A Bird’s-Eye View of Cell Sources for Cell-Based Therapies in Blood Cancers

 , , ,
, , ,
Abstract
:1. Introduction
2. T Lymphocytes
3. Natural Killer (NK) Cells
4. Dendritic Cells (DCs)
5. Macrophages
6. Platelets
7. Stem Cells and Progenitor Cells
7.1. Hematopoietic Stem Cells (HSCs)
7.2. Mesenchymal Stem Cells (MSCs)
7.3. Endothelial Progenitor Cells (EPCs)
7.4. Induced Pluripotent Stem Cells (iPSCs)
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| AP-1 | Activator protein 1 |
| B16 | Name of murine melanoma cell line |
| BCL | B cell lymphoma |
| BCMA | B-cell maturation antigen |
| CAR-T | Chimeric antigen receptor T-cell |
| CARs | Chimeric antigen receptors |
| Cas9 | CRISPR associated protein 9 |
| CCL25 | C-C motif chemokine ligand 25 |
| CCR2 | C-C motif chemokine receptor 2; cluster of differentiation 192 |
| CCR9 | C-C motif chemokine receptor 9 |
| CD137 ζ | Cluster of differentiation 137 ζ |
| CD138 | Syndecan-1; Cluster of differentiation 138 |
| CD14 | Cluster of differentiation 14 |
| CD16 | Cluster of differentiation 16 |
| CD19 | Cluster of differentiation 19 |
| CD20 | Cluster of differentiation 20 |
| CD22 | Cluster of differentiation 22 |
| CD244 | Cluster of differentiation 244 |
| CD28 | Cluster of differentiation 28 |
| CD3 | Cluster of differentiation 3 |
| CD30 | Tumor necrosis factor receptor superfamily member 8; Cluster of differentiation 30 |
| CD33 | Cluster of differentiation |
| CD3ζ | Cluster of differentiation 3ζ |
| CD4 | Cluster of differentiation 4 |
| CD8 | Cluster of differentiation 8 |
| CLL | Chronic lymphocytic leukemia |
| CML | Chronic myelogenous leukemia |
| CR | Complete response |
| CRISPR | Clustered regularly interspaced palindromic repeats |
| CRS | Cytokine release syndrome |
| DCs | Dendritic cells |
| DLBCL | Diffuse large B-cell lymphoma |
| EG7 | Name of cell line derived from murine T-cell lymphoma |
| EGFRt | Epidermal growth factor receptor |
| EPCs | Endothelial progenitor cells |
| FcRγ | Fc receptor gamma |
| FDA | Food and Drug Administration |
| GvHD | Graft versus host disease |
| GvT | Graft versus tumor effect |
| HLA | Human leukocyte antigen |
| HSCs | Hematopoietic stem cells |
| IFN-γ | Human interferon gamma |
| IL-2 | Interleukin 2 |
| iPS-MEF-Ng-20D-17 | Name of mouse induced pluripotent stem cell line |
| iPSCs | Induced pluripotent stem cells |
| K562 | Human erytroleukemic cell line |
| Klf4 | Kruppel-like factor 4 |
| L1210 | Name of murine cancer cell line derived from lymphoblasts |
| LeY | Lewis Y antigen |
| M1 | Macrophage phenotype 1 |
| M2 | Macrophage phenotype 2 |
| MDS | Myelodysplastic syndrome |
| Megf10 | Multiple epidermal growth factor-like domains 10 |
| MHC | Major histocompatibility complex |
| MM | Multiple myeloma |
| MRD | Minimal residual disease |
| mRNA | Messenger ribonucleic acid |
| MSCs | Mesenchymal stem cells |
| Myc | Master regulator of cell cycle entry and proliferative metabolism |
| Nalm6 | Human B cell precursor leukemia cell line |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFAT | Nuclear factor of activated T-cells |
| NHL | Non-Hodgkin’s lymphoma |
| NK | Natural killer |
| NK-92 | Name of human NK cell line |
| NK1.1 | NK cell-associated marker 1.1; cluster of differentiation 161 |
| NKG2D | Natural killer group 2D receptor |
| NKp46 | Natural cytotoxicity triggering receptor 1; cluster of differentiation 335 |
| NKT | Natural killer T cells |
| NT | Neurotoxicity |
| NY-ESO-1 | Cancer-testis antigen |
| Oct3/4 | Octamer-binding transcription factor 4 |
| OP9 | Name of murine embryonic cell line |
| OR | Objective response |
| PD | Progressive disease |
| PDGF-BB | Platelet-derived growth factor BB |
| PR | Partial response |
| RD114 | Name of envelope glycoprotein of lentiviral vectors |
| RPMI8226 | Name of multiple myeloma human cancer cell line derived from B lymphocytes |
| scFv | Single-chain variable fragment |
| SCR | Sustained complete response |
| SCs | Stem cells |
| SD | Stable disease |
| SDF-1 | Stromal cell derived factor 1 |
| Sox2 | Sex determining region Y-box 2 |
| sTRAIL | Soluble TNF-related apoptosis-inducing ligand |
| SUPRA | Split Universal Programmable |
| TAAs | Tumor-associated antigens |
| TCR | T cell receptor |
| TGF-β | Transforming growth factor β |
| TNF-α | Tumor necrosis factor alpha |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| TRUCKs | T cells redirected for universal cytokine-mediated killing |
| U-266 | Name of human cancer cell line derived from B lymphocyte |
| VEGF | Vascular endothelial growth factor |
| VGPR | Very good partial remission |
| VSV-G | Vesicular stomatitis virus G |
| WT1 | Wilms’ tumor 1 |
| 4T1 | Murine mammary tumor cell line |
References
- SEER Cancer Statistics Review (CSR) 1975-2014. Available online: https://seer.cancer.gov (accessed on 22 May 2020).
