Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy
by
 ,
,
Flávia Alves Verza
1 ,
,
Umashankar Das
2, 
Ana Lúcia Fachin
1,3,
Jonathan R. Dimmock
2,* and
Mozart Marins
1,2,3,4,* 1
Biotechnology Unit, University of Ribeirão Preto, Ribeirão Preto SP CEP 14096-900, Brazil
2
College of Pharmacy and Nutrition, University of Saskatchewan, 110 Science Place, Saskatoon, SK S7N 5C9, Canada
3
Medicine School, University of Ribeirão Preto, Ribeirão Preto SP CEP 14096-900, Brazil
4
Pharmaceutical Sciences School, University of Ribeirão Preto, Ribeirão Preto SP CEP 14096-900, Brazil
*
Authors to whom correspondence should be addressed.
Cancers 2020, 12(6), 1664; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061664
Submission received: 10 April 2020
/
Revised: 19 May 2020
/
Accepted: 19 May 2020
/
Published: 23 June 2020
Abstract
:Histones are the main structural proteins of eukaryotic chromatin. Histone acetylation/ deacetylation are the epigenetic mechanisms of the regulation of gene expression and are catalyzed by histone acetyltransferases (HAT) and histone deacetylases (HDAC). These epigenetic alterations of DNA structure influence the action of transcription factors which can induce or repress gene transcription. The HATs catalyze acetylation and the events related to gene transcription and are also responsible for transporting newly synthesized histones from the cytoplasm to the nucleus. The activity of HDACs is mainly involved in silencing gene expression and according to their specialized functions are divided into classes I, II, III and IV. The disturbance of the expression and mutations of HDAC genes causes the aberrant transcription of key genes regulating important cancer pathways such as cell proliferation, cell-cycle regulation and apoptosis. In view of their role in cancer pathways, HDACs are considered promising therapeutic targets and the development of HDAC inhibitors is a hot topic in the search for new anticancer drugs. The present review will focus on HDACs I, II and IV, the best known inhibitors and potential alternative inhibitors derived from natural and synthetic products which can be used to influence HDAC activity and the development of new cancer therapies.
1. Introduction
The mechanisms regulating gene expression involve a series of molecular modifications in DNA and chromatin and are responsible for the response to any type of physiological signaling in the organism [1]. This constant maintenance requires specific levels of control, and is undertaken by transcriptional regulators which bind to selected sequences in the DNA, inducing the production of proteins responsible for the structural modifications in chromatin and the consequent binding of regulators to DNA, causing epigenetic modifications [2]. Chromatin consists of the association of double-stranded DNA and proteins whose basic unit is the nucleosome. Chromatin can be divided into euchromatin, which corresponds to transcriptionally active DNA, and heterochromatin, which consists of inactive DNA and appears to exert structural functions during the cell cycle, allowing the cell to have control of the genes present in its nucleus [3,4]. The nucleosome core consists of 146 bp of DNA wrapped around a histone octamer. Histones are the main structural proteins associated with DNA in eukaryotic cells [5]. These proteins are divided into two groups: core histones, including H2A, H2B, H3 and H4, and linker histones (H1/H5) [6]. The four core histones H2A, H2B, H3 and H4 are assembled into H2A/H2B dimers and H3/H4 tetramers, forming the octamer complex composed of two H2A/H2B dimers and one H3/H4 tetramer, while the linker histone H1 binds to the DNA entry or exit sites on the surface of the nucleosomal core particle and completes the nucleosome [6,7]. These nuclear histones are small basic proteins composed of a large number of amino acids, mostly lysines and arginines [8]. Histones possess side chains that can be the target of covalent modifications such as acetylation, the mono-, di- and tri-methylation of lysins and the phosphorylation of serines [9]. Several of these side chain modifications can occur in the N-terminal tail domains of histones located outside the nucleosome, thereby weakening the binding between histones and DNA [10] and between regulatory proteins and histones [11,12]. This weakened binding can facilitate the action of transcription factors in this altered region, inducing or repressing gene transcription [13]. Specific modifications can also occur in the side chains of the internal histones of the nucleosome [14]. Histone modifications catalyzed by enzymes use the energy of ATP hydrolysis to modify nucleosomes [15] and this recruitment of complexes with specific enzymatic activities can influence gene transcription, replication, repair and recombination [14]. The tail domains of core histones can be modified by acetylation, phosphorylation, methylation, ubiquitination and sumoylation and less frequently, by citrullination and ADP-ribosylation [16]. These post-translational modifications alter the interactions between DNA and histones or the binding of proteins and transcription factors to chromatin [17]. Histone acetylation is a post-translational modification with functional implications for different cellular processes [18]. The presence of acetylated lysine in the histone tail results in a relaxed chromatin state, allowing the activation of gene transcription in that region. On the other hand, the deacetylation of lysine residues is associated with condensed chromatin, which impedes the transcription of genes present in that chromatin region [19,20]. The acetylation and deacetylation of lysine residues is controlled by two enzymes with opposite activities involved in gene regulation [21]. These reactions are catalyzed by enzymes with “histone acetyltransferase” (HAT) or “histone deacetylase” (HDAC) activity, which add or remove acetyl groups, respectively [22] (see Figure 1).
