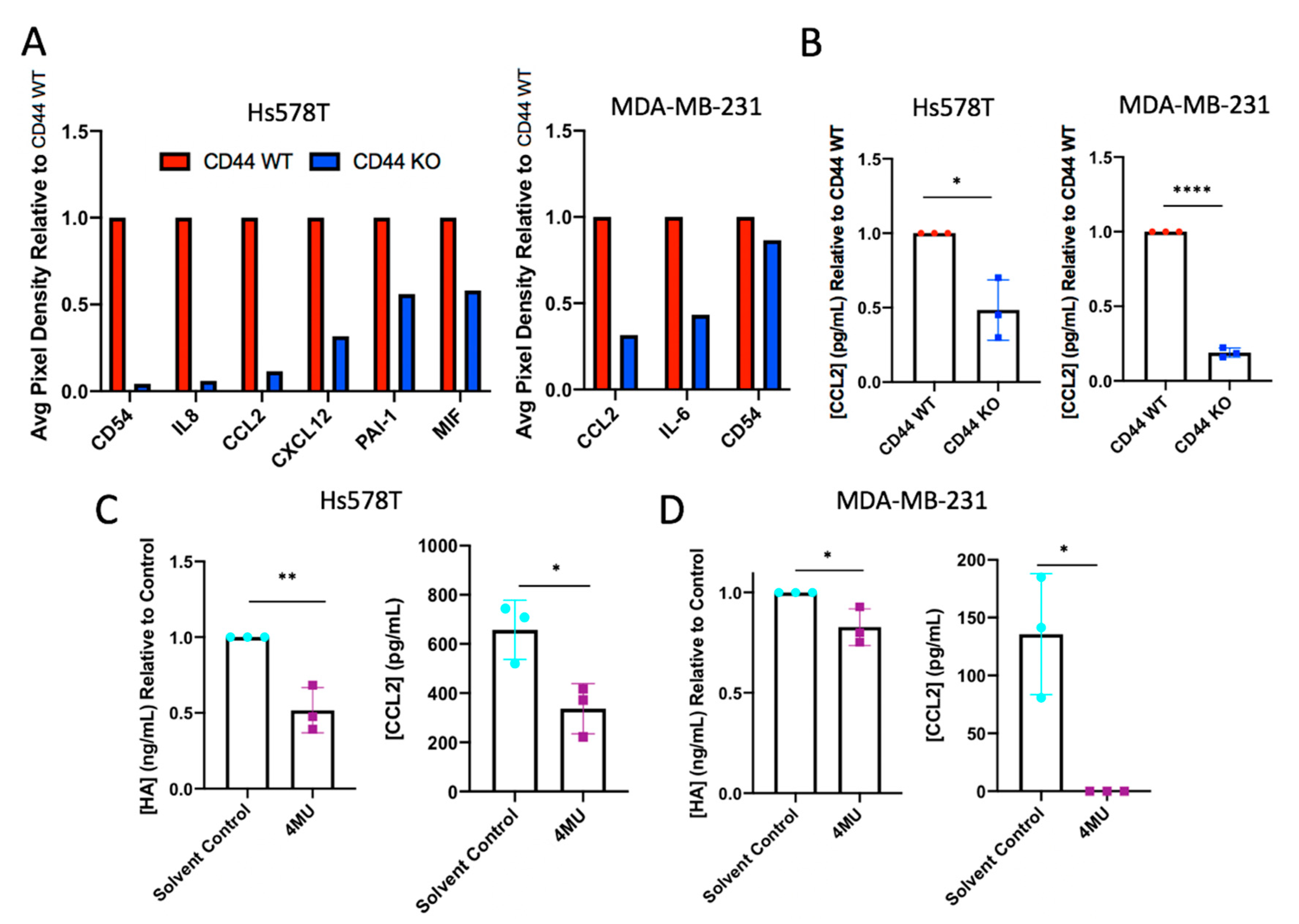
2.3. CD44 Knockout or Hyaluronan Depletion from Breast Cancer Cells Decreases Tumor Cell Production of Inflammatory Cytokines
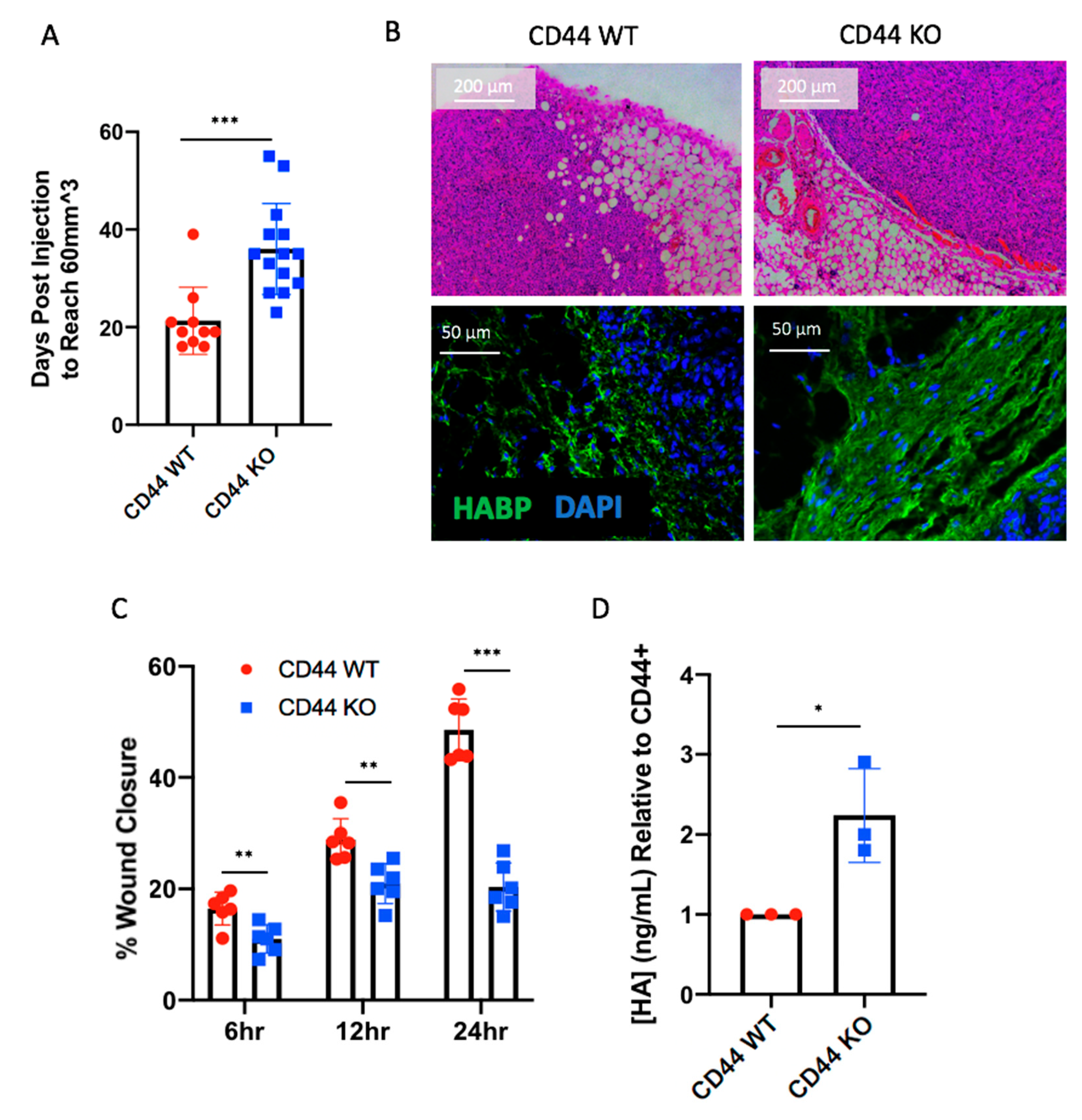
2.4. CD44 Deletion in Breast Cancer Cells Delays Early Tumor Formation
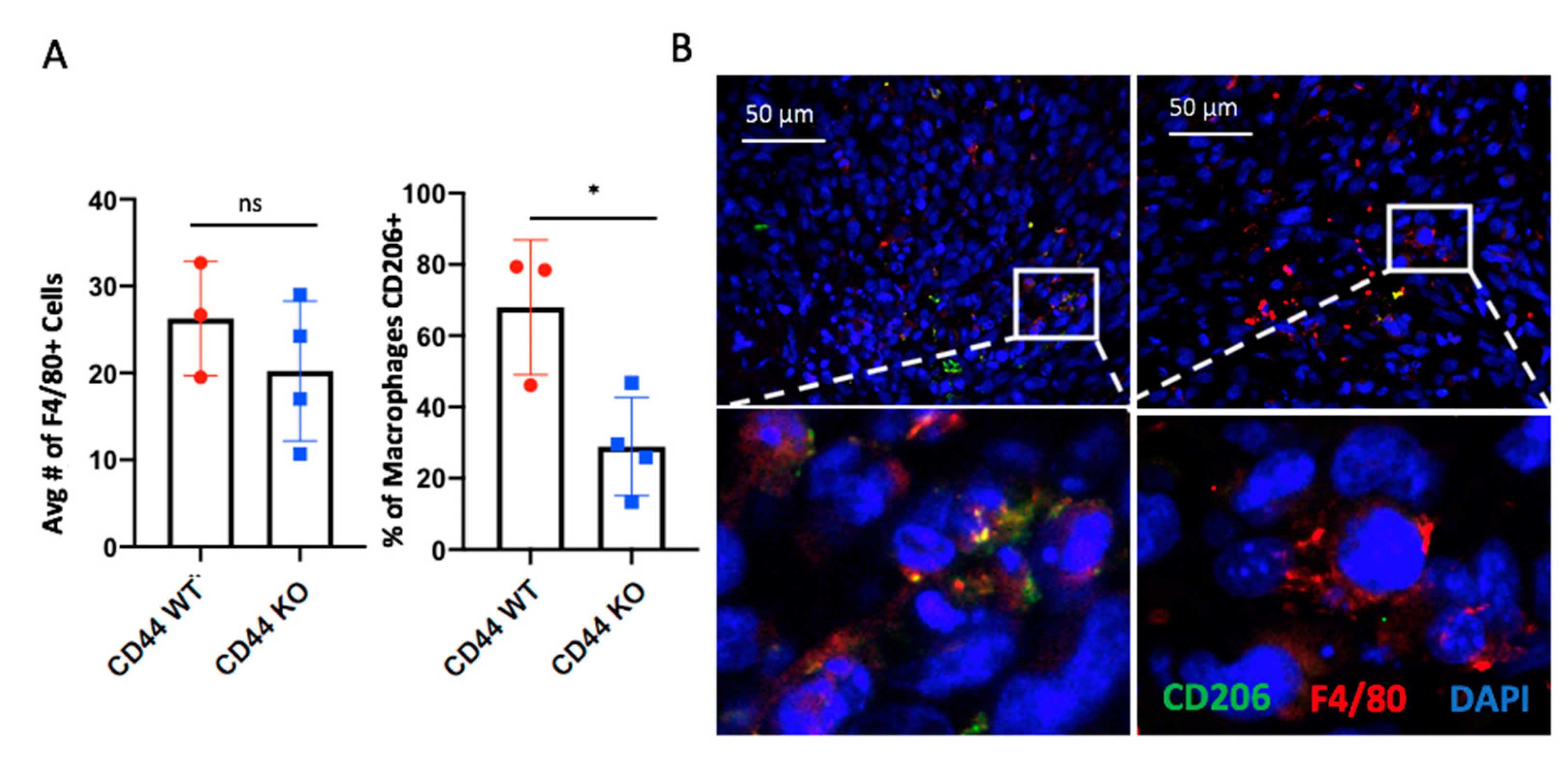
2.5. CD44 Deletion in Hs578T Tumors Decreases the Number of Infiltrating CD206+ Macrophages
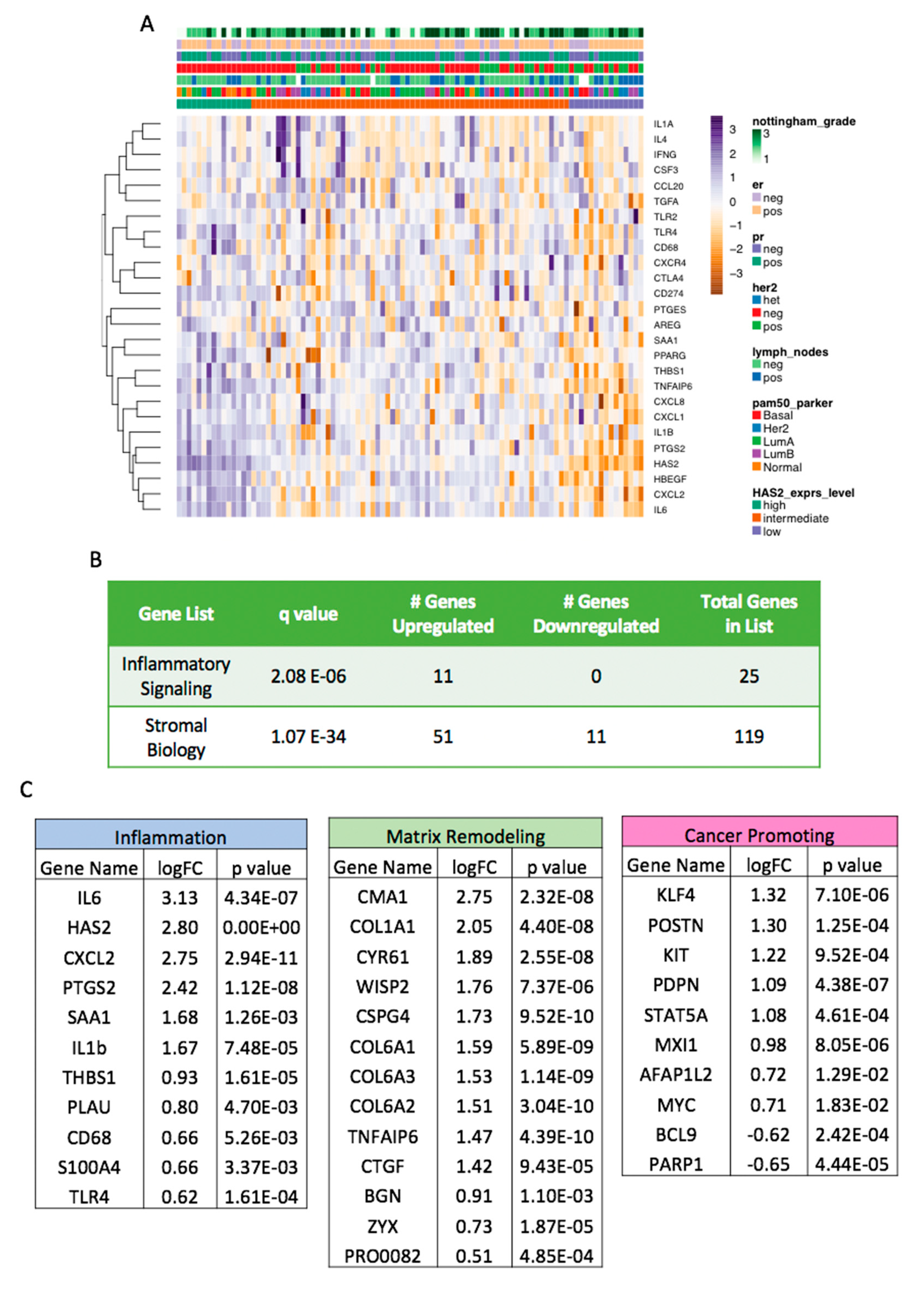
2.6. Increased HAS2 Gene Expression is Associated with Inflammatory and Stromal Biology Gene Signatures in Human Cases of Breast Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Mice
4.3. Nanostring Analysis of Human Breast Cancer Samples
4.4. CD44 Knockout vis CRISPR/Cas9
4.4.1. Target Sequence Design (5′ to 3′)
4.4.2. gRNA Cloning Vector
4.4.3. Cas9 Plasmid and Lentivirus Generation
4.4.4. Cas9 Transduction
4.4.5. CD44 Knockout
4.5. RNA Extraction and Sequencing
RNA-Seq Differential Gene Expression and Pathway Analysis
4.6. Inflammatory Cytokine Array
4.7. ELISAs
4.8. Flow Cytometry
4.9. Molecular Mass Analysis of HA
4.10. MTT Assay
4.11. Inhibition of HA Synthesis
4.12. Scratch Assay
4.13. In Vivo Experiments
4.13.1. Effects of CD44 on Animal Survival
4.13.2. Effects of CD44 on Early Tumor Formation
4.13.3. Effects of CD44 on Lung Colonization
4.14. Immunofluorescence
4.15. Microscope Imaging
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, B.A.; Sherman, R.L.; Howlader, N.; Jemal, A.; Ryerson, A.B.; Henry, K.A.; Boscoe, F.P.; Cronin, K.A.; Lake, A.; Noone, A.-M.; et al. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. JNCI J. Natl. Cancer Inst. 2015. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics. CA Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, G.M.; Quah, S.; Sampath, P. Cancer: The dark side of wound healing. FEBS J. 2018, 285, 4516–4534. [Google Scholar] [CrossRef]
- Schwertfeger, K.L.; Cowman, M.K.; Telmer, P.G.; Turley, E.A.; McCarthy, J.B. Hyaluronan, inflammation, and breast cancer progression. Front. Immunol. 2015, 6, 236. [Google Scholar] [CrossRef]
- Auvinen, P.; Tammi, R.; Parkkinen, J.; Tammi, M.; Ågren, U.; Johansson, R.; Hirvikoski, P.; Eskelinen, M.; Kosma, V.-M. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 2000, 156, 529–536. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Brown, T.J.; Heldin, P. Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells. Int. J. Cancer 2007, 120, 2557–2567. [Google Scholar] [CrossRef]