- DeSantis, C.E.; Miller, K.D.; Dale, W.; Mohile, S.G.; Cohen, H.J.; Leach, C.R.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Cancer statistics for adults aged 85 years and older, 2019. CA Cancer J. Clin. 2019, 69, 452–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostindie, S.C.; van der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; van den Brink, E.N.; van den Brakel, J.H.N.; Rademaker, H.J.; van Kessel, B.; van den Noort, J.; et al. DuoHexaBody-CD37((R)), a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Bonello, F.; D’Agostino, M.; Moscvin, M.; Cerrato, C.; Boccadoro, M.; Gay, F. CD38 as an immunotherapeutic target in multiple myeloma. Expert Opin. Biol. Ther. 2018, 18, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Salles, G.; Barrett, M.; Foa, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottardi, M.; Mosna, F.; de Angeli, S.; Papayannidis, C.; Candoni, A.; Clavio, M.; Tecchio, C.; Piccin, A.; dell’Orto, M.C.; Benedetti, F.; et al. Clinical and experimental efficacy of gemtuzumab ozogamicin in core binding factor acute myeloid leukemia. Hematol. Rep. 2017, 9, 7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, N.; Kumar, S.K. Daratumumab in untreated newly diagnosed multiple myeloma. Ther. Adv. Hematol. 2019, 10, 2040620719894871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Choi, I.; Yoshimoto, G.; Muta, T.; Yamasaki, S.; Tanimoto, K.; Kamimura, T.; Iwasaki, H.; Ogawa, R.; Akashi, K.; et al. Phase I/II study of bortezomib, lenalidomide, and dexamethasone treatment for relapsed and refractory multiple myeloma. Int. J. Hematol. 2020, 111, 673–680. [Google Scholar] [CrossRef]
- Fathi, E.; Farahzadi, R.; Sheervalilou, R.; Sanaat, Z.; Vietor, I. A general view of CD33(+) leukemic stem cells and CAR-T cells as interesting targets in acute myeloblatsic leukemia therapy. Blood Res. 2020, 55, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.R.; Baek, K.H. Role of natural killer cells for immunotherapy in chronic myeloid leukemia (Review). Oncol. Rep. 2019, 41, 2625–2635. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Versteven, M.; Lichtenegger, F.S.; Roex, G.; Campillo-Davo, D.; Lion, E.; Subklewe, M.; Van Tendeloo, V.F.; Berneman, Z.N.; Anguille, S. Dendritic Cell-Based Immunotherapy of Acute Myeloid Leukemia. J. Clin. Med. 2019, 8, 579. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zhang, M. Use of CAR-T cell therapy, PD-1 blockade, and their combination for the treatment of hematological malignancies. Clin. Immunol. 2020, 214, 108382. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Abate-Daga, D.; Davila, M.L. CAR models: Next-generation CAR modifications for enhanced T-cell function. Mol. Ther. Oncolytics 2016, 3, 16014. [Google Scholar] [CrossRef] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J. Hematol. Oncol. 2019, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef] [Green Version]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 2017, 7, e1368604. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Riviere, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- MacKay, M. CARGlobalTrials. Available online: https://carglobaltrials.com/ (accessed on 22 May 2020).
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des. Devel. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Wang, Y.; Guo, Y.L.; Dai, H.R.; Yang, Q.M.; Zhang, Y.J.; Zhang, Y.; Chen, M.X.; Wang, C.M.; Feng, K.C.; et al. Treatment of CD20-directed Chimeric Antigen Receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: An early phase IIa trial report. Signal Transduct. Target. Ther. 2016, 1, 16002. [Google Scholar] [CrossRef]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Ballard, B.; Zhang, H.; Dakhova, O.; Gee, A.P.; Mei, Z.; Bilgi, M.; Wu, M.F.; Liu, H.; Grilley, B.; et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J. Clin. Invest. 2017, 127, 3462–3471. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Honemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Picanco-Castro, V.; Moco, P.D.; Mizukami, A.; Vaz, L.D.; de Souza Fernandes Pereira, M.; Silvestre, R.N.; de Azevedo, J.T.C.; de Sousa Bomfim, A.; de Abreu Neto, M.S.; Malmegrim, K.C.R.; et al. Establishment of a simple and efficient platform for car-t cell generation and expansion: From lentiviral production to in vivo studies. Hematol. Transfus. Cell Ther. 2019. [Google Scholar] [CrossRef]
