From CENTRAL to SENTRAL (SErum aNgiogenesis cenTRAL): Circulating Predictive Biomarkers to Anti-VEGFR Therapy

 , , ,
, , , 
 , , add
Show full author list
, , add
Show full author list
Abstract
:1. Introduction
2. Results
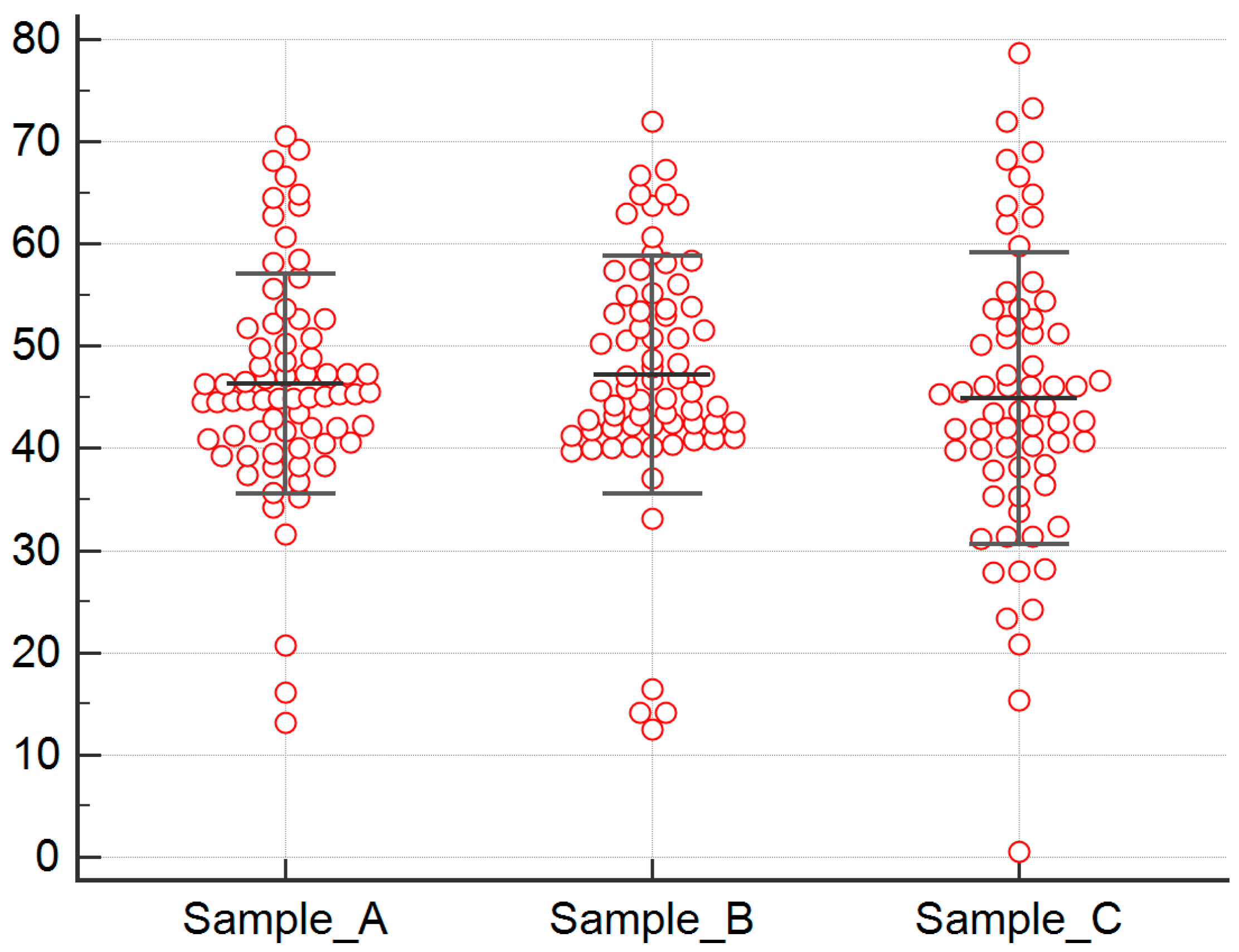
2.1. Serum Biomarkers’ Analysis
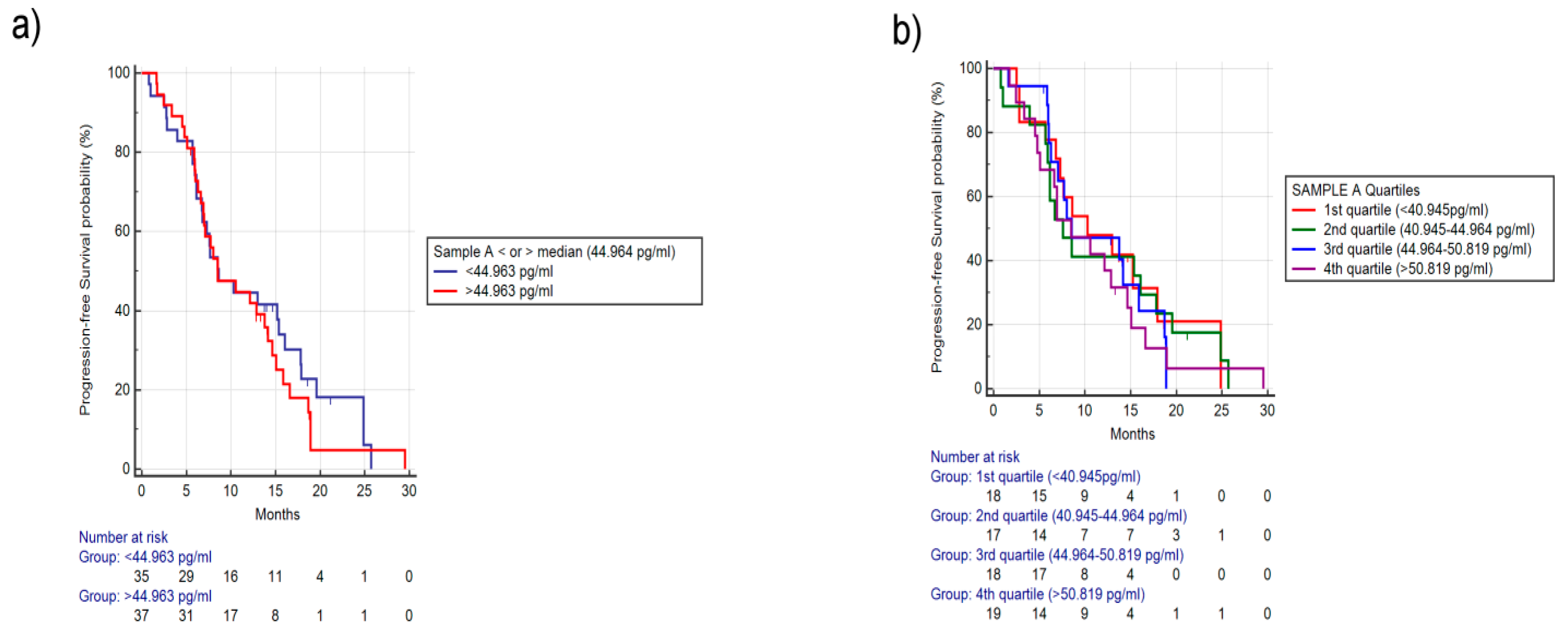
2.1.1. Analysis for Sample A FGF-2 Levels
2.1.2. Analysis for Differences in FGF-2 Concentrations between Samples A and B
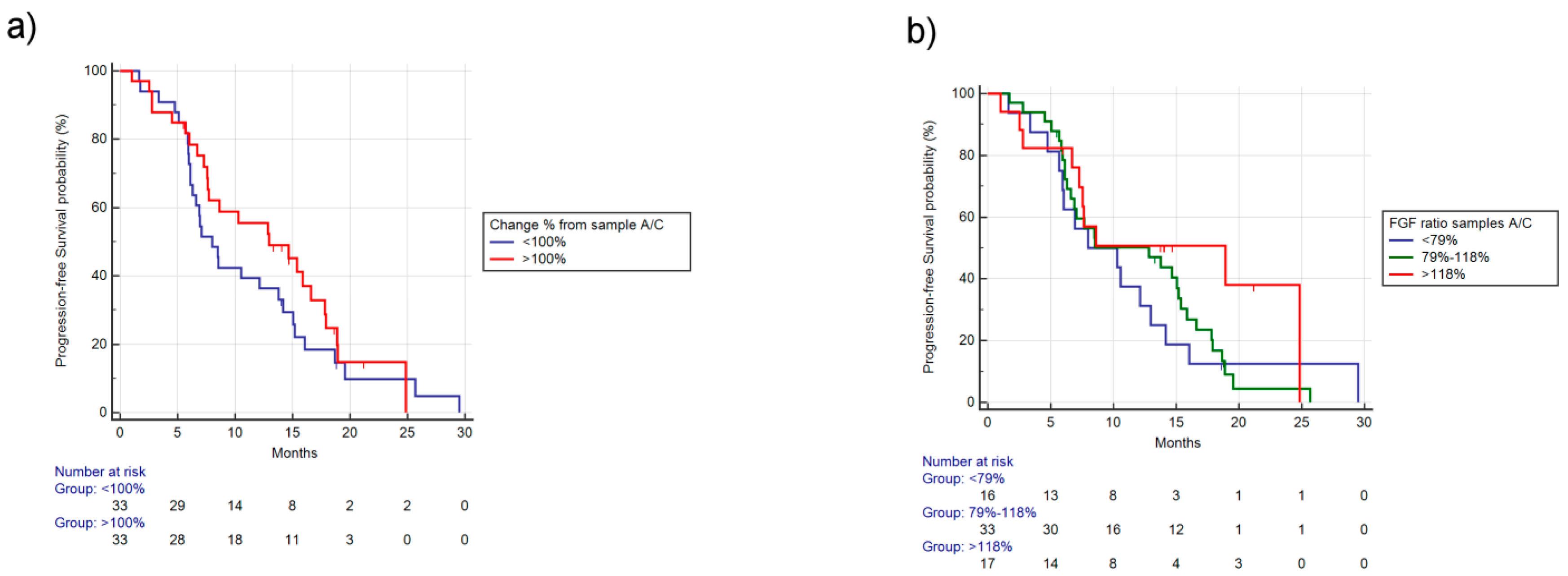
2.1.3. Analysis for Differences in Concentrations of FGF-2 between Samples A and C
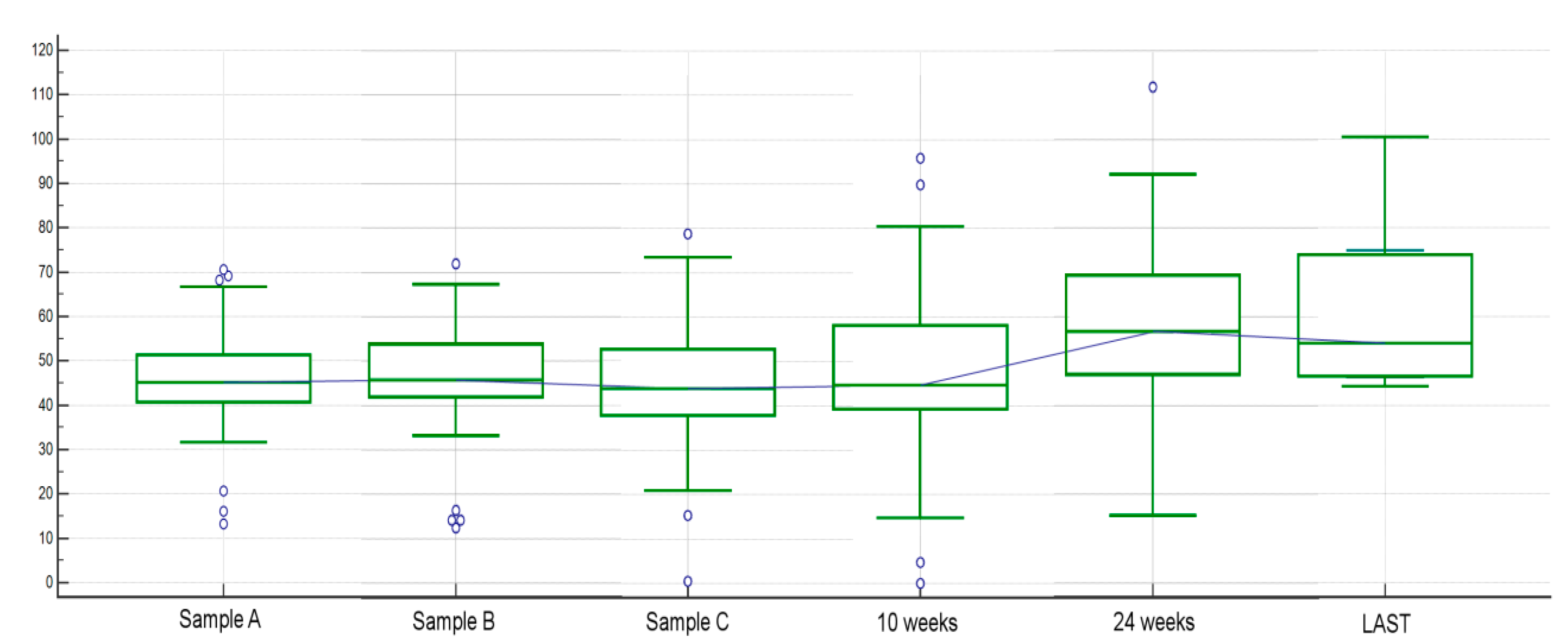
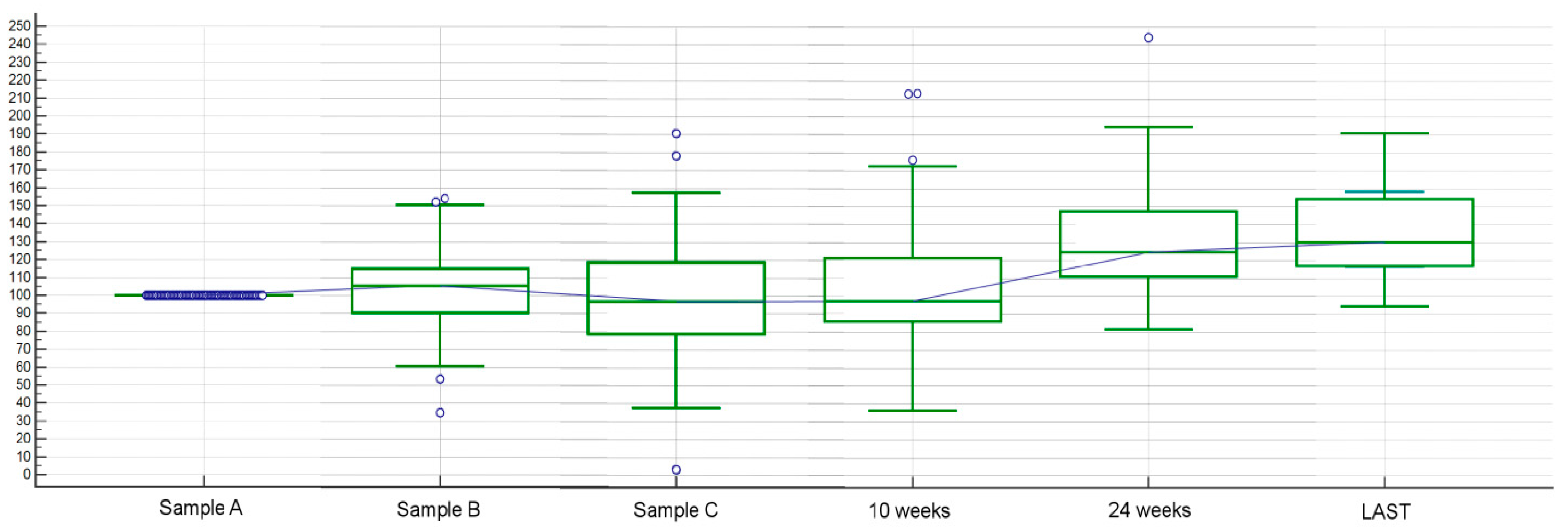
2.1.4. Other Analyses Concerning Circulating FGF-2 among Different Time Points and Clinical Factors
3. Discussion
4. Patients and Methods
4.1. Patients
4.2. Statistical Considerations
4.3. Analysis of Serum Pro-Angiogenic Factors
4.4. Data Availability
4.5. Statement of Ethics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [Green Version]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth. factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Allegra, C.J.; Yothers, G.; O’Connell, M.J.; Sharif, S.; Petrelli, N.J.; Colangelo, L.H.; Atkins, J.N.; Seay, T.E.; Fehrenbacher, L.; Goldberg, R.M.; et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: Results of NSABP protocol C-08. J. Clin. Oncol. 2011, 29, 11–16. [Google Scholar] [CrossRef]
- Allegra, C.J.; Yothers, G.; O’Connell, M.J.; Sharif, S.; Petrelli, N.J.; Lopa, S.H.; Wolmark, N. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J. Clin. Oncol. 2013, 31, 359–364. [Google Scholar] [CrossRef]