- Lien, H.-C.; Lee, Y.-H.; Jeng, Y.-M.; Lin, C.-H.; Lu, Y.-S.; Yao, Y.-T. Differential expression of hyaluronan synthase 2 in breast carcinoma and its biological significance. Histopathology 2014, 65, 328–339. [Google Scholar] [CrossRef]
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: Their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.W.; Singleton, P.A.; Diedrich, F.; Stern, R.; Gilad, E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J. Biol. Chem. 2004. [Google Scholar] [CrossRef]
- Lepperdinger, G.; Strobl, B.; Kreil, G. HYAL2, a human gene expressed in many cells, encodes a lysosomal hyaluronidase with a novel type of specificity. J. Biol. Chem. 1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stair-Nawy, S.; Csóka, A.B.; Stern, R. Hyaluronidase expression in human skin fibroblasts. Biochem. Biophys. Res. Commun. 1999, 266, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Afify, A.; McNiel, M.A.; Braggin, J.; Bailey, H.; Paulino, A.F. Expression of CD44s, CD44v6, and hyaluronan across the spectrum of normal-hyperplasia-carcinoma in breast. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikström, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013. [Google Scholar] [CrossRef]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef]
- Tammi, M.I.; Oikari, S.; Pasonen-Seppänen, S.; Rilla, K.; Auvinen, P.; Tammi, R.H. Activated hyaluronan metabolism in the tumor matrix—Causes and consequences. Matrix Biol. 2019, 78–79, 147–164. [Google Scholar] [CrossRef]
- Ooki, T.; Murata-Kamiya, N.; Takahashi-Kanemitsu, A.; Wu, W.; Hatakeyama, M. High-molecular-weight hyaluronan is a hippo pathway ligand directing cell density-dependent growth inhibition via PAR1b. Dev. Cell 2019, 49, 590–604. [Google Scholar] [CrossRef]
- Gao, F.; Liu, Y.; He, Y.; Yang, C.; Wang, Y.; Shi, X.; Wei, G. Hyaluronan oligosaccharides promote excisional wound healing through enhanced angiogenesis. Matrix Biol. 2010, 29, 107–116. [Google Scholar] [CrossRef]
- Yamawaki, H.; Hirohata, S.; Miyoshi, T.; Takahashi, K.; Ogawa, H.; Shinohata, R.; Demircan, K.; Kusachi, S.; Yamamoto, K.; Ninomiya, Y. Hyaluronan receptors involved in cytokine induction in monocytes. Glycobiology 2008, 19, 83–92. [Google Scholar] [CrossRef]