- Pampusch, M.S.; Haran, K.P.; Hart, G.T.; Rakasz, E.G.; Rendahl, A.K.; Berger, E.A.; Connick, E.; Skinner, P.J. Rapid Transduction and Expansion of Transduced T Cells with Maintenance of Central Memory Populations. Mol. Ther. Methods Clin. Dev. 2020, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated With Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Invest. 2016, 126, 3363–3376. [Google Scholar] [CrossRef] [PubMed]
- Morita, D.; Nishio, N.; Saito, S.; Tanaka, M.; Kawashima, N.; Okuno, Y.; Suzuki, S.; Matsuda, K.; Maeda, Y.; Wilson, M.H.; et al. Enhanced Expression of Anti-CD19 Chimeric Antigen Receptor in piggyBac Transposon-Engineered T Cells. Mol. Ther. Methods Clin. Dev. 2018, 8, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 2008, 118, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel) 2016, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Griffioen, M.; van Egmond, E.H.; Kester, M.G.; Willemze, R.; Falkenburg, J.H.; Heemskerk, M.H. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica 2009, 94, 1316–1320. [Google Scholar] [CrossRef] [Green Version]
- Paszkiewicz, P.J.; Frassle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 2016, 126, 4262–4272. [Google Scholar] [CrossRef]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schonfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, F.; Perea, F.; Bernal, M.; Sanchez-Palencia, A.; Aptsiauri, N.; Ruiz-Cabello, F. The Escape of Cancer from T Cell-Mediated Immune Surveillance: HLA Class I Loss and Tumor Tissue Architecture. Vaccines (Basel) 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.R.; Zaman, S.; Nedunchezhian, M.; Chakrabarti, A.; Bhakuni, P.; Ahmed, M.; Sharma, K.; Rawat, S.; O’Donnell, P.; Chakrabarti, S. CD56-enriched donor cell infusion after post-transplantation cyclophosphamide for haploidentical transplantation of advanced myeloid malignancies is associated with prompt reconstitution of mature natural killer cells and regulatory T cells with reduced incidence of acute graft versus host disease: A pilot study. Cytotherapy 2017, 19, 531–542. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; denHollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [Green Version]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef]
- Masuyama, J.; Murakami, T.; Iwamoto, S.; Fujita, S. Ex vivo expansion of natural killer cells from human peripheral blood mononuclear cells co-stimulated with anti-CD3 and anti-CD52 monoclonal antibodies. Cytotherapy 2016, 18, 80–90. [Google Scholar] [CrossRef]
- Herrera, L.; Santos, S.; Vesga, M.A.; Anguita, J.; Martin-Ruiz, I.; Carrascosa, T.; Juan, M.; Eguizabal, C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci. Rep. 2019, 9, 18729. [Google Scholar] [CrossRef] [Green Version]
- Herrera, L.; Salcedo, J.M.; Santos, S.; Vesga, M.A.; Borrego, F.; Eguizabal, C. OP9 Feeder Cells Are Superior to M2-10B4 Cells for the Generation of Mature and Functional Natural Killer Cells from Umbilical Cord Hematopoietic Progenitors. Front. Immunol. 2017, 8, 755. [Google Scholar] [CrossRef] [Green Version]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (iPSC)-Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Topfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Muller, N.; Lindemann, D.; Bachmann, M.; Fussel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Childs, R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef] [Green Version]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Romero, H.; Selleri, S.; Beland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P.; et al. Human CAR NK Cells: A New Non-viral Method Allowing High Efficient Transfection and Strong Tumor Cell Killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lupo, K.B.; Chambers, A.M.; Matosevic, S. Purinergic targeting enhances immunotherapy of CD73(+) solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J. Immunother. Cancer 2018, 6, 136. [Google Scholar] [CrossRef]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, J.M.; Czerwinski, D.K.; Davis, T.A.; Hsu, F.J.; Benike, C.; Hao, Z.M.; Taidi, B.; Rajapaksa, R.; Caspar, C.B.; Okada, C.Y.; et al. Idiotype-pulsed dendritic cell vaccination for B-cell lymphoma: Clinical and immune responses in 35 patients. Blood 2002, 99, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Kokhaei, P.; Choudhury, A.; Mahdian, R.; Lundin, J.; Moshfegh, A.; Osterborg, A.; Mellstedt, H. Apoptotic tumor cells are superior to tumor cell lysate, and tumor cell RNA in induction of autologous T cell response in B-CLL. Leukemia 2004, 18, 1810–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, V.T.; Vanneman, M.; Kim, H.; Sasada, T.; Kang, Y.J.; Pasek, M.; Cutler, C.; Koreth, J.; Alyea, E.; Sarantopoulos, S.; et al. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 15825–15830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Driessche, A.; Van de Velde, A.L.; Nijs, G.; Braeckman, T.; Stein, B.; De Vries, J.M.; Berneman, Z.N.; Van Tendeloo, V.F. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy 2009, 11, 653–668. [Google Scholar] [CrossRef]
- Roddie, H.; Klammer, M.; Thomas, C.; Thomson, R.; Atkinson, A.; Sproul, A.; Waterfall, M.; Samuel, K.; Yin, J.; Johnson, P.; et al. Phase I/II study of vaccination with dendritic-like leukaemia cells for the immunotherapy of acute myeloid leukaemia. Br. J. Haematol. 2006, 133, 152–157. [Google Scholar] [CrossRef]
- Tel, J.; Anguille, S.; Waterborg, C.E.; Smits, E.L.; Figdor, C.G.; de Vries, I.J. Tumoricidal activity of human dendritic cells. Trends Immunol. 2014, 35, 38–46. [Google Scholar] [CrossRef]
- Zhou, L.J.; Tedder, T.F. CD14+ blood monocytes can differentiate into functionally mature CD83+ dendritic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 2588–2592. [Google Scholar] [CrossRef] [Green Version]
- Fidler, I.J. Inhibition of pulmonary metastasis by intravenous injection of specifically activated macrophages. Cancer Res. 1974, 34, 1074–1078. [Google Scholar]
- Andreesen, R.; Hennemann, B.; Krause, S.W. Adoptive immunotherapy of cancer using monocyte-derived macrophages: Rationale, current status, and perspectives. J. Leukoc. Biol. 1998, 64, 419–426. [Google Scholar] [CrossRef]
- Stevenson, H.C.; Keenan, A.M.; Woodhouse, C.; Ottow, R.T.; Miller, P.; Steller, E.P.; Foon, K.A.; Abrams, P.G.; Beman, J.; Larson, S.M.; et al. Fate of gamma-interferon-activated killer blood monocytes adoptively transferred into the abdominal cavity of patients with peritoneal carcinomatosis. Cancer Res. 1987, 47, 6100–6103. [Google Scholar] [PubMed]
- Quillien, V.; Moisan, A.; Lesimple, T.; Leberre, C.; Toujas, L. Biodistribution of 111indium-labeled macrophages infused intravenously in patients with renal carcinoma. Cancer Immunol. Immunother. 2001, 50, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squadrito, M.L.; De Palma, M. Macrophage regulation of tumor angiogenesis: Implications for cancer therapy. Mol. Aspects Med. 2011, 32, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Kan, O.; Day, D.; Iqball, S.; Burke, F.; Grimshaw, M.J.; Naylor, S.; Binley, K. Genetically modified macrophages expressing hypoxia regulated cytochrome P450 and P450 reductase for the treatment of cancer. Int. J. Mol. Med. 2011, 27, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Basel, M.T.; Balivada, S.; Shrestha, T.B.; Seo, G.M.; Pyle, M.M.; Tamura, M.; Bossmann, S.H.; Troyer, D.L. A cell-delivered and cell-activated SN38-dextran prodrug increases survival in a murine disseminated pancreatic cancer model. Small 2012, 8, 913–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.D.; Choi, W.; Yoon, T.H.; Lee, K.J.; Lee, J.S.; Joo, J.H.; Lee, M.G.; Yim, H.S.; Choi, K.M.; Kim, B.; et al. In vivo photothermal treatment by the peritumoral injection of macrophages loaded with gold nanoshells. Biomed. Opt. Express 2016, 7, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. Elife 2018, 7. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin. Cancer Res. 2013, 19, 3640–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. 2018, 67, 1505–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Lavergne, M.; Janus-Bell, E.; Schaff, M.; Gachet, C.; Mangin, P.H. Platelet Integrins in Tumor Metastasis: Do They Represent a Therapeutic Target? Cancers (Basel) 2017, 9, 133. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, G.; Wei, J.; Nie, G. Platelet membrane-based and tumor-associated platelettargeted drug delivery systems for cancer therapy. Front. Med. 2018, 12, 667–677. [Google Scholar] [CrossRef]