Different forms of HAT and HDAC have been identified, including coactivators that can interact with transcription regulators [23]. Deacetylation increases the ionic interactions between positively charged histones and negatively charged DNA, which produces a more compact chromatin structure, repressing gene transcription by preventing the access of the transcription machinery to the site. On the other hand, histone acetylation has been associated with other functions of the genome, such as producing a looser chromatin assembly that influences DNA repair and recombination [24,25]. The modifications in the histone chains define the compaction or relaxation of chromatin through the recruitment and binding of various specific proteins, functioning as an epigenetic machinery [26]. This epigenetic information is an important component in the regulation of gene expression since the breakdown of epigenetic integrity has been associated with different diseases, including cancer [27,28]. The addition of an acetyl group to a lysine residue neutralizes its positive charge on the nitrogen atom, alters the structure of the amino acid and blocks other modifications at this specific site [29]. This epigenetic alteration is related to disorders and diseases because of its participation in various physiological processes, acting in synergy with transcription factors, oncoproteins and kinases, affecting protein stabilization and activating or inhibiting gene transcription and DNA repair [30].
The HATs are divided into two classes (type A and type B) according to their localization and function in the cell. Their activity can be modulated by several protein–protein interactions, protein cofactors and autoacetylation. Type A HATs are nuclear enzymes which contain acetyl-CoA binding sites. This class of HATs probably catalyzes acetylation and events related to gene transcription. Type B HATs are believed to have a maintenance role in cell functioning as cytoplasmic enzymes that modify free histones in the cytoplasm after their synthesis and transport them to the nucleus, where they can be deacetylated and incorporated into chromatin [31]. In addition, HATs can be divided into four families based on their primary structure: GNAT (Gcn5, PCAF, Hat1, Elp3 and Hpa2); p300/CBP (p300 and CBP); MYST (Esa1, MOF, Sas2, Sas3, MORF, Tip60 and Hbo1) and Rtt109 [31,32]. One specific HAT can acetylate several lysine residues of a histone. Many HATs contain bromodomains for the recognition of the acetylated lysine. The acetyl lysine residues in the histone tails form bromodomain binding sites where the adjacent amino acids determine specificity. This indicates that acetylation, like many protein phosphorylation events, and creates a new binding surface to recruit other proteins to the nucleosome [33]. Histone acetylation occurs throughout the cell cycle and differs from histone methylation, which has greater activity in the G2 phase [34]. The acetylation pattern of certain lysine residues in the histone tails appears to result from the opposite activities of HATs and HDACs. These changes in the acetyl group can be quickly reversed in some chromatin environments, suggesting that the ‘transient’ nature of gene expression may be linked to the degree of acetylation. Even though acetylation and histone methylation are dynamic and involved in several biological processes, little is known about how the different functional domains of chromatin are established and maintained [35]. Finally, the activity and specificity of these acetyltransferases can be altered by factors such as autoacetylation proteins and chaperones [36,37].
