DFIQ, a Novel Quinoline Derivative, Shows Anticancer Potential by Inducing Apoptosis and Autophagy in NSCLC Cell and In Vivo Zebrafish Xenograft Models

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
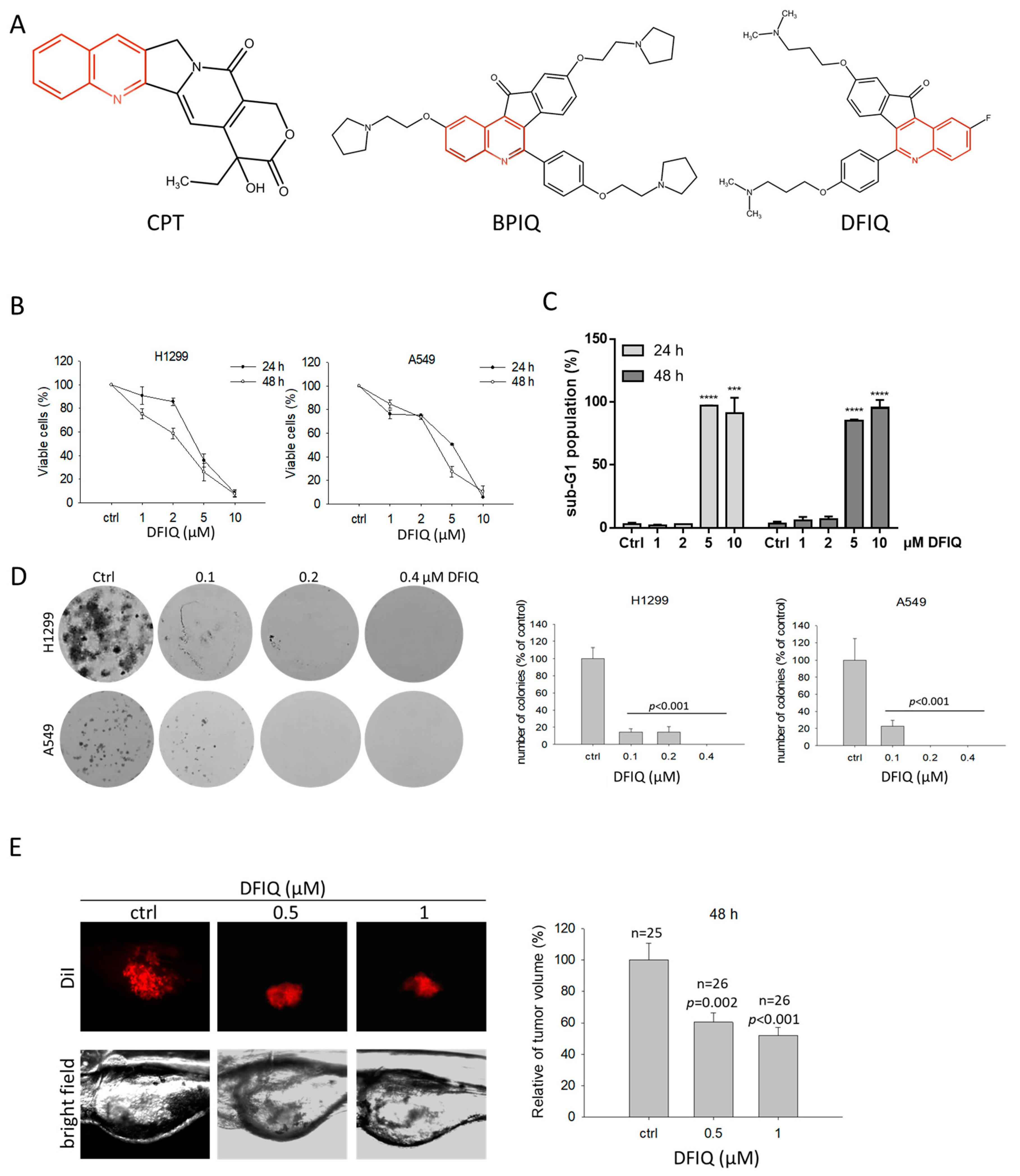
2.1. DFIQ Shows Anti-NSCLC Potential
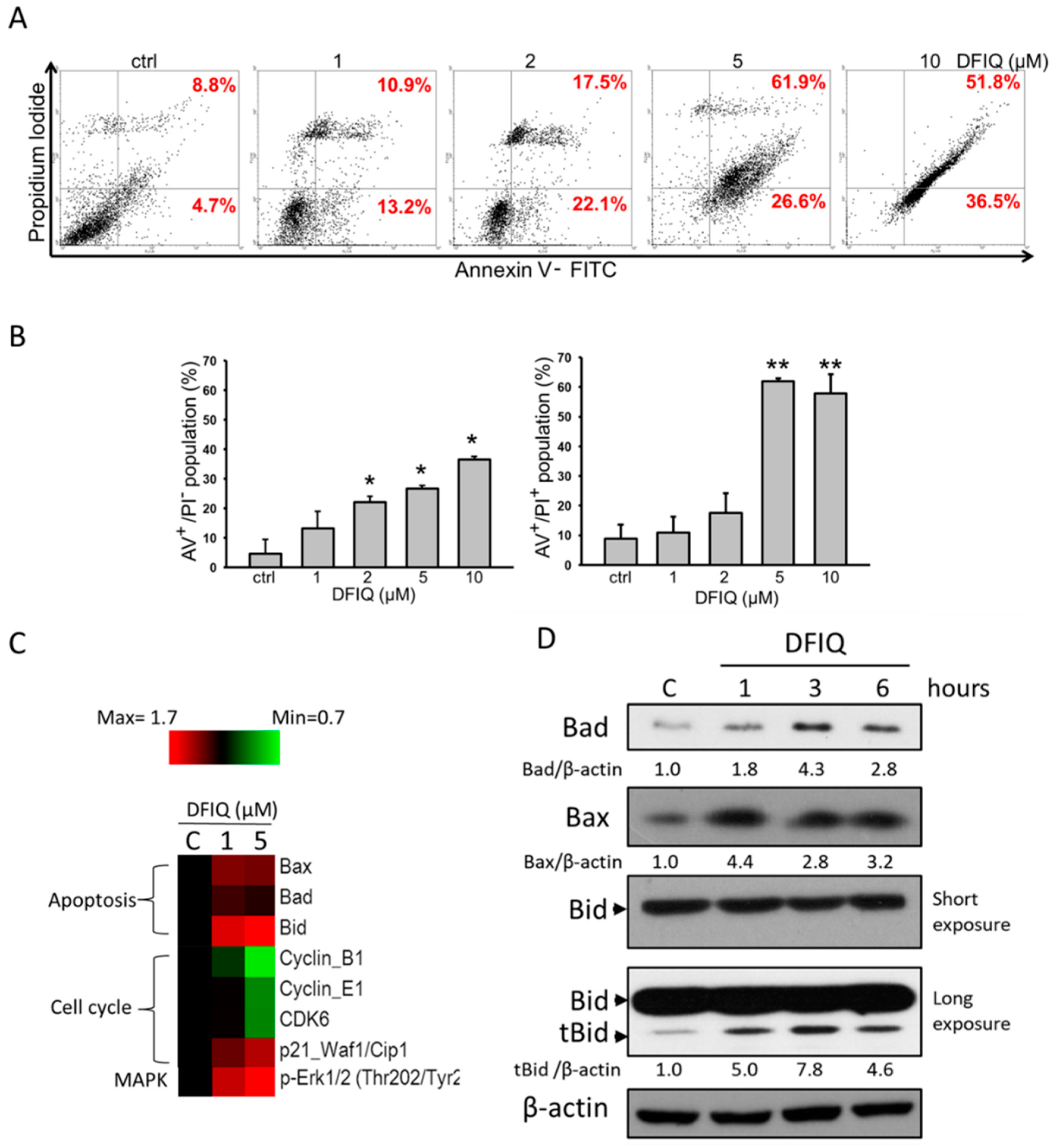
2.2. Apoptosis is Associated with DFIQ-Induced Cell Death
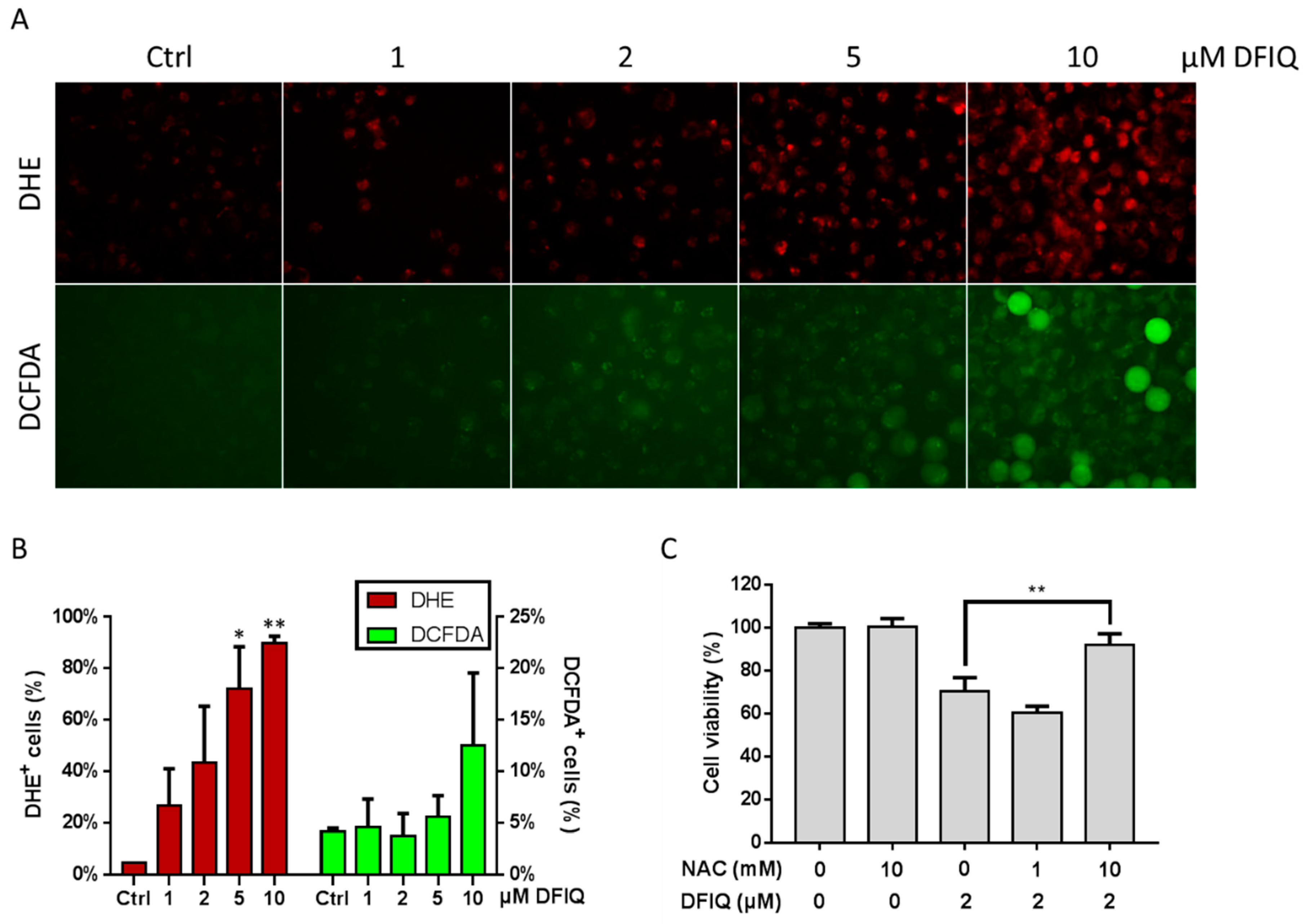
2.3. DFIQ Disrupted the Metabolic ROS Clearance Axis and Induced Cell Apoptosis
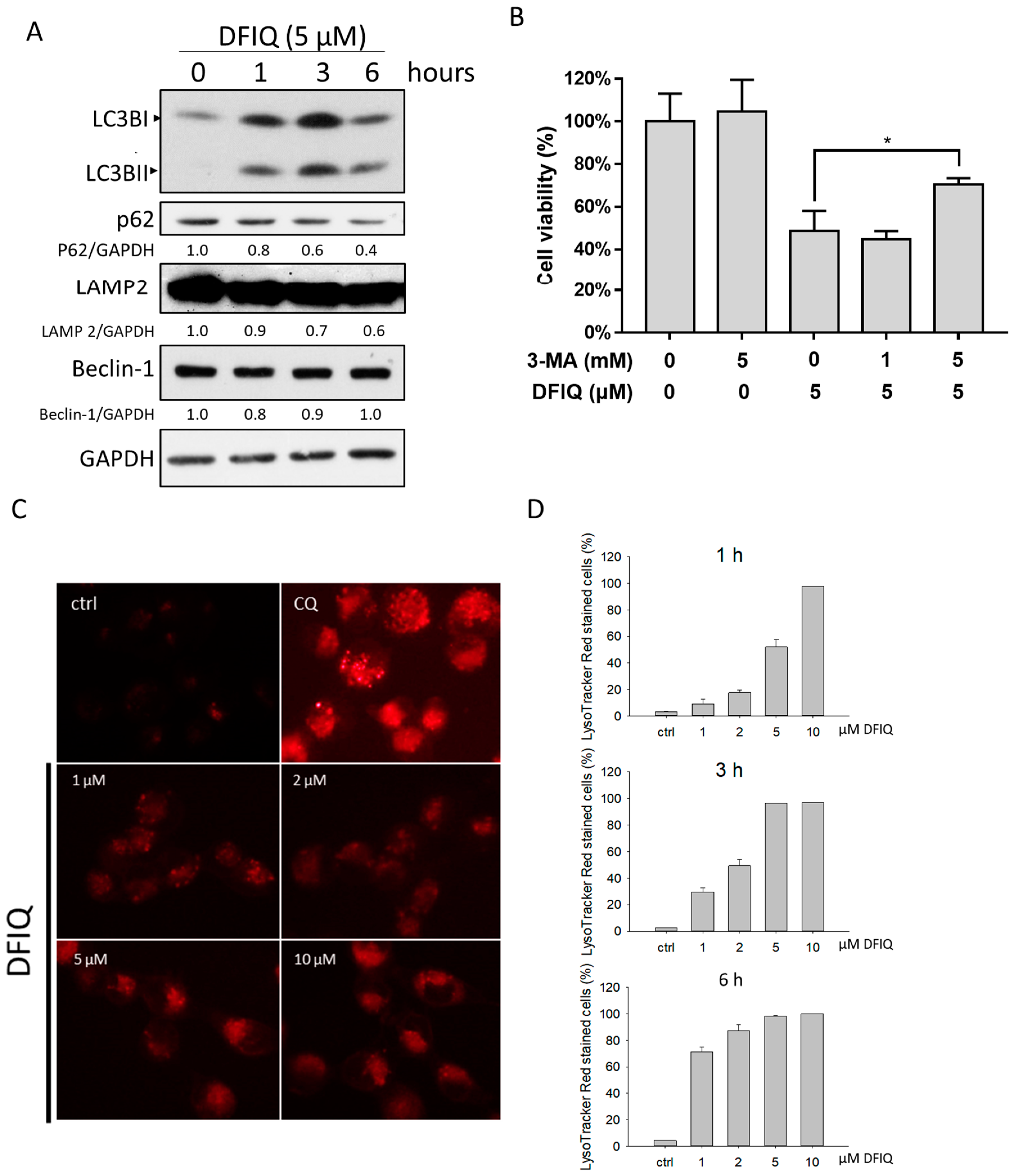
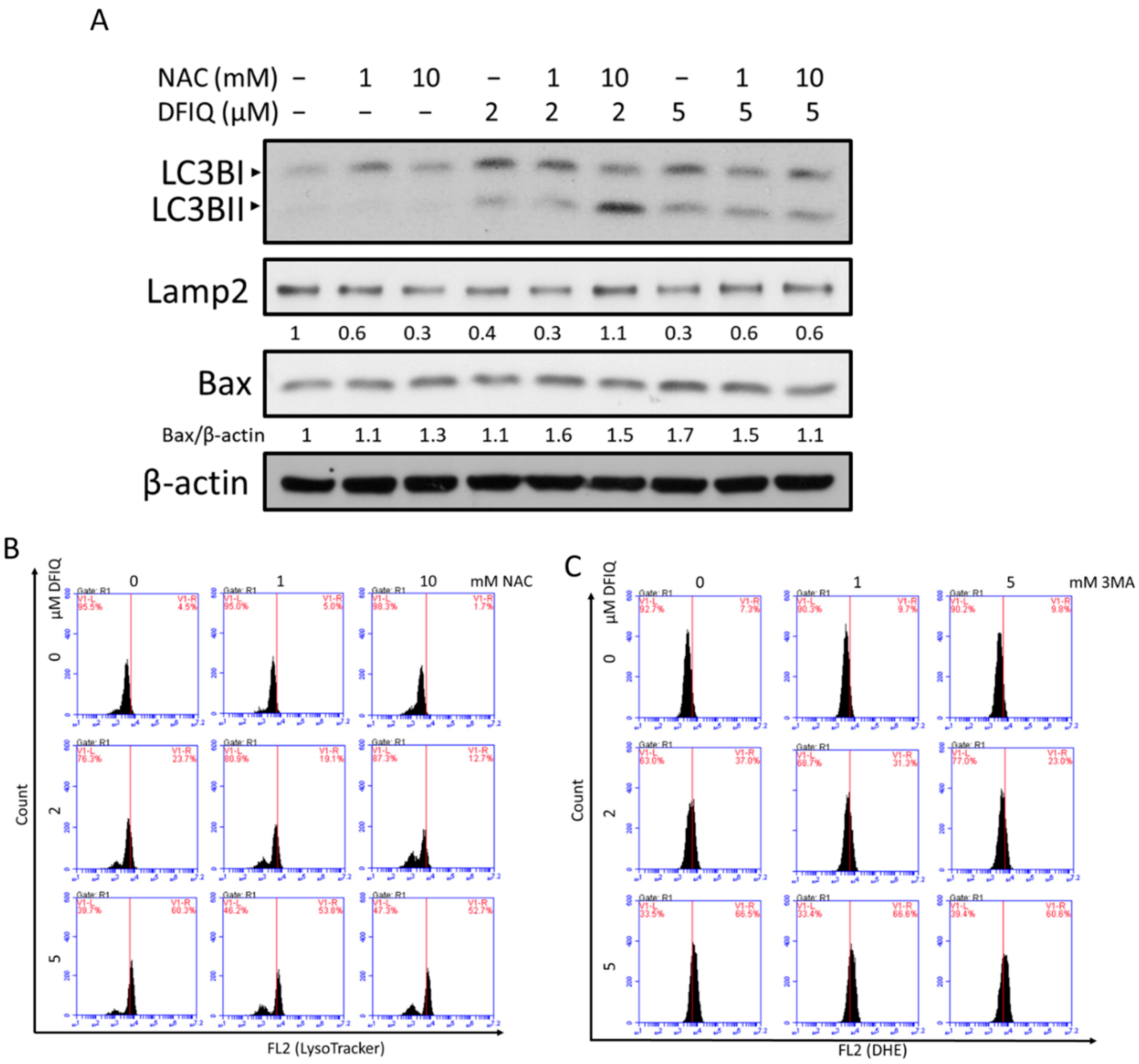
2.4. DFIQ-Induced Apoptosis is Initiated by Autophagy
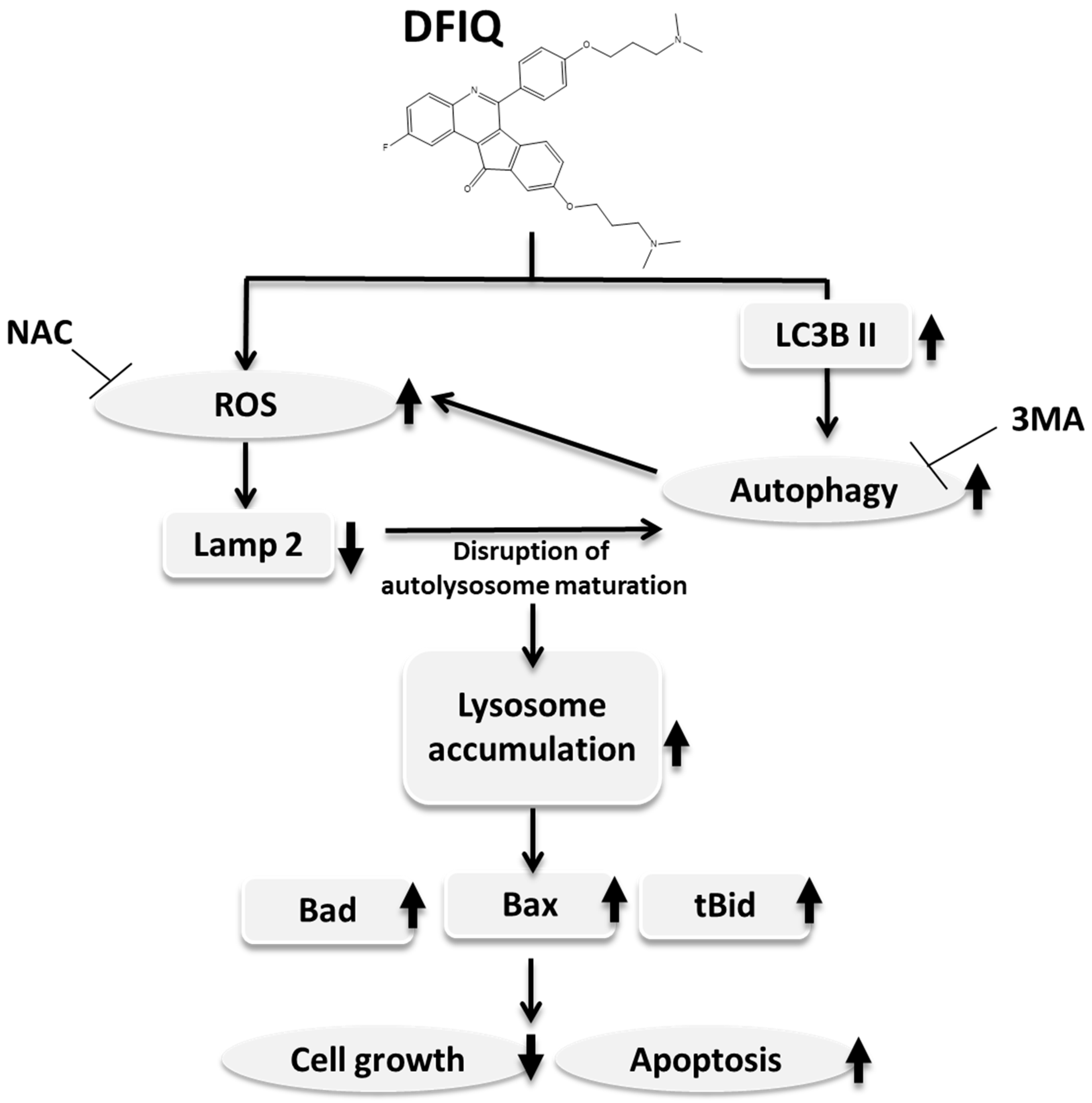
2.5. Crosstalk between DFIQ-Induced Autophagy and ROS Production
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability
4.3. Colony Formation Assay
4.4. Apoptosis Determination
4.5. Micro-Western Blot Array
4.6. Western Blot Analysis
4.7. ROS Detection
4.8. LysoTracker Red Assay
4.9. Statistical Analysis
4.10. Zebrafish Xenograft Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA 2019, 322, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.D.; Yang, G.Y. The role of chemotherapy and radiation in the treatment of locally advanced non-small cell lung cancer (NSCLC). Curr. Drug Targets 2010, 11, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Osmani, L.; Askin, F.; Gabrielson, E.; Li, Q.K. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin. Cancer Boil. 2017, 52, 103–109. [Google Scholar] [CrossRef]