- De Gramont, A.; Van Cutsem, E.; Schmoll, H.J.; Tabernero, J.; Clarke, S.; Moore, M.J.; Cunningham, D.; Cartwright, T.H.; Hecht, J.R.; Rivera, F.; et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): A phase 3 randomised controlled trial. Lancet Oncol. 2012, 13, 1225–1233. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, A.; Goto, H.; Saijo, A.; Trung, V.T.; Aono, Y.; Ogino, H.; Kuramoto, T.; Tabata, S.; Uehara, H.; Izumi, K.; et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 2015, 6, 1–15. [Google Scholar]
- Batchelor, T.T.; Sorensen, A.G.; di Tomaso, E.; Zhang, W.T.; Duda, D.G.; Cohen, K.S.; Kozak, K.R.; Cahill, D.P.; Chen, P.J.; Zhu, M.; et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 2007, 11, 83–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of anti-angiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdenakker, G.; Froyen, G.; Fiten, P.; Proost, P.; Van Damme, J. Human monocyte chemotactic protein-3 (MCP-3): Molecular cloning of the cDNA and comparison with other chemokines. Biochem. Biophys. Res. Commun. 1993, 191, 535–542. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 Mediates Resistance to Antiangiogenic Agent Sunitinib in Renal Cell Carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar] [CrossRef]
- Tille, J.C.; Wood, J.; Mandriota, S.J.; Schnell, C.; Ferrari, S.; Mestan, J.; Zhu, Z.; Witte, L.; Pepper, M.S. Vascular endothelial growth factor (VEGF) receptor-2 antagonists inhibit VEGF-and basic fibroblast growth factor-induced angiogenesis In Vivo and In Vitro. J. Pharmacol. Exp. Ther. 2001, 299, 1073–1085. [Google Scholar]