HDACs are a family of enzymes that play important roles in different biological processes, mainly because of their gene transcription-repressing activity [38]. These enzymes are able to remove acetyl groups (O=C-CH3) from an ε-N-acetyl lysine in a histone, an event that confers to histones the capacity to compact DNA [39]. The enzymatic activity that catalyzes the deacetylation of histones was discovered for the first time in 1969 [40]. HDACs possess a catalytic domain that requires a Zn2+ ion (classes I, II and IV) [41] or NAD+ (class III) [41] (see Figure 2).
Eighteen HDACs have been identified in humans (Table 1), which are divided into four classes: class I Rpd3-like proteins (HDAC1, HDAC2, HDAC3, and HDAC8) which have a nuclear localization. Class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and IIb (HDAC6 and HDAC10) Hda1-like proteins which show a specific expression in tissue and can be transported between the nucleus and cytoplasm, suggesting the involvement of this HDAC class in the acetylation of non-histone proteins [42]. Class III are Sir2-type proteins (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7). The specific expression pattern of this class is unknown, and its mechanisms differ from those of the other two classes and are not in the scope of this review. The protein of class IV (HDAC11) shows homology to both class I and class II members [43]. Table 1 summarizes these three related classes. Evidence suggests that HDACs can also deacetylate non-histone proteins such as hormone receptors, chaperones and cytoskeletal proteins that regulate cell proliferation and death [43,44,45,46]. The HDACs can form gene silencing complexes with nuclear receptors when a specific ligand is absent [47]. Studies have indicated that HDACs can regulate the expression of various genes through the interaction with transcription factors such as E2f, Stat3, p53, NF-κB and TFIIE [48]. The absence of HDAC1 leads to reduced deacetylase activity and the hyperacetylation of other histones. In addition, an increase in HDAC2 and HDAC3 expression is observed in HDAC1-deficient cells, which is unable to compensate for the loss of HDAC1, suggesting a unique function of this enzyme [49]. HDAC4 has been described as an important regulator of chondrocyte hypertrophy during skeletogenesis and a general role of class II HDACs in the control of cellular hypertrophy has also been suggested [50,51].
HDAC1 and HDAC2 were discovered in 1996 [69,70], while HDAC3 was described in 1997 [71]. HDAC4, 5 and 6 were characterized in the same year, HDAC7 in 1999 [72] and HDAC8 in 2000 [73,74]. Many different forms of HDACs can arise from nucleotide polymorphisms or alternative splicing. For example, different isoforms of HDAC9 have been described [75]. Although HDAC4, HDAC5, HDAC7 and HDAC9 have similar functions in the regulation of cytoplasmic-nuclear transport and DNA binding, they are encoded by different genes and are not isoforms [41,76]. HDAC10, on the other hand, has no association with other proteins forming complexes, which indicates that the involvement in the transcription control occurs by other means [77]. Like HDAC10, HDAC11 was discovered in 2002 [78] and so far they are the least studied and understood HDACs.
2. Class I HDACs
Class I HDACs are multiprotein complexes (except for HDAC8) that can be expressed simultaneously at different sites [39]. Enzymes of this class are involved in cell proliferation and survival [38,79]. HDACs 1 and 2 are found in the complex that represses the expression of neuronal genes in non-neuronal tissues [80]. HDAC1 was found to exert a protective function against the formation of teratomas with malignant potential in mice and human patients [81]. HDAC2 negatively regulates memory formation and synaptic plasticity [82]. HDAC1 and HDAC2 repress the expression of proteins p21 and p57 which regulate the transition from the G1 to the S phase of the cell cycle in fibroblasts [83]. HDAC3 also has a repressive function when it interacts with other molecules forming an enzyme complex, in addition to playing an important role among class I HDACs in gene expression in inflammation [84]. The activity of HDAC8 was found to be dependent on oxidoreduction reactions [85].