- Sekgota, K.C.; Majumder, S.; Isaacs, M.; Mnkandhla, D.; Hoppe, H.; Khanye, S.D.; Kriel, F.H.; Coates, J.; Kaye, P.T. Application of the Morita-Baylis-Hillman reaction in the synthesis of 3-[( N -cycloalkylbenzamido)methyl]-2-quinolones as potential HIV-1 integrase inhibitors. Bioorganic Chem. 2017, 75, 310–316. [Google Scholar] [CrossRef]
- Chokkar, N.; Kalra, S.; Chauhan, M.; Kumar, R. A Review on Quinoline Derived Scaffolds as Anti-HIV Agents. Mini-Rev. Med. Chem. 2019, 19, 510–526. [Google Scholar] [CrossRef]
- Fan, Y.; Cheng, X.-W.; Wu, J.-B.; Liu, M.; Zhang, F.; Xu, Z.; Feng, L.-S. Antiplasmodial and antimalarial activities of quinolone derivatives: An overview. Eur. J. Med. Chem. 2018, 146, 1–14. [Google Scholar] [CrossRef]
- Gebreyohanns, E.A.; Bhagavathula, A.; Seid, M.A.; Tegegn, H. Anti-malarial treatment outcomes in Ethiopia: A systematic review and meta-analysis. Malar. J. 2017, 16, 269. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, J.; Yang, X.; Feng, X.; Li, X.; Huang, L.; Chan, A.S.C. Design, Synthesis, and Evaluation of Orally Bioavailable Quinoline–Indole Derivatives as Innovative Multitarget-Directed Ligands: Promotion of Cell Proliferation in the Adult Murine Hippocampus for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2018, 61, 1871–1894. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, X.; Wang, T.; Xiao, J. Quinolone hybrids and their anti-cancer activities: An overview. Eur. J. Med. Chem. 2019, 165, 59–79. [Google Scholar] [CrossRef]
- Pun, I.H.Y.; Chan, D.; Chan, S.H.; Chung, P.-Y.; Zhou, Y.Y.; Law, S.; Lam, A.K.-Y.; Chui, C.H.; Chan, A.S.C.; Lam, K.H.; et al. Anti-cancer Effects of a Novel Quinoline Derivative 83b1 on Human Esophageal Squamous Cell Carcinoma through Down-Regulation of COX-2 mRNA and PGE2. Cancer Res. Treat. 