- Xu, P.; Zuo, H.; Chen, B.; Wang, R.; Ahmed, A.; Hu, Y.; Ouyang, J. Corrigendum: Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7, 44974. [Google Scholar] [CrossRef] [Green Version]
- Grozovsky, R.; Giannini, S.; Falet, H.; Hoffmeister, K.M. Regulating billions of blood platelets: Glycans and beyond. Blood 2015, 126, 1877–1884. [Google Scholar] [CrossRef] [Green Version]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef]
- Gasic, G.J.; Gasic, T.B.; Galanti, N.; Johnson, T.; Murphy, S. Platelet-tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int. J. Cancer 1973, 11, 704–718. [Google Scholar] [CrossRef]
- Cho, M.S.; Bottsford-Miller, J.; Vasquez, H.G.; Stone, R.; Zand, B.; Kroll, M.H.; Sood, A.K.; Afshar-Kharghan, V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012, 120, 4869–4872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radziwon-Balicka, A.; Medina, C.; O’Driscoll, L.; Treumann, A.; Bazou, D.; Inkielewicz-Stepniak, I.; Radomski, A.; Jow, H.; Radomski, M.W. Platelets increase survival of adenocarcinoma cells challenged with anticancer drugs: Mechanisms and implications for chemoresistance. Br. J. Pharmacol. 2012, 167, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Damaser, M.S. Stem cells as drug delivery methods: Application of stem cell secretome for regeneration. Adv. Drug. Deliv. Rev. 2015, 82–83, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavari, S.L.; Shah, K. Engineered stem cells targeting multiple cell surface receptors in tumors. Stem Cells 2020, 38, 34–44. [Google Scholar] [CrossRef] [Green Version]
- van Rood, J.J.; Scaradavou, A.; Stevens, C.E. Indirect evidence that maternal microchimerism in cord blood mediates a graft-versus-leukemia effect in cord blood transplantation. Proc. Natl. Acad. Sci. USA 2012, 109, 2509–2514. [Google Scholar] [CrossRef] [Green Version]
- Pessina, A.; Cocce, V.; Pascucci, L.; Bonomi, A.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Ciusani, E.; Crovace, A.; Falchetti, M.L.; et al. Mesenchymal stromal cells primed with Paclitaxel attract and kill leukaemia cells, inhibit angiogenesis and improve survival of leukaemia-bearing mice. Br. J. Haematol. 2013, 160, 766–778. [Google Scholar] [CrossRef]
- Ciavarella, S.; Grisendi, G.; Dominici, M.; Tucci, M.; Brunetti, O.; Dammacco, F.; Silvestris, F. In vitro anti-myeloma activity of TRAIL-expressing adipose-derived mesenchymal stem cells. Br. J. Haematol. 2012, 157, 586–598. [Google Scholar] [CrossRef]
- Bock, A.M.; Knorr, D.; Kaufman, D.S. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and and induced pluripotent stem cells (iPSCs). J. Vis. Exp. 2013, e50337. [Google Scholar] [CrossRef] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Thomas, E.D.; Lochte, H.L., Jr.; Lu, W.C.; Ferrebee, J.W. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N. Engl. J. Med. 1957, 257, 491–496. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. Allogeneic Stem Cell Transplantation: A Historical and Scientific Overview. Cancer Res. 2016, 76, 6445–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bair, S.M.; Brandstadter, J.D.; Ayers, E.C.; Stadtmauer, E.A. Hematopoietic stem cell transplantation for blood cancers in the era of precision medicine and immunotherapy. Cancer 2020, 126, 1837–1855. [Google Scholar] [CrossRef] [PubMed]
- Panch, S.R.; Szymanski, J.; Savani, B.N.; Stroncek, D.F. Sources of Hematopoietic Stem and Progenitor Cells and Methods to Optimize Yields for Clinical Cell Therapy. Biol. Blood Marrow Transplant. 2017, 23, 1241–1249. [Google Scholar] [CrossRef]
- Khaddour, K.; Mewawalla, P. Hematopoietic Stem Cell Transplantation. In StatPearls StatPearls [Internet], Treasure Island (FL); StatPearls Publishing: Petersburg, FL, USA, 2020. [Google Scholar]
- Gschweng, E.; De Oliveira, S.; Kohn, D.B. Hematopoietic stem cells for cancer immunotherapy. Immunol. Rev. 2014, 257, 237–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig-Saus, C.; Parisi, G.; Garcia-Diaz, A.; Krystofinski, P.E.; Sandoval, S.; Zhang, R.; Champhekar, A.S.; McCabe, J.; Cheung-Lau, G.C.; Truong, N.A.; et al. IND-Enabling Studies for a Clinical Trial to Genetically Program a Persistent Cancer-Targeted Immune System. Clin. Cancer Res. 2019, 25, 1000–1011. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Gimble, J.M.; Guilak, F.; Nuttall, M.E.; Sathishkumar, S.; Vidal, M.; Bunnell, B.A. In vitro Differentiation Potential of Mesenchymal Stem Cells. Transfus. Med. Hemother. 2008, 35, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Li, T.S. Mini review: Recent advances in the cell-based therapies for cardiac regeneration. Curr. Stem Cell Res. Ther. 2020. [Google Scholar] [CrossRef]