2.1. The HDAC1/2 Functional Complex
HDAC1 and HDAC2 are homologous proteins (82–85% similar in human proteins) and are part of stable multiprotein complexes [86,87]. These complexes account for about 50% of all deacetylase activity in embryonic stem cells [88] and T cells [89]. HDAC1/2 cannot bind directly to DNA without interacting with other specific molecules and its activity was reduced in the absence of this binding [90]. These molecules can be transcription regulators, transcription factors and DNA-binding factors, as well as coactivator and corepressor complexes that alter the chromatin structure [91]. Corepressors act on transcriptional silencing by recruiting promoters of chromatin remodeling that can inhibit or silence the basal transcription mechanism [92]. Some of the known HDAC1/2 corepressor complexes are Sin3A, nucleosome remodeling and deacetylase (NuRD), CoREST, mitotic deacetylase (MiDAC) and SMRT/NCor, which are recruited to chromatin by transcription factors [2,9,93,94,95]. As described below, the structures of Sin3A and NuRD exemplify the diversity of sizes and the number of subunits of these complexes. This diversity and the incorporation of sequence-specific DNA-binding proteins can modulate their function and cellular context activity [96]. It is extremely important that the machinery involved in the deacetylation of histones is not only mechanically regulated but also with high specificity. Even today, little is known about how this specificity is achieved; however, studies have indicated that different multiprotein complexes are involved in this regulation.
2.2. Sin3A Complex
The Sin3/HDAC corepressor complex is a multiprotein complex that mediates gene repression by recruiting HDACs (class I, especially HDAC1 and HDAC2) and other chromatin-modifying enzymes [97] and also acts as a coactivator and general transcription factor [98]. The Sin3A core complex includes HDAC1, HDAC2, Sin3a, RbAp46 and RbAp48, RbAp4, RbAp7, SAP30, SAP18 and SDS3 [96,99,100]. Besides these core proteins, several other proteins have been associated to this core complex and include SAP180 [101], RBP1 [102], BRMS1 [103], SAP130 [101], SAP25 [104], MeCP2 [105] and ING1/2 [100] (see Figure 3A). These factors are essential for the Sin3A complex and exert their function through different types of interaction mediated by amphipathic helical domains that contain a polar and an apolar end and conserved segments [106]. Although structurally similar, the amphipathic helical domains and conserved segments have binding specificity [107]. The Sin3A complex has also been associated with other enzymatic activities depending on the molecules of the interaction [87,108,109]. This complex plays an important role in maintaining the pluripotency of embryonic stem cells [110]. On the other hand, the acetylation of STAT3 and its association with Sin3A contribute to the oncogenic potential of STAT3 [111]. The absence of the Sin3 complex can result in positive and negative gene regulation [112,113]; however, its interaction mode with these regulators still remains unknown.
2.3. NuRD Complex
The nucleosome remodeling deacetylase complex (NuRD), also known as Mi-2, according to stoichiometry data [114,115], is composed of one copy of the CHD3 (Mi2α) or CHD4 (Mi2β) proteins (chromodomain, helicase, DNA binding domain), one HDAC1 or HDAC2, three MTA1/2/3 (metastasis associated), one copy of MBD2 and MDB3 proteins (Methylated CpGBinding), six copies of RbAp46/48 proteins (retinoblastoma associated protein), two GATAD2a/b (p66a/b) and two DOC-1 (deleted in oral cancer) [115]. Other studies have also indicated that four molecules of RBBP4/7 (4/7 retinoblastoma binding protein) integrate the NuRD core complex [116,117] (see Figure 3B). This protein complex is conserved in animals and is widely expressed in most tissues, influencing gene transcription, chromatin assembly, cell cycle progression and genomic stability [118]. The role of the NuRD complex is determined by the combination of the six main protein subunits that make up this complex [119]. Several functional differences exist between the enzymes that form the NuRD complex, a fact that interferes with the specialized functions of the complex, which can act in different types of cells and biological systems. For example, MBD2 and MBD3 are related proteins with a methyl-CpG consensus binding domains that are found exclusively in NuRD complexes [120]. MBD2 recognizes and binds to methylated DNA, while MBD3 contains an amino acid alteration that prevents this binding [121,122]. In addition to their functions within the NuRD complex, some subunits of this complex such as MBD3 can serve as a protein interaction domain and bind to other protein complexes, for example the JUN oncoprotein [123]. The NuRD complex also regulates how DNA is read in different cells. This feature is extremely important to transform adult cells into induced pluripotent stem cells (iPSCs), an epigenetic change that may be used to treat different diseases [124].