- Miletti-González, K.E.; Murphy, K.; Kumaran, M.N.; Ravindranath, A.K.; Wernyj, R.P.; Kaur, S.; Miles, G.D.; Lim, E.; Chan, R.; Chekmareva, M.; et al. Identification of function for CD44 intracytoplasmic domain (CD44-ICD): Modulation of matrix metalloproteinase 9 (mmp-9) transcription via novel promoter response element. J. Biol. Chem. 2012, 287, 18995–19007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orian-Rousseau, V.; Sleeman, J. Chapter nine—CD44 is a multidomain signaling platform that integrates extracellular matrix cues with growth factor and cytokine signals. In Hyaluronan Signaling and Turnover; Simpson, M.A., Heldin, P., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 123, pp. 231–254. [Google Scholar]
- Freeman, S.A.; Vega, A.; Riedl, M.; Collins, R.F.; Ostrowski, P.P.; Woods, E.C.; Bertozzi, C.R.; Tammi, M.I.; Lidke, D.S.; Johnson, P.; et al. Transmembrane pickets connect cyto- and pericellular skeletons forming barriers to receptor engagement. Cell 2018, 172, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udabage, L.; Brownlee, G.R.; Nilsson, S.K.; Brown, T.J. The over-expression of HAS2, hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp. Cell Res. 2005, 310, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xiang, T.; Li, H.; Li, Q.; Yang, B.; Huang, J.; Zhang, X.; Shi, Y.; Tan, J.; Ren, G. Hyaluronan synthase 2 overexpression is correlated with the tumorigenesis and metastasis of human breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12101–12114. [Google Scholar]
- Wu, M.; Cao, M.; He, Y.; Liu, Y.; Yang, C.; Du, Y.; Wang, W.; Gao, F. A novel role of low molecular weight hyaluronan in breast cancer metastasis. FASEB J. 2015. [Google Scholar] [CrossRef]
- Ryoo, I.; Choi, B.; Ku, S.-K.; Kwak, M.-K. High CD44 expression mediates P62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- Morath, I.; Hartmann, T.N.; Orian-Rousseau, V. CD44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173. [Google Scholar] [CrossRef]