2017, 49, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamma, M.; Georgiev, V.S. Camptothecin. J. Pharm. Sci. 1974, 63, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Wani, M.C. Camptothecin and taxol: From discovery to clinic. J. Ethnopharmacol. 1996, 51, 239–254, discussion 253–234. [Google Scholar] [CrossRef]
- De Man, F.; Goey, A.; Van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef] [Green Version]
- Oh, I.; Kim, K.-S.; Park, C.; Kim, Y.-C.; Lee, K.-H.; Jeong, J.-H.; Kim, S.-Y.; Lee, J.E.; Shin, K.-C.; Jang, T.-W.; et al. Belotecan/cisplatin versus etoposide/cisplatin in previously untreated patients with extensive-stage small cell lung carcinoma: A multi-center randomized phase III trial. BMC Cancer 2016, 16, 690. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.-H.; Chen, Y.-L.; Chung, K.-Y.; Cheng, C.-M.; Wang, C.-H.; Tzeng, C.-C. Synthesis and antiproliferative evaluation of 6-arylindeno[1,2-c]quinoline derivatives. Bioorganic Med. Chem. 2009, 17, 7465–7476. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Tzeng, C.-C.; Yang, C.-L.; Lu, P.-J.; Chen, H.-L.; Li, H.-Y.; Chuang, Y.-C.; Yang, C.-N.; Chen, Y.-L. Synthesis and Antiproliferative Evaluation of Certain Indeno[1,2-c]quinoline Derivatives. Part 2. J. Med. Chem. 2010, 53, 6164–6179. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Chou, H.-L.; Chen, B.-H.; Chang, K.-F.; Tseng, C.-H.; Fong, Y.; Fu, T.-F.; Chang, H.; Wu, C.-Y.; Tsai, E.-M.; et al. BPIQ, a novel synthetic quinoline derivative, inhibits growth and induces mitochondrial apoptosis of lung cancer cells in vitro and in zebrafish xenograft model. BMC Cancer 2015, 15, 962. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.-C.; Hung, C.-T.; Chen, K.; Wu, W.-C.; Suen, J.; Chang, C.-H.; Lu, C.-Y.; Tseng, C.-H.; Chen, Y.-L.; Chiu, C.-C. Quinoline-Based Compound BPIQ Exerts Anti-Proliferative Effects on Human Retinoblastoma Cells via Modulating Intracellular Reactive Oxygen Species. Arch. Immunol. Ther. Exp. 2015, 64, 139–147. [Google Scholar] [CrossRef]
- Chang, W.-T.; Fong, Y.; Chuang, S.-C.; Chou, C.-K.; Chou, H.-L.; Yang, C.-F.; Tseng, C.-H.; Chen, Y.-L.; Chiu, C.-C. 9-bis[2-(pyrrolidin-1-yl)ethoxy]-6-{4-[2-(pyrrolidin-1-yl)ethoxy]phenyl}-11H-indeno[1,2-c]quinolin-11-one (BPIQ), a quinoline derivative inhibits human hepatocellular carcinoma cells by inducing ER stress and apoptosis. Anti-Cancer Agents Med. Chem. 2017, 17, 692–700. [Google Scholar] [CrossRef]
- Fong, Y.; Wu, C.-Y.; Chang, K.-F.; Chen, B.-H.; Chou, W.-J.; Tseng, C.-H.; Chen, Y.-C.; Wang, H.-M.D.; Chen, Y.-L.; Chiu, C.-C. Dual roles of extracellular signal-regulated kinase (ERK) in quinoline compound BPIQ-induced apoptosis and anti-migration of human non-small cell lung cancer cells. Cancer Cell Int. 2017, 17, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytom. Part A 2007, 71, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.-J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewson, G.; Kluck, R.M. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 2009, 122, 2801–2808. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S.A. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T. ROS signalling in the biology of cancer. Semin. Cell Dev. Boil. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2014, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Diwanji, N.; Bergmann, A. An unexpected friend—ROS in apoptosis-induced compensatory proliferation: Implications for regeneration and cancer. Semin. Cell Dev. Boil. 2018, 80, 74–82. [Google Scholar] [CrossRef]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Gu, L.; Smerin, D.; Mao, S.; Xiong, X. The Interrelation between Reactive Oxygen Species and Autophagy in Neurological Disorders. Oxidative Med. Cell. Longev. 2017, 2017, 8495160. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Boil. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.-M. Dual Role of 3-Methyladenine in Modulation of Autophagy via Different Temporal Patterns of Inhibition on Class I and III Phosphoinositide 3-Kinase*. J. Boil. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [Green Version]
- Mohan, N.; Sorokina, E.M.; Verdeny, I.; Alvarez, A.S.; Lakadamyali, M. Detyrosinated microtubules spatially constrain lysosomes facilitating lysosome–autophagosome fusion. J. Cell Boil. 2018, 218, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Horita, N.; Yamamoto, M.; Sato, T.; Tsukahara, T.; Nagakura, H.; Tashiro, K.; Shibata, Y.; Watanabe, H.; Nagai, K.; Inoue, M.; et al. Topotecan for Relapsed Small-cell Lung Cancer: Systematic Review and Meta-Analysis of 1347 Patients. Sci. Rep. 2015, 5, 15437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Chandra, V.; Jain, P.K.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef] [Green Version]
- Zastre, J.; Anantha, M.; Ramsay, E.; Bally, M. Irinotecan–cisplatin interactions assessed in cell-based screening assays: Cytotoxicity, drug accumulation and DNA adduct formation in an NSCLC cell line. Cancer Chemother. Pharmacol. 2006, 60, 91–102. [Google Scholar] [CrossRef]