These complexes show a diversity in the sizes and numbers of subunits. Larger complex formation is required for better activity. The MiDAC and CoREST complexes also contain HDAC I and II enzymes. MiDAC is a tetrameric complex that is composed of HDAC1/2, DNTTIP1 (deoxynucleotidyltransferase terminally interacting protein 1) and the protein co-repressor MIDEAS (SANT domain associated with mitotic deacetylase) [125,126]. The CoREST complex contains a single copy of CoREST1/2/3, LSD1 demethylase (specific lysine demethylase 1) and HDAC1/2 proteins [127]. NCoR/SMRT is associated with HDAC3 and contains transducin β-like protein 1 (TBL1)/TBL1-related protein 1 (TBLR1) and G-protein pathway suppressor 2 (GPS2) [96]. Class II HDAC activity is dependent on the interaction with the SMRT/NCoR–HDAC3 complex [128].
3. Class II HDACs
Class II HDACs (HDAC4, 5, 6, 7, 9 and 10) are found in the nucleus and cytoplasm (see Table 1) and can freely shuttle between these two compartments, exhibiting specific functions in tissues [38,79]. For example, HDACs 4, 5 and 7 regulate cell differentiation according to a specific signal, which results in changes in gene expression [39]. This class of HDACs can be subdivided into class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10) [38]. Class II HDACs do not only act as transcription repressors but also interact with non-histone substrates, inducing autophagy and regulating the microtubules of the cytoskeleton [129].
Class IIa HDACs have low enzymatic activity but can recruit other protein complexes, exerting deacetylase function [128]. HDACs of this class can induce the conversion of cell signaling by presenting conservative serine residues in the regulatory N-terminal domains, with reversible phosphorylation [130]. This phosphorylation leads to the activation of several kinases and phosphatases which, when functioning downstream of biological pathways, regulate the transit of HDACs between the cytoplasm and nucleus, as well as their binding to DNA [131]. The phosphorylation of class IIa HDACs is crucial for the determination of their localization and transcriptional repression capacity in the nucleus. For example, in the nucleus, HDAC9 represses proteins such as myocyte enhancer factor-2 (MEF2) until a myogenic differentiation signal causes its export to the cytoplasm [130]. Class IIa HDACs have been shown to exert their transcriptional repressive function in different tissues such as skeletal, cardiac and smooth muscles, bone, immune system, vascular system and brain [132,133]. Most known HDAC inhibitors do not affect class IIa HDACs [134].
Class IIb HDACs (HDAC6 and HDAC10) possess duplicated catalytic domains and are usually localized in the cytoplasm [134]. HDAC10 is very similar to HDAC6, with both forms containing a second catalytic domain that is not found in other HDACs. However, in HDAC10, this domain has no known function [135]. HDAC10 together with HDAC9 has been shown to be necessary for homologous recombination activity, but it remains unclear whether they are direct participants in this process or act through transcriptional control [136]. HDAC6 catalyzes the deacetylation of α-tubulin and promotes microtubule- and actin-dependent cell motility. In addition, this enzyme plays a critical role in the clearance of misfolded proteins by inducing autophagy [137]. HDAC6 contains two catalytic sites and a ubiquitin-binding domain (see Figure 1). The latter is important for the response to cytotoxic protein aggregates [138]. HDAC6 is an important potential therapeutic target for the treatment of diseases such as Alzheimer’s and cancer [139].
Class I and II HDACs show great sequence similarity in proteins of the same class, directed to the catalytic site. For example, HDACs 1, 2 and 3 have a homologous sequence and can be expressed together, but only HDACs 1 and 2 together form other co-repressor complexes in the cell while HDAC3 joins other proteins to exert its repressive activity [140]. Except for HDAC8, all other HDAC isoforms are always found associated with other proteins, or other HDAC isoforms, in multiprotein complexes [141]. In the same cell, there may be similar isoforms being expressed that may be part of different DNA binding complexes. The homologous proteins of HDAC1, HDAC2 and HDAC3 are regulated in the absence of HDAC1, but mammalian cells need to maintain specific levels of deacetylase activities to ensure the uninterrupted and efficient cell cycle progression. We can also observe that the loss of HDAC1 results in an increase in acetylation at its specific activity site, which can affect the expression of specific genes [49].