- Pályi-Krekk, Z.; Barok, M.; Isola, J.; Tammi, M.; Szöllo“si, J.; Nagy, P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur. J. Cancer 2007, 43, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Mohammadalipour, A.; Burdick, M.M.; Tees, D.F.J. Deformability of breast cancer cells in correlation with surface markers and cell rolling. FASEB J. 2018, 32, 1806–1817. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; He, Y.; Zhang, H.; Liu, Y.; Wang, W.; Du, Y.; Gao, F. Selective killing of breast cancer cells expressing activated CD44 using CD44 ligand-coated nanoparticles in vitro and in vivo. Oncotarget 2015. [Google Scholar] [CrossRef] [Green Version]
- Klingbeil, P.; Natrajan, R.; Everitt, G.; Vatcheva, R.; Marchio, C.; Palacios, J.; Buerger, H.; Reis-Filho, J.S.; Isacke, C.M. CD44 is overexpressed in basal-like breast cancers but is not a driver of 11p13 amplification. Breast Cancer Res. Treat. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernert, B.; Porsch, H.; Heldin, P. Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1). J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.-F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Lei, Z.; Zhao, J.; Gong, W.; Liu, J.; Chen, Z.; Liu, Y.; Li, D.; Yuan, Y.; Zhang, G.-M.; et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007, 252, 86–92. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Toi, M.; Saji, H.; Muta, M.; Bando, H.; Kuroi, K.; Koike, M.; Inadera, H.; Matsushima, K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 2000, 6, 3282–3289. [Google Scholar]
- Thomas, N.K.; Brown, T.J. ABC transporters do not contribute to extracellular translocation of hyaluronan in human breast cancer in vitro. Exp. Cell Res. 2010, 316, 1241–1253. [Google Scholar] [CrossRef]
- Hanioka, N.; Iwabu, H.; Hanafusa, H.; Nakada, S.; Narimatsu, S. Expression and inducibility of UDP-glucuronosyltransferase 1As in MCF-7 human breast carcinoma cells. Basic Clin. Pharmacol. Toxicol. 2012, 110, 253–258. [Google Scholar] [CrossRef]
- Karn, T.; Pusztai, L.; Holtrich, U.; Iwamoto, T.; Shiang, C.Y.; Schmidt, M.; Müller, V.; Solbach, C.; Gaetje, R.; Hanker, L.; et al. Homogeneous datasets of triple negative breast cancers enable the identification of novel prognostic and predictive signatures. PLoS ONE 2011, 6, e28403. [Google Scholar] [CrossRef] [Green Version]