- Valtola, R.; Salven, P.; Heikkilä, P.; Taipale, J.; Joensuu, H.; Rehn, M.; Pihlajaniemi, T.; Weich, H.; deWaal, R.; Alitalo, K. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am. J. Pathol. 1999, 154, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Benest, A.V.; Harper, S.J.; Herttuala, S.Y.; Alitalo, K.; Bates, D.O. VEGF-C induced angiogenesis preferentially occurs at a distance from lymphangiogenesis. Cardiovasc. Res. 2008, 78, 315–323. [Google Scholar] [CrossRef]
- Lieu, C.H.; Tran, H.; Jiang, Z.Q.; Mao, M.; Overman, M.J.; Lin, E.; Eng, C.; Morris, J.; Ellis, L.; Heymach, J.V.; et al. The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. PLoS ONE 2013, 8, e77117. [Google Scholar] [CrossRef]
- Weickhardt, A.J.; Williams, D.S.; Lee, C.K.; Chionh, F.; Simes, J.; Murone, C.; Wilson, K.; Parry, M.M.; Asadi, K.; Scott, A.M.; et al. Vascular endothelial growth factor D expression is a potential biomarker of bevacizumab benefit in colorectal cancer. Br. J. Cancer 2015, 113, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampieri, R.; Puzzoni, M.; Daniele, B.; Ferrari, D.; Lonardi, S.; Zaniboni, A.; Cavanna, L.; Rosati, G.; Pella, N.; Zampino, M.G.; et al. First-line FOLFIRI and bevacizumab in patients with advanced colorectal cancer prospectively stratified according to serum LDH: Final results of the GISCAD (Italian Group for the Study of Digestive Tract Cancers) CENTRAL (ColorEctalavastiNTRiAlLdh) trial. Br. J. Cancer 2017, 117, 1099–1104. [Google Scholar] [CrossRef] [Green Version]
- Giampieri, R.; Salvatore, L.; Del Prete, M.; Prochilo, T.; D’Anzeo, M.; Loretelli, C.; Loupakis, F.; Aprile, G.; Maccaroni, E.; Andrikou, K.; et al. Angiogenesis genotyping and clinical outcome during regorafenib treatment in metastatic colorectal cancer patients. Sci. Rep. 2016, 6, 25195. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausová, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. RAISE Study Investigators. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Yi, J.; Ince, W.; Novotny, W.F.; Rosen, O. The clinical benefit of bevacizumab in metastatic colorectal cancer is independent of K-ras mutation status: Analysis of a phase III study of bevacizumab with chemotherapy in previously untreated metastatic colorectal cancer. Oncologist 2009, 14, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Bagri, A.; Berry, L.; Gunter, B.; Singh, M.; Kasman, I.; Damico, L.A.; Xiang, H.; Schmidt, M.; Fuh, G.; Hollister, B.; et al. Effects of anti-VEGF treatment duration on tumor growth, tumor regrowth, and treatment efficacy. Clin. Cancer Res. 2010, 16, 3887–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loupakis, F.; Ruzzo, A.; Salvatore, L.; Cremolini, C.; Masi, G.; Frumento, P.; Schirripa, M.; Catalano, V.; Galluccio, N.; Canestrari, E.; et al. Retrospective exploratory analysis of VEGF polymorphisms in the prediction of benefit from first-line FOLFIRI plus bevacizumab in metastatic colorectal cancer. BMC Cancer 2011, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loupakis, F.; Cremolini, C.; Yang, D.; Salvatore, L.; Zhang, W.; Wakatsuki, T.; Bohanes, P.; Schirripa, M.; Benhaim, L.; Lonardi, S.; et al. Prospective validation of candidate SNPs of VEGF/VEGFR pathway in metastatic colorectal cancer patients treated with first-line FOLFIRI plus bevacizumab. PLoS ONE 2013, 8, e66774. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.A.; Kim, S.H.; Oh, S.Y.; Lee, S.; Han, J.Y.; Kim, K.H.; Goh, R.Y.; Choi, H.J.; Park, K.J.; Roh, M.S.; et al. Clinical significance of preoperative serum vascular endothelial growth factor, interleukin-6, and C-reactive protein level in colorectal cancer. BMC Cancer 2010, 10, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jürgensmeier, J.M.; Schmoll, H.J.; Robertson, J.D.; Brooks, L.; Taboada, M.; Morgan, S.R.; Wilson, D.; Hoff, P.M. Prognostic and predictive value of VEGF, sVEGFR-2 and CEA in mCRC studies comparing cediranib, bevacizumab and chemotherapy. Br. J. Cancer 2013, 108, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyanchandani, R.; Alves, M.V.; Myers, J.N.; Kim, S. A proangiogenic signature is revealed in FGF-mediated bevacizumab-resistant head and neck squamous cell carcinoma. Mol. Cancer Res. 2013, 11, 1585–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, K.; Miyano, S.W.; Minoshima, Y.; Matsui, J.; Funahashi, Y. Activated FGF2 signaling pathway in tumor vasculature is essential for acquired resistance to anti-VEGF therapy. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Guerrouehan, B.S.; Pasquier, J.; Kaoud, N.A.; Maleki, M.; Beauchamp, M.C.; Yasmeen, A.; Ghiabi, P.; Lis, R.; Vidal, F.; Saleh, A.; et al. Akt-activated endothelium constitutes the niche for residual disease and resistance to bevacizumab in ovarian cancer. Mol. Cancer Ther. 2014, 13, 3123–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, C.V.; Steffensen, K.D.; Olsen, D.A.; Waldstrøm, M.; Smerde, M.; Adimi, P.; Brandslund, I.; Jakobsen, A. Serial measurements of serum PDGF-AA, PDGF-BB, FGF2, and VEGF in multiresistant ovarian cancer patients treated with bevacizumab. J. Ovarian Res. 2012, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Poon, R.T.P.; Ng, I.O.L.; Lau, C.; Yu, W.C.; Fan, S.T.; Wong, J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am. J. Surg. 2001, 182, 298–304. [Google Scholar] [CrossRef]