Specific patterns of histone acetylation and deacetylation do not occur by chance, they are influenced by other modifications of histones. These post-translational modifications together generate a ‘histone code’ [142]. For example, the acetylation of histone H3-K9 and the methylation of H3-K4 are associated with active transcription. The loss of the acetylation of histone H3-K9 and the gain of the methylation of H3-K9 and H3-K27 are indicative of heterochromatin [143]. In many cases, the modifications of a single residue are mutated exclusively. The presence of a modification can induce additional modifications in nearby amino acids, expanding the change in the protein coding information. Gene promoters suppressed by the HDAC inhibitors generally contain hypermethylated DNA, indicating interference between the histone acetylation/methylation and DNA methylation. Each of these events can have profound implications for gene expression in normal and cancerous cells [144].
4. Class IV HDACs
HDAC11 is the only class IV HDAC. The expression of this enzyme was observed in some tissues such as the brain and heart, but little is known about its function [78,145]. HDAC11 is related to immune system regulation since the suppression of this deacetylase promoted the expression of IL-10 in animals [146].
HDACs have been studied extensively, especially because of their role in cancer. In contrast, no clinical applications have been described for HATs. However, the latter may have important functions in inflammatory diseases, cancer and neurological disorders since histone acetylation results in less condensed DNA and increased gene transcription [147,148]. HATs can act on different cell substrates such as histones, transcription factors, enzymes and nuclear receptors. Despite its potential, the development of HAT inhibitors proved to be challenging and a large gap remains between the biological activity of HAT inhibitors in in vivo studies and their use as therapeutic agents, since many HAT gene knockout mutants are incompatible with life in mice [149].
5. Participation of Transcription Factors in the Mechanism of Regulation by HDACs
HDACs can exert direct effects on physiological processes such as apoptosis, differentiation, metabolism and inflammation through the deacetylation of non-histone proteins, affecting their functions, cellular localization and protein–protein interactions [150]. Some proteins such as p53, NF-κB, STAT3, Hsp90, SCL/TAL1, OCT1, YY1, Akt and Ku70 are modified by HDACs, with consequent changes in embryonic development and cell proliferation, differentiation and death [46] (see Figure 4).
Other proteins that serve as transcription factors can also be modulated by chromatin status, limiting their capacity to bind to DNA and to activate transcription. Some transcription factors are specifically expressed in certain tissues and regulate specialized cellular functions; thus, the silencing of these transcription factors may result in a functional blockade. The deletion of the SCL/TAL1 transcription factor, for example, resulted in the inability to generate hematopoietic precursors and caused embryo death in mice [151]. Likewise, the silencing of the gene encoding transcription factor organic cation transporter 1 (OCT1) also caused disturbances in embryonic development [152]. The Yin Yang 1 (YY1) transcription factor can regulate different genes and has specific DNA-binding activity and transcriptional repression [153]; however, this transcription factor requires co-activators or co-repressors for its full function, thus interacting with HATs (CBP and p300) and class I HDACs (HDAC1, 2 and 3) [154]. Several transcription factors participate in the regulation of these pathways. These factors directly or indirectly regulate DNA repair genes which are also part of the DNA repair machinery [155]. The tumor suppressor protein p53 is an important transcription factor, particularly for the response to stress and cellular homeostasis. Under normal physiological conditions, this protein is maintained at low levels by its negative regulator MDM2 [156]. In the absence of MDM2, the levels of p53 increase, causing embryonic lethality in mice [157]. The acetylation of p53 at different lysine residues is mediated by p300/CBP, increasing its DNA-binding capacity and the consequent transcription of its target genes [158,159]. This even occurs in response to cell damage but is transitory and reversible, as the post-translational modifications maintain p53 acetylation under control [160].