- Valeta-Magara, A.; Gadi, A.; Volta, V.; Walters, B.; Arju, R.; Giashuddin, S.; Zhong, H.; Schneider, R.J. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network. Cancer Res. 2019, 79, 3360–3371. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalaf, H.H.; Al-Harbi, B.; Al-Sayed, A.; Arafah, M.; Tulbah, A.; Jarman, A.; Al-Mohanna, F.; Aboussekhra, A. Interleukin-8 activates breast cancer-associated adipocytes and promotes their angiogenesis- and tumorigenesis-promoting effects. Mol. Cell. Biol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhou, B.P. Inflammation: A driving force speeds cancer metastasis. Cell Cycle 2009, 8, 3267–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anttila, M.A.; Tammi, R.H.; Tammi, M.I.; Syrjänen, K.J.; Saarikoski, S.V.; Kosma, V.-M. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000, 60, 150–155. [Google Scholar]
- Ropponen, K.; Tammi, M.; Parkkinen, J.; Eskelinen, M.; Tammi, R.; Lipponen, P.; Ågren, U.; Alhava, E.; Kosma, V.-M. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 1998, 58, 342–347. [Google Scholar]
- Setälä, L.P.; Tammi, M.I.; Tammi, R.H.; Eskelinen, M.J.; Lipponen, P.K.; Ågren, U.M.; Parkkinen, J.; Alhava, E.M.; Kosma, V.-M. Hyaluronan expression in gastric cancer cells is associated with local and nodal spread and reduced survival rate. Br. J. Cancer 1999, 79, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Pirinen, R.; Tammi, R.; Tammi, M.; Hirvikoski, P.; Parkkinen, J.J.; Johansson, R.; Böhm, J.; Hollmén, S.; Kosma, V.-M. Prognostic value of hyaluronan expression in non-small-cell lung cancer: Increased stromal expression indicates unfavorable outcome in patients with adenocarcinoma. Int. J. Cancer 2001, 95, 12–17. [Google Scholar] [CrossRef]
- Bhilocha, S.; Amin, R.; Pandya, M.; Yuan, H.; Tank, M.; LoBello, J.; Shytuhina, A.; Wang, W.; Wisniewski, H.-G.; de la Motte, C.; et al. Agarose and polyacrylamide gel electrophoresis methods for molecular mass analysis of 5- to 500-KDa hyaluronan. Anal. Biochem. 2011, 417, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Tempero, M.A.; van Cutsem, E.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulia, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109–301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20)+ nab-paclitaxel/gemcitabine (AG) in patients (Pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinom. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Wolf, K.J.; Shukla, P.; Springer, K.; Lee, S.; Coombes, J.D.; Choy, C.J.; Kenny, S.J.; Xu, K.; Kumar, S. A mode of cell adhesion and migration facilitated by CD44-dependent microtentacles. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [PubMed]