- Rozier, P.; Maria, A.; Goulabchand, R.; Jorgensen, C.; Guilpain, P.; Noel, D. Mesenchymal Stem Cells in Systemic Sclerosis: Allogenic or Autologous Approaches for Therapeutic Use? Front. Immunol. 2018, 9, 2938. [Google Scholar] [CrossRef] [Green Version]
- Conrad, S.; Weber, K.; Walliser, U.; Geburek, F.; Skutella, T. Stem Cell Therapy for Tendon Regeneration: Current Status and Future Directions. Adv. Exp. Med. Biol. 2019, 1084, 61–93. [Google Scholar] [CrossRef]
- Bago, J.R.; Soler-Botija, C.; Casani, L.; Aguilar, E.; Alieva, M.; Rubio, N.; Bayes-Genis, A.; Blanco, J. Bioluminescence imaging of cardiomyogenic and vascular differentiation of cardiac and subcutaneous adipose tissue-derived progenitor cells in fibrin patches in a myocardium infarct model. Int. J. Cardiol. 2013, 169, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of Mesenchymal Stem Cells for Therapeutic Agent Delivery in Anti-tumor Treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Bago, J.R.; Pegna, G.J.; Okolie, O.; Hingtgen, S.D. Fibrin matrices enhance the transplant and efficacy of cytotoxic stem cell therapy for post-surgical cancer. Biomaterials 2016, 84, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Menu, E.; De Becker, A.; Van Camp, B.; Vanderkerken, K.; Van Riet, I. Bone marrow-derived mesenchymal stromal cells are attracted by multiple myeloma cell-produced chemokine CCL25 and favor myeloma cell growth in vitro and in vivo. Stem Cells 2012, 30, 266–279. [Google Scholar] [CrossRef]
- Klopp, A.H.; Spaeth, E.L.; Dembinski, J.L.; Woodward, W.A.; Munshi, A.; Meyn, R.E.; Cox, J.D.; Andreeff, M.; Marini, F.C. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007, 67, 11687–11695. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, Y.; Huang, W.; Xu, H.; Chen, X.; Geng, Q.; Fan, H.; Tan, Y.; Xue, G.; Jiang, X. In vitro effect of adenovirus-mediated human Gamma Interferon gene transfer into human mesenchymal stem cells for chronic myelogenous leukemia. Hematol. Oncol. 2006, 24, 151–158. [Google Scholar] [CrossRef]
- Bonomi, A.; Steimberg, N.; Benetti, A.; Berenzi, A.; Alessandri, G.; Pascucci, L.; Boniotti, J.; Cocce, V.; Sordi, V.; Pessina, A.; et al. Paclitaxel-releasing mesenchymal stromal cells inhibit the growth of multiple myeloma cells in a dynamic 3D culture system. Hematol. Oncol. 2017, 35, 693–702. [Google Scholar] [CrossRef]
- Cocce, V.; Farronato, D.; Brini, A.T.; Masia, C.; Gianni, A.B.; Piovani, G.; Sisto, F.; Alessandri, G.; Angiero, F.; Pessina, A. Drug Loaded Gingival Mesenchymal Stromal Cells (GinPa-MSCs) Inhibit In Vitro Proliferation of Oral Squamous Cell Carcinoma. Sci. Rep. 2017, 7, 9376. [Google Scholar] [CrossRef]
- Dwyer, R.M.; Khan, S.; Barry, F.P.; O’Brien, T.; Kerin, M.J. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res. Ther. 2010, 1, 25. [Google Scholar] [CrossRef] [Green Version]
- Klingemann, H.; Matzilevich, D.; Marchand, J. Mesenchymal Stem Cells—Sources and Clinical Applications. Transfus. Med. Hemother. 2008, 35, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.D.; Fong, C.Y.; Biswas, A.; Choolani, M.; Bongso, A. Human Umbilical Cord Wharton’s Jelly Stem Cell Conditioned Medium Induces Tumoricidal Effects on Lymphoma Cells Through Hydrogen Peroxide Mediation. J. Cell. Biochem. 2016, 117, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Park, Y.J.; Kim, D.S.; Park, H.J.; Jung, H.L.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Human Adipose Tissue Stem Cells Promote the Growth of Acute Lymphoblastic Leukemia Cells in NOD/SCID Mice. Stem Cell Rev. Rep. 2018, 14, 451–460. [Google Scholar] [CrossRef]