- Loprevite, M.; Favoni, R.E.; De Cupis, A.; Pirani, P.; Merlo, D.F.; Grossi, F.; Ardizzoni, A. Pre-clinical evaluation of new antineoplastic agents in NSCLC cell lines: Evidence of histological subtype-dependent cytotoxicity. Int. J. Oncol. 1999, 15, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Zander, S.A.; Kersbergen, A.; Van Der Burg, E.; De Water, N.; Van Tellingen, O.; Gunnarsdottir, S.; Jaspers, J.E.; Pajic, M.; Nygren, A.O.; Jonkers, J.; et al. Sensitivity and Acquired Resistance of BRCA1;p53-Deficient Mouse Mammary Tumors to the Topoisomerase I Inhibitor Topotecan. Cancer Res. 2010, 70, 1700–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letrado, P.; De Miguel, I.; Lamberto, I.; Díez-Martínez, R.; Oyarzabal, J. Zebrafish: Speeding Up the Cancer Drug Discovery Process. Cancer Res. 2018, 78, 6048–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Shibuya, S.; Koyama, H.; Ozawa, Y.; Toda, T.; Yokote, K.; Shimizu, T. Sod1 Loss Induces Intrinsic Superoxide Accumulation Leading to p53-Mediated Growth Arrest and Apoptosis. Int. J. Mol. Sci. 2013, 14, 10998–11010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.; Shin, D.; Getzoff, E.; Tainer, J.A. The structural biochemistry of the superoxide dismutases. Biochim. Biophys. Acta 2009, 1804, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Li, Y.; Wang, S.; Wang, X.; Liao, C.; Hu, X.; Fan, L.; Kang, Q.; Zeng, Y.; Wu, X.; et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy 2016, 12, 1593–1613. [Google Scholar] [CrossRef] [Green Version]