- Bartolazzi, A.; Nocks, A.; Aruffo, A.; Spring, F.; Stamenkovic, I. Glycosylation of CD44 is implicated in CD44-mediated cell adhesion to hyaluronan. J. Cell Biol. 1996. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Glynn, D.J.; Hodson, L.J.; Huo, C.; Britt, K.; Thompson, E.W.; Woolford, L.; Evdokiou, A.; Pollard, J.W.; Robertson, S.A.; et al. CCL2-Driven inflammation increases mammary gland stromal density and cancer susceptibility in a transgenic mouse model. Breast Cancer Res. 2017, 19, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karalis, T.T.; Heldin, P.; Vynios, D.H.; Neill, T.; Buraschi, S.; Iozzo, R.V.; Karamanos, N.K.; Skandalis, S.S. Tumor-suppressive functions of 4-MU on breast cancer cells of different ER status: Regulation of hyaluronan/HAS2/CD44 and specific matrix effectors. Matrix Biol. 2019, 78, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Tolg, C.; Hamilton, S.R.; Nakrieko, K.-A.; Kooshesh, F.; Walton, P.; McCarthy, J.B.; Bissell, M.J.; Turley, E.A. Rhamm−/− fibroblasts are defective in CD44-mediated ERK1,2 motogenic signaling, leading to defective skin wound repair. J. Cell Biol. 2006, 175, 1017–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, R.F.; Legg, J.W.; Isacke, C.M. The role of the CD44 transmembrane and cytoplasmic domains in Co-ordinating adhesive and signalling events. J. Cell Sci. 2004, 117, 373–380. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, I.; Kawano, Y.; Murakami, D.; Sasayama, T.; Araki, N.; Miki, T.; Wong, A.J.; Saya, H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J. Cell Biol. 2001, 155, 755–762. [Google Scholar] [CrossRef]
- Lavender, N.; Yang, J.; Chen, S.-C.; Sai, J.; Johnson, C.A.; Owens, P.; Ayers, G.D.; Richmond, A. The Yin/Yan of CCL2: A minor role in neutrophil anti-tumor activity in vitro but a major role on the outgrowth of metastatic breast cancer lesions in the lung in vivo. BMC Cancer 2017, 17, 88. [Google Scholar] [CrossRef] [Green Version]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Enninga, E.A.L.; Chatzopoulos, K.; Butterfield, J.T.; Sutor, S.L.; Leontovich, A.A.; Nevala, W.K.; Flotte, T.J.; Markovic, S.N. CD206-positive myeloid cells bind galectin-9 and promote a tumor-supportive microenvironment. J. Pathol. 2018, 245, 468–477. [Google Scholar] [CrossRef]