During the response to genotoxic stress, p53 functions as a transcription factor and regulates effector genes such as GADD45A and p21 [161]. The high expression of Gadd45A can reduce the efficiency of DNA repair [162]. HDACs can decrease the activity of p53; for example, the overexpression of HDAC1 was found to reduce p53 acetylation in vivo [163]. MDM2 can recruit HDAC1, promoting the deacetylation of p53, and this silencing can more quickly interrupt the function of this protein when its target genes are no longer necessary [164].
The transcription factor Sp1 (specificity protein 1) is found in all animal cells where it directly or indirectly regulates the expression of genes such as Gadd45A and MGMT. However, in response to DNA damage, Sp1 is phosphorylated and recruited to the sites of DNA double-strand breaks where it possibly mediates the recruitment of chromatin remodeling factors involved in the repair of these breaks [165]. Sp1 can define the binding of Gadd45A at a specific site of DNA damage [166]. Sp1 is a specificity protein that belongs to the family of Krüppel transcription factors (Sp/KLF), which currently has 26 members [167]. The DNA-binding domain consists of three Cys2His2 zinc finger proteins (81 amino acids per protein), which are responsible for recognizing the GC (GGGGCGGGG) and GT/CACC (GGTGTGGGG) sequences in DNA [168]. Post-translational modifications in Sp1 modulate chromatin remodeling factors, DNA, the transcription machinery and other transcription factors to induce or repress expression [169,170]. The high expression of Sp1 has been observed in several types of cancer [170]. Sp1 can increase the activity of a gene promoter or recruit other protein complexes to exert activating or repressing functions. In breast cancer, human epidermal growth factor receptor type 2 (HER2) signaling phosphorylates Sp1, which recruits HDAC1 to the sites of gene regulation, forming a protein complex that can involve other regulators [171]. GM2 synthase is an enzyme that produces glycosphingolipids. The high expression of this enzyme is associated with a poor tumor prognosis. Its activation is regulated by histone acetylation and a reduction in the Sp1-HDAC1 repressor complex [172]. In multiple myeloma, the inhibition of HDACs was associated with the down-regulation of Sp1, indicating that the effects of HDAC expression might be mediated by Sp1 [173].
HDACs are involved in the dysregulation of pathways and transcription factors in the various phases of cancer cells. The exacerbated expression of HDACs may be one of the factors responsible for the worst prognosis for patients, with stomach and ovarian cancer [54], neuroblastoma [66] and multiple myeloma (MM) [174] for example.
Epigenetic dysregulation can nurture the onset and progression of various human diseases. HDACs induce several cellular and molecular effects through the hyperacetylation of histone and non-histone protein substrates, which are involved in the regulation of cell cycle, apoptosis, DNA-damage response, metastasis, angiogenesis, autophagy and other cellular processes being able to influence the onset or progression of diseases such as cancer [175]. However, the contribution of HDACs to this pathology may not be related to the level of expression of these proteins. HDACs can function as catalytic subunits of large protein complexes and can be recruited in ways that alter the expression of several protein genes in order to induce a tumor. The molecular and biological consequences of inhibiting HDACs need to be analyzed in this context. HDACs can be targeted using small molecules and more selective agents [176]. The inhibition of HDAC10 in combination with doxorubicin treatment decimates neuroblastoma, but not healthy cells, preventing the efflux of drugs, as well as improving DNA damage [177], and the combined genetic deletion of HDAC1 and HDAC2 results in the activation of a senescent program and the death of transformed cells [178]. HDAC1 has oncogenic activity in tumor cells, but can have different functions in different subpopulations, but the combined genetic deletion of HDAC1 and HDAC2 results in accelerated leukemogenesis. One study noted that in the pre-leukemic phase, HDAC1 blocks differentiation; compromises genomic stability; and increases self-renewal in hematopoietic progenitors, all events affected by the reduction of HDAC1 levels. The short-term treatment of pre-leukemic mice with an HDAC inhibitor (HDACi) accelerated leukemogenesis. On the other hand, the absence of HDAC1 in mice led to a longer survival time for the animals. Thus, HDAC1 has a dual role in tumorigenesis: oncosuppressive in the early stages and oncogenic in established tumor cells [179]. The authors suggest that the inhibition of HDACs may block the intrinsic antitumor functions of these proteins. However, further studies are extremely important for a greater understanding of the role of HDACs alone and together, in different stages of carcinogenesis and in different types of tumor cells.