- Song, N.; Gao, L.; Qiu, H.; Huang, C.; Cheng, H.; Zhou, H.; Lv, S.; Chen, L.; Wang, J. Mouse bone marrow-derived mesenchymal stem cells inhibit leukemia/lymphoma cell proliferation in vitro and in a mouse model of allogeneic bone marrow transplant. Int. J. Mol. Med. 2015, 36, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, R.; Lam, E.W.; Soeiro, I.; Tisato, V.; Bonnet, D.; Dazzi, F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: Impact on in vivo tumor growth. Leukemia 2007, 21, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Sarmadi, V.H.; Tong, C.K.; Vidyadaran, S.; Abdullah, M.; Seow, H.F.; Ramasamy, R. Mesenchymal stem cells inhibit proliferation of lymphoid origin haematopoietic tumour cells by inducing cell cycle arrest. Med. J. Malaysia 2010, 65, 209–214. [Google Scholar] [PubMed]
- Ning, H.; Yang, F.; Jiang, M.; Hu, L.; Feng, K.; Zhang, J.; Yu, Z.; Li, B.; Xu, C.; Li, Y.; et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: Outcome of a pilot clinical study. Leukemia 2008, 22, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar] [CrossRef] [Green Version]
- Nwabo Kamdje, A.H.; Mosna, F.; Bifari, F.; Lisi, V.; Bassi, G.; Malpeli, G.; Ricciardi, M.; Perbellini, O.; Scupoli, M.T.; Pizzolo, G.; et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 2011, 118, 380–389. [Google Scholar] [CrossRef]
- Di Ianni, M.; Del Papa, B.; De Ioanni, M.; Moretti, L.; Bonifacio, E.; Cecchini, D.; Sportoletti, P.; Falzetti, F.; Tabilio, A. Mesenchymal cells recruit and regulate T regulatory cells. Exp. Hematol. 2008, 36, 309–318. [Google Scholar] [CrossRef]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Roorda, B.D.; ter Elst, A.; Kamps, W.A.; de Bont, E.S. Bone marrow-derived cells and tumor growth: Contribution of bone marrow-derived cells to tumor micro-environments with special focus on mesenchymal stem cells. Crit. Rev. Oncol. Hematol. 2009, 69, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Tian, C.; Guo, S.; Zhang, L.; Zhao, D.; Qu, F.; Zhao, W.; Wang, Y.; Wu, X.; Da, W.; et al. c-Myc plays part in drug resistance mediated by bone marrow stromal cells in acute myeloid leukemia. Leuk. Res. 2015, 39, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef]
- Lee, M.W.; Ryu, S.; Kim, D.S.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Mesenchymal stem cells in suppression or progression of hematologic malignancy: Current status and challenges. Leukemia 2019, 33, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Barber, C.L.; Iruela-Arispe, M.L. The ever-elusive endothelial progenitor cell: Identities, functions and clinical implications. Pediatr. Res. 2006, 59, 26R–32R. [Google Scholar] [CrossRef]
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into Endothelial Progenitor Cells: Origin, Classification, Potentials, and Prospects. Stem Cells Int. 2018, 2018, 9847015. [Google Scholar] [CrossRef]
- Laurenzana, A.; Margheri, F.; Chilla, A.; Biagioni, A.; Margheri, G.; Calorini, L.; Fibbi, G.; Del Rosso, M. Endothelial Progenitor Cells as Shuttle of Anticancer Agents. Hum. Gene Ther. 2016, 27, 784–791. [Google Scholar] [CrossRef]
- Keighron, C.; Lyons, C.J.; Creane, M.; O’Brien, T.; Liew, A. Recent Advances in Endothelial Progenitor Cells Toward Their Use in Clinical Translation. Front. Med. (Lausanne) 2018, 5, 354. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Liu, H.Q.; Li, J.; Liu, X.L. Endothelial progenitor cells promote tumor growth and progression by enhancing new vessel formation. Oncol. Lett. 2016, 12, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, F.; Haque, R.; Weiler, L.; Vrana, K.E.; Song, J. T lineage differentiation from induced pluripotent stem cells. Cell Immunol. 2009, 260, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Watarai, H.; Fujii, S.; Yama da, D.; Rybouchkin, A.; Sakata, S.; Nagata, Y.; Iida-Kobayashi, M.; Sekine-Kondo, E.; Shimizu, K.; Shozaki, Y.; et al. Murine induced pluripotent stem cells can be derived from and differentiate into natural killer T cells. J. Clin. Invest. 2010, 120, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhao, W.; Zhu, Y.; Sun, Z.; Zhang, H.; et al. Induced Pluripotent Stem Cell-derived CAR-Macrophage Cells with Antigen-dependent Anti-Cancer Cell Functions for Liquid and Solid Tumors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yasuda, S.; Kusakawa, S.; Kuroda, T.; Miura, T.; Tano, K.; Takada, N.; Matsuyama, S.; Matsuyama, A.; Nasu, M.; Umezawa, A.; et al. Tumorigenicity-associated characteristics of human iPS cell lines. PLoS ONE 2018, 13, e0205022. [Google Scholar] [CrossRef] [PubMed] [Green Version]