- Polo, R.-A.G.; Boya, P.; Pauleau, A.-L.; Jalil, A.; Larochette, N.; Souquèxre, S.; Eskelinen, E.-L.; Pierron, G.; Saftig, P.; Kroemer, G. The apoptosis/autophagy paradox: Autophagic vacuolization before apoptotic death. J. Cell Sci. 2005, 118, 3091–3102. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, I.; Vosoughi, A.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2019, 287, 1005–1034. [Google Scholar] [CrossRef]
- Xie, H.; Xu, Q.; Jia, J.; Ao, G.; Sun, Y.; Hu, L.; Alkayed, N.J.; Wang, C.; Cheng, J. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem. Biophys. Res. Commun. 2015, 458, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; DeSantis, C.; Virgo, K.; Stein, K.; Mariotto, A.; Smith, T.; Cooper, D.; Gansler, T.; Lerro, C.; Fedewa, S.; et al. Cancer treatment and survivorship statistics, 2012. CA A Cancer J. Clin. 2012, 62, 220–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sun, D.; Hu, Y.; Xu, X.; Jiang, W.; Shang, H.; Cui, D. The roles of oxidative stress and Beclin-1 in the autophagosome clearance impairment triggered by cardiac arrest. Free Radic. Boil. Med. 2019, 136, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Jin, Y.; Yang, L.; Hou, Z.; Liu, Y.; Sun, T.; Pei, J.; Li, J.; Yao, C.; Wang, X.; et al. Promotion of SIRT1 protein degradation and lower SIRT1 gene expression via reactive oxygen species is involved in Sb-induced apoptosis in BEAS-2b cells. Toxicol. Lett. 2018, 296, 73–81. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [Green Version]
- Ciaccio, M.F.; Wagner, J.P.; Chuu, C.-P.; Lauffenburger, U.A.; Jones, R.B. Systems analysis of EGF receptor signaling dynamics with microwestern arrays. Nat. Methods 2010, 7, 148–155. [Google Scholar] [CrossRef]