- Shen, L.; Li, H.; Shi, Y.; Wang, D.; Gong, J.; Xun, J.; Zhou, S.; Xiang, R.; Tan, X. M2 tumour-associated macrophages contribute to tumour progression via legumain remodelling the extracellular matrix in diffuse large B cell lymphoma. Sci. Rep. 2016, 6, 30347. [Google Scholar] [CrossRef] [PubMed]
- Tiainen, S.; Masarwah, A.; Oikari, S.; Rilla, K.; Hämäläinen, K.; Sudah, M.; Sutela, A.; Vanninen, R.; Ikonen, J.; Tammi, R.; et al. Tumor microenvironment and breast cancer survival: Combined effects of breast fat, M2 macrophages and hyaluronan create a dismal prognosis. Breast Cancer Res. Treat. 2020, 179, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Spadea, A.; Rios de la Rosa, J.M.; Tirella, A.; Ashford, M.B.; Williams, K.J.; Stratford, I.J.; Tirelli, N.; Mehibel, M. Evaluating the efficiency of hyaluronic acid for tumor targeting via CD44. Mol. Pharm. 2019, 16, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Rios de la Rosa, J.M.; Pingrajai, P.; Pelliccia, M.; Spadea, A.; Lallana, E.; Gennari, A.; Stratford, I.J.; Rocchia, W.; Tirella, A.; Tirelli, N. Binding and internalization in receptor-targeted carriers: The complex role of CD44 in the uptake of hyaluronic acid-based nanoparticles (SiRNA delivery). Adv. Healthc. Mater. 2019, 8, 1901182. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Mouse models for tumor metastasis. In Rational Drug Design: Methods and Protocols; Zheng, Y., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 221–228. [Google Scholar]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiratsuka, S.; Ishibashi, S.; Tomita, T.; Watanabe, A.; Akashi-Takamura, S.; Murakami, M.; Kijima, H.; Miyake, K.; Aburatani, H.; Maru, Y. Primary tumours modulate innate immune signalling to create pre-metastatic vascular hyperpermeability foci. Nat. Commun. 2013, 4, 1853. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8–serum amyloid A3–TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R.; Badve, S.; Nakshatri, H. CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006, 8, R59. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Hu, H.; Tan, H.; Chow, L.W.C.; Yip, A.Y.S.; Loo, W.T.Y. Relationship of CD44+CD24−/low breast cancer stem cells and axillary lymph node metastasis. J. Transl. Med. 2012, 10, S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K.; et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012, 72, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Cordoba, S.; Meneghini, E.; Sant, M.; Iorio, V.M.; Sfondrini, L.; Paolini, B.; Agresti, R.; Tagliabue, E.; Bianchi, F. Decoding immune heterogeneity of triple negative breast cancer and its association with systemic inflammation. Cancers 2019, 11, 911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2010. [Google Scholar]
- Yang, L.; Mali, P.; Kim-Kiselak, C.; Church, G. CRISPR-CAS-mediated targeted genome editing in human cells. In Gene Correction: Methods and Protocols; Storici, F., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 245–267. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2018, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes Among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2018, 47, D330–D338. [Google Scholar]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, N.; Idiyatullin, D.; Corum, C.; Weber, J.; Garwood, M.; Sachdev, D. SWIFT MRI enhances detection of breast cancer metastasis to the lung. Magn. Reson. Med. 2015, 73, 1812–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Witschen, P.M.; Chaffee, T.S.; Brady, N.J.; Huggins, D.N.; Knutson, T.P.; LaRue, R.S.; Munro, S.A.; Tiegs, L.; McCarthy, J.B.; Nelson, A.C.; et al. Tumor Cell Associated Hyaluronan-CD44 Signaling Promotes Pro-Tumor Inflammation in Breast Cancer. Cancers 2020, 12, 1325. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051325
Witschen PM, Chaffee TS, Brady NJ, Huggins DN, Knutson TP, LaRue RS, Munro SA, Tiegs L, McCarthy JB, Nelson AC, et al. Tumor Cell Associated Hyaluronan-CD44 Signaling Promotes Pro-Tumor Inflammation in Breast Cancer. Cancers. 2020; 12(5):1325. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051325
Chicago/Turabian StyleWitschen, Patrice M., Thomas S. Chaffee, Nicholas J. Brady, Danielle N. Huggins, Todd P. Knutson, Rebecca S. LaRue, Sarah A. Munro, Lyubov Tiegs, James B. McCarthy, Andrew C. Nelson, and et al. 2020. "Tumor Cell Associated Hyaluronan-CD44 Signaling Promotes Pro-Tumor Inflammation in Breast Cancer" Cancers 12, no. 5: 1325. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051325