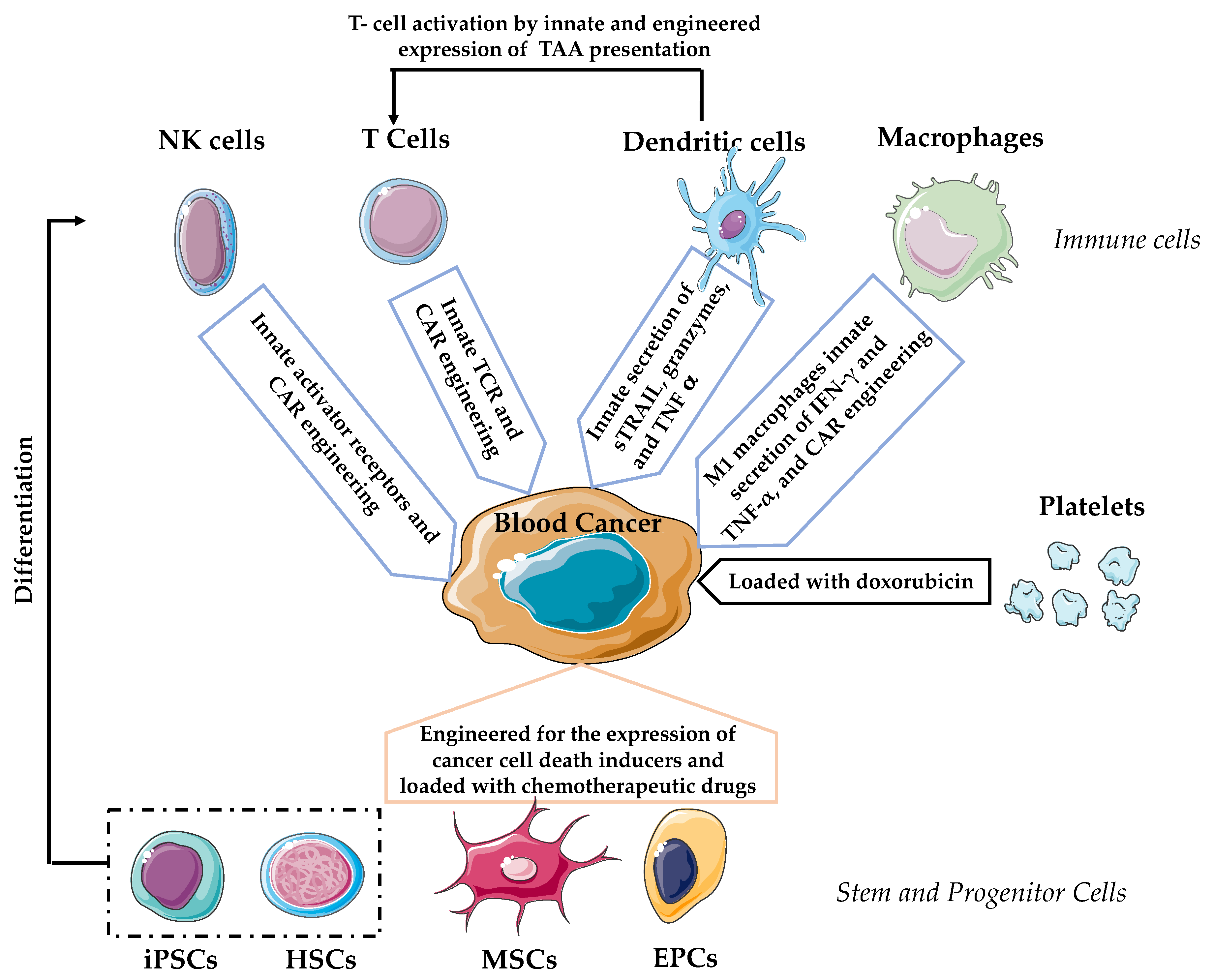{kind=link}
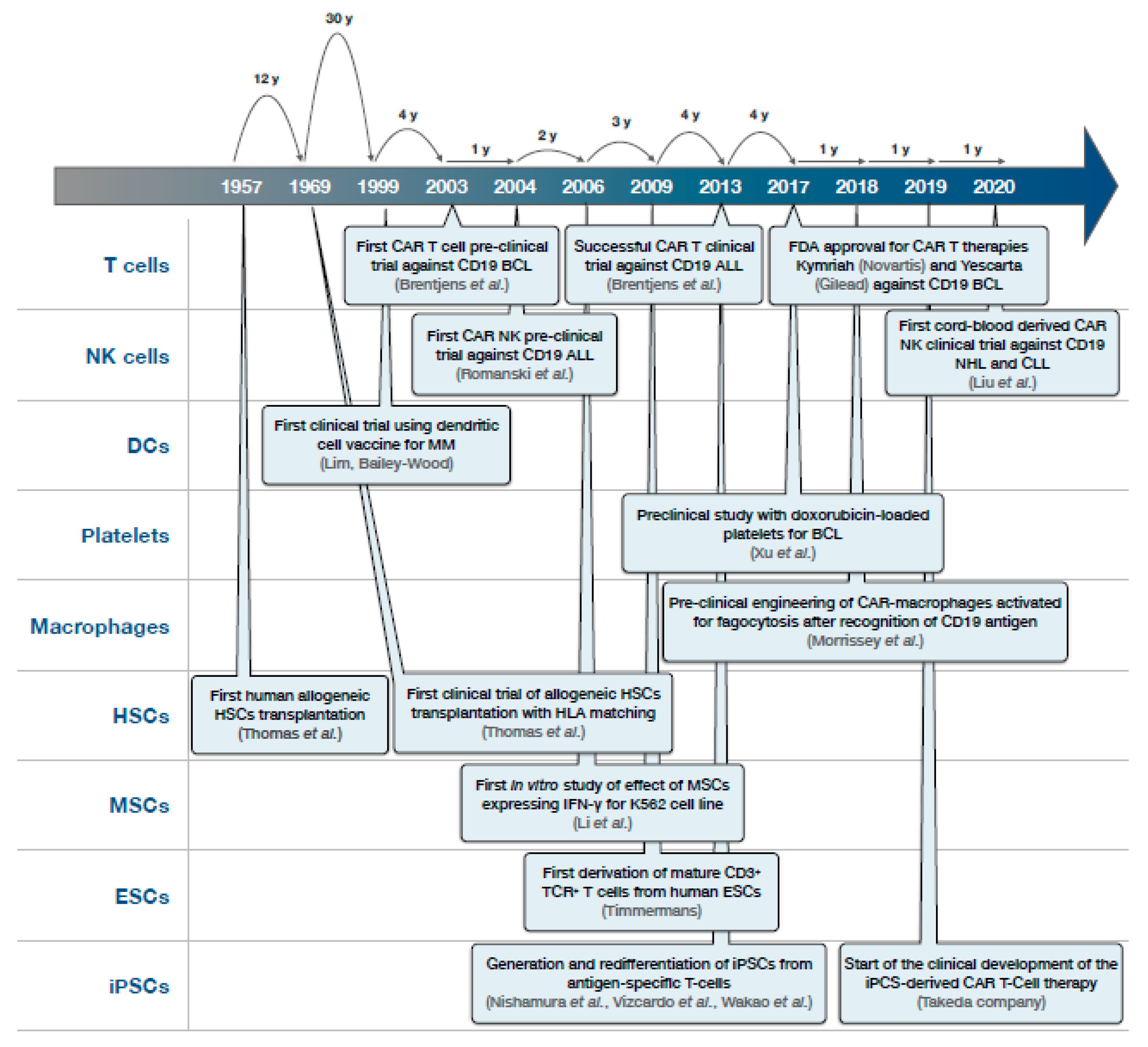{kind=link}
| Year | Cell Source | Target | Engineering Method | No. of Patients | Age of Patients (Mean) | Dose | Outcomes | Cytotoxic Effects | Additional Notes | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 2010 | Autologous DCs | ALL | WT1 mRNA electroporation | 10 | 31–83 (61) | 4 shots 2 weeks interval 5, 10, or 20 × 106 | 2CR | – | [92] | |
| 2013 | Autologous DCs | CD38+ and CD138+ MM | Fusion with whole MM cell | 100 | 35–70 | 3–4 shots 2 × 106/kg per shot | 78% R 47% VGPR 31% CR | – | 57 % two-year progression-free survival | [93] |
| 2018 | Allogenic DCs | AML | Differentiated from AML cell line | 12 | 57–74 (68) | 4 shots 2 weeks interval 10, 25 or 50 × 106 | 5 CR 4 PD | – | Post-remission treatment | [94] |
| 2013 | Autologous T cells | CD19+ ALL | 2nd gen CAR (+CD28) | 16 | 23–74 (50) | One shot 1.5–3 × 106/kg | 14 OR 10 CR 13 MRD- | 2 CRS4 2 CRS3 | [17] | |
| 2014 | Autologous T cells | CD19+ B-ALL and T-ALL | 2nd gen CAR (+4-1BB) | 30 | 5–60 | One shot 0.76–20 × 106/kg | 27 CR 19 SCR | Severe in 8 patients | FDA approved for ALL and DLBCL Produced by Novartis under the commercial name Kymriah | [25] |
| 2016 | Autologous T cells | CD20+ B-cell NHL | 2nd gen CAR (+CD137ζ) | 11 | 25–70 (59) | One shot 0.5–1.5 × 106/kg | 6 CR 3 PR 2 SD | 2 CRS3 | [28] | |
| 2017 | Autologous T cells | CD19+ B-cell lymphomas | 2nd gen CAR (+CD28) | 111 | 23–76 | One shot 2 × 106/kg | 82% OR 54% CR | 95% CRS3+ | FDA-approved for B-cell lymphoma Produced by Gilead under the commercial name Yescarta | [95] |
| 2017 | Autologous T cells | CD22+ lymphoma and leukemia | 2nd gen CAR | 21 | 7–30 (19) | One shot 0.3–3 × 106/kg | 12 CR 9 MRD- | 1 CRS4 1 CRS3 | 17 patients were resistant to CAR anti-CD19 in the past | [29] |
| 2019 | Autologous T cells | CD19+ and BCMA+ MM | 2nd gen CAR | 21 | 49–61 (58) | One shot 1 × 106 + 1 × 106/kg | 12 CR 8 PR 1 SD 17 MRD- | 1CRS3 2 NT | [96] | |
| 2018 | NK92 cell line | CD33+ AML | 3rd gen CAR (+CD28 and 4-1BB) | 3 | 14–49 | Three shots 2 days interval 300, 600, 1000 × 106 | No response | 2CRS1 | One patient died of GvHD after chemotherapy and donor lymphocyte infusion | [65] |
| 2020 | Allogenic NK cells from cord blood | CD19+ lymphoma and leukemia | 2nd gen CAR (+CD28) | 11 | 23–66 (52) | One shot 0.1, 1 or 10 × 106/kg | 8 OR 7 CR | No GvHD No CRS | No GvHD despite some HLA mismatch; Toxic events related to lymphodepletion | [66] |
| Cell Source | Target | Mode of Action | Outcomes | Reference |
|---|---|---|---|---|
| HSCs from cord blood | Patients with AML or ALL | GvT | Decrease in leukemic relapse | [107] |
| MSCs from bone marrow | Mouse models with leukemia cell lines MOLT-4 and L1210 | Loaded with paclitaxel | Antileukemia activity in vitro and in the mouse model | [108] |
| MSCs from adipose tissue | MM cell lines in vitro: RPMI-8226 U-266, MMCAR-1, LIG-1, and MCC-2 | Engineered with the expression of TRAIL | Antimyeloma activity in vitro | [109] |
| iPSCs from murine embryonic fibroblasts | Murine T-cell lymphoma | Differentiation to NKT cells | Tumor growth suppression in the mouse model | [110] |
| iPSCs from murine embryonic fibroblasts | Raji human Burkitt lymphoma cell line | Differentiation to T cells expressing CD19 CAR | Tumor growth inhibition in the mouse model | [111] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motais, B.; Charvátová, S.; Hrdinka, M.; Šimíček, M.; Jelínek, T.; Ševčíková, T.; Kořístek, Z.; Hájek, R.; Bagó, J.R. A Bird’s-Eye View of Cell Sources for Cell-Based Therapies in Blood Cancers. Cancers 2020, 12, 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051333
Motais B, Charvátová S, Hrdinka M, Šimíček M, Jelínek T, Ševčíková T, Kořístek Z, Hájek R, Bagó JR. A Bird’s-Eye View of Cell Sources for Cell-Based Therapies in Blood Cancers. Cancers. 2020; 12(5):1333. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051333
Chicago/Turabian StyleMotais, Benjamin, Sandra Charvátová, Matouš Hrdinka, Michal Šimíček, Tomáš Jelínek, Tereza Ševčíková, Zdeněk Kořístek, Roman Hájek, and Juli R. Bagó. 2020. "A Bird’s-Eye View of Cell Sources for Cell-Based Therapies in Blood Cancers" Cancers 12, no. 5: 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051333