- Marques, I.J.; Weiss, F.-U.; Vlecken, D.H.; Nitsche, C.; Bakkers, J.; Lagendijk, A.K.; Partecke, L.I.; Heidecke, C.-D.; Lerch, M.M.; Bagowski, C.P. Metastatic behaviour of primary human tumours in a zebrafish xenotransplantation model. BMC Cancer 2009, 9, 128. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (μM) |
|---|---|
| H1299 | 24 h: 4.16 μM |
| 48 h: 2.81 μM | |
| A549 | 24 h: 5.06 μM |
| 48 h: 3.53 μM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.-W.; Bow, Y.-D.; Wang, C.-Y.; Chen, Y.-C.; Fu, P.-R.; Chang, K.-F.; Wang, T.-W.; Tseng, C.-H.; Chen, Y.-L.; Chiu, C.-C. DFIQ, a Novel Quinoline Derivative, Shows Anticancer Potential by Inducing Apoptosis and Autophagy in NSCLC Cell and In Vivo Zebrafish Xenograft Models. Cancers 2020, 12, 1348. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051348
Huang H-W, Bow Y-D, Wang C-Y, Chen Y-C, Fu P-R, Chang K-F, Wang T-W, Tseng C-H, Chen Y-L, Chiu C-C. DFIQ, a Novel Quinoline Derivative, Shows Anticancer Potential by Inducing Apoptosis and Autophagy in NSCLC Cell and In Vivo Zebrafish Xenograft Models. Cancers. 2020; 12(5):1348. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051348
Chicago/Turabian StyleHuang, Hurng-Wern, Yung-Ding Bow, Chia-Yih Wang, Yen-Chun Chen, Pei-Rong Fu, Kuo-Feng Chang, Tso-Wen Wang, Chih-Hua Tseng, Yeh-Long Chen, and Chien-Chih Chiu. 2020. "DFIQ, a Novel Quinoline Derivative, Shows Anticancer Potential by Inducing Apoptosis and Autophagy in NSCLC Cell and In Vivo Zebrafish Xenograft Models" Cancers 12, no. 5: 1348. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12051348