- Faridi, A.; Rudlowski, C.; Biesterfeld, S.; Schuh, S.; Rath, W.; Schröder, W. Long-term follow-up and prognostic significance of angiogenic basic fibroblast growth factor (bFGF) expression in patients with breast cancer. Pathol. Res. Pract. 2002, 198, 1–5. [Google Scholar] [CrossRef]
- George, M.; Tutton, M.; Abulafi, A.M.; Eccles, S.A.; Swift, R.I. Plasma basic fibroblast growth factor levels in colorectal cancer: A clinically useful assay? Clin. Exp. Metastasis 2002, 19, 735–738. [Google Scholar] [CrossRef]
- Akl, M.R.; Nagpal, P.; Ayoub, N.M.; Ayoub, N.M.; Tai, B.; Prabhu, S.A.; Capac, C.M.; Gliksman, M.; Goy, A.; Suh, K.S. Molecular and clinical significance of fibroblast growth factor 2 (FGF2/bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget 2016, 7, 44735–44762. [Google Scholar] [CrossRef] [Green Version]
- Gunsilius, E.; Petzer, A.; Stockhammer, G.; Nussbaumer, W.; Schumacher, P.; Clausen, J.; Gastl, G. Thrombocytes are the major source for soluble vascular endothelial growth factor in peripheral blood. Oncology 2000, 58, 169–174. [Google Scholar] [CrossRef]
- Zimmermann, R.; Ringwald, J.; Eckstein, R. EDTA plasma is unsuitable for in vivo determinations of platelet-derived angiogenic cytokines. J. Immunol. Methods 2009, 347, 91–92. [Google Scholar] [CrossRef]
- Hoying, J.B.; Williams, S.K. Effects of basic fibroblast growth factor on human microvessel endothelial cell migration on collagen I correlates inversely with adhesion and is cell density dependent. J. Cell. Physiol. 1996, 168, 294–304. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol. Cancer Res. 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [Green Version]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Sulaiman, A.; Wang, L. Bridging the divide: Preclinical research discrepancies between triple-negative breast cancer cell lines and patient tumors. Oncotarget 2017, 8, 113269–113281. [Google Scholar] [CrossRef] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Nissen, L.J.; Cao, R.; Hedlund, E.M.; Wang, Z.; Zhao, X.; Wetterskog, D.; Funa, K.; Bråkenhielm, E.; Cao, Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J. Clin. Investig. 2007, 117, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- Wildiers, H.; Guetens, G.; De Boeck, G.; Verbeken, E.; Landuyt, B.; Landuyt, W.; de Bruijn, E.A.; van Oosterom, A.T. Effect of antivascular endothelial growth factor treatment on the intratumoral uptake of CPT-11. Br. J. Cancer 2003, 88, 1979–1986. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Gamez, S.; Hill, A.L.; Rosenbloom, D.I.; Petrov, D.A.; Nowak, M.A.; Pennings, P.S. Imperfect drug penetration leads to spatial monotherapy and rapid evolution of multidrug resistance. Proc. Natl. Acad. Sci. USA 2015, 112, E2874–E2883. [Google Scholar] [CrossRef] [Green Version]
- Eisenhauera, E.A.; Therasseb, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gender | 41 (57%) Males 31 (43%) Females |
| Age | 60 Y.O. (Median) 33–76 Y.O. (Range) 19 (26%) > 70 Y.O. |
| Sidedness | 39 (54%) right-32 (46%) left sided (14 rectal cancer and 18 sigmoid cancer) |
| ECOG PS at treatment start | 59 (82%) ECOG PS: 0 13 (18%) ECOG PS: 1 |
| Synchronous/Metachronous metastatic involvement | 39 (54%) had synchronous metastatic involvement 33 (46%) had metachronous metastatic involvement |
| LDH value | 55 (76%) had LDH value lower than 1.17xULN (as per CENTRAL) 17 (24%) had LDH value higher than 1.17xULN (as per CENTRAL) |
| K-RAS/B-RAF/N-RAS status | 42 (58%) patients were K-RAS mutant 25 (35%) patients were RAS/B-RAF wild type 4 (5%) patients were B-RAF mutant 1 (1%) patient was N-RAF mutant |
| Severe (>G3 NCI CTCAE) toxicities under treatment | 58 (80%) did not experience G3 NCI CTCAE toxicities or higher 14 (20%) did experience G3 NCI CTCAE toxicities or higher |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giampieri, R.; Ziranu, P.; Daniele, B.; Zizzi, A.; Ferrari, D.; Lonardi, S.; Zaniboni, A.; Cavanna, L.; Rosati, G.; Casagrande, M.; et al. From CENTRAL to SENTRAL (SErum aNgiogenesis cenTRAL): Circulating Predictive Biomarkers to Anti-VEGFR Therapy. Cancers 2020, 12, 1330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051330
Giampieri R, Ziranu P, Daniele B, Zizzi A, Ferrari D, Lonardi S, Zaniboni A, Cavanna L, Rosati G, Casagrande M, et al. From CENTRAL to SENTRAL (SErum aNgiogenesis cenTRAL): Circulating Predictive Biomarkers to Anti-VEGFR Therapy. Cancers. 2020; 12(5):1330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051330
Chicago/Turabian StyleGiampieri, Riccardo, Pina Ziranu, Bruno Daniele, Antonio Zizzi, Daris Ferrari, Sara Lonardi, Alberto Zaniboni, Luigi Cavanna, Gerardo Rosati, Mariaelena Casagrande, and et al. 2020. "From CENTRAL to SENTRAL (SErum aNgiogenesis cenTRAL): Circulating Predictive Biomarkers to Anti-VEGFR Therapy" Cancers 12, no. 5: 1330. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051330