6. Inhibitors of HDACs
HDAC proteins are a promising class for drug targets due to the importance of these enzymes in a variety of processes, including cell cycle regulation, proliferation, survival, differentiation, metabolism, protein trafficking, DNA repair and angiogenesis. In recent decades, a class of inhibitors that block HDAC activity have been discovered. These inhibitors are able to inhibit gene silencing through the hyperacetylation of histones, acting on the regulation of gene expression and influencing cell growth and differentiation and the induction of apoptosis in neoplasms [180]. Numerous synthetic or natural molecules that aim at classes I, II and IV enzymes have been developed and characterized, although interest in the class III Sirtuin family is increasing. Class I, II and IV exhibit their Zn2+-dependent deacetylase activity. The binding of HDACi to this ion, found in the active site of HDACs, alters the deacetylase activity of these proteins and damages their enzymatic function [181]. As the expression of HDACs is not organized in several types of cancer, the disrupted balance between HATs and HDACs in neoplastic cells can contribute to carcinogenesis and the reversible modulation function of HDACs makes these proteins interesting targets for cancer treatment [41]. HDACi can neutralize the abnormal acetylation status of proteins found in cancer cells and can reactivate the expression of tumor suppressors. Cancer cells may also be more sensitive to HDACi-induced apoptosis than normal cells, enhancing the therapeutic potential of HDACi [182].
The effect of HDAC inhibitors (HDACis) is not restricted to histone proteins. These inhibitors can also target non-histone proteins, transcription factors, regulators, signal transduction mediators, DNA repair proteins and chaperones [183,184,185] (see Figure 5). There has been increasing interest in producing these drugs to better understand the functions of HDACs and to investigate the anticancer potential of these inhibitors [186].
Most HDACs have a Zn2+-dependent active site that can be inhibited by compounds with the ability to chelate this ion [187]. The currently used HDACis possess a pharmacophore that can bind to this active site and block it [188]. The preclinical efficacy of HDACis is associated with the gene activation mediated by these drugs, which promote the hyperacetylation of the N-terminal tails of histones, facilitating the access of transcription factors to gene promoters [180]. HDAC inhibitors can be natural or synthetic compounds that differ in terms of their target specificity [189]. Many structurally diverse compounds can bind to HDACs and inhibit their enzymatic activity. These compounds are classified into two large classes, isoform-selective inhibitors and pan-inhibitors, which act against all class I HDACs [134,190].
These inhibitors are divided into five subgroups based on their chemical structure: short-chain fatty acids that include hydroxamic acids, benzamides, cyclic peptides, and sirtuin inhibitors [19,176,191]. The functions of immune system cells with excessive accumulation of acetylated histones may be altered and it is therefore of the utmost importance to carefully select HDACis for the treatment of diseases such as cancer [175]. The inhibitors of HDACs render cancer cells more sensitive to immunotherapy, increasing the expression of antigens present in the tumor and thus acting as immunomodulators [192]. The anticancer activity of HDACis comprises different molecular and physiological events such as the inhibition of the vascular endothelial growth factor (VEGF), endothelial nitric oxide synthase (eNOS) and TGFβ1 [193,194,195]. The apoptosis of tumor cells induced by HDACis is associated with their ability to selectively regulate proapoptotic pathways [196], which does not occur in normal cells [197]. There are a number of different classes of HDACis which are available to treat cancers.
First, there are the drugs which contain a hydroxamic acid group such as vorinostat [198], whose structure, and that of other HDACis, are presented in Figure 6. The function of the hydroxamic acid group is to chelate with zinc which is located in the active site of the enzymes. Another HDACi which contains the hydroxamic acid group is trichostatin A which is a highly toxic compound [199]. These compounds can inhibit all classes of HDACs. A second cluster of drugs which also interacts with zinc are benzamide derivatives, as exemplified by entinostat, which inhibits class I HDACs. A third group of HDACs are short chain fatty acids such as valproic acid which has been widely used in treating epilepsy. It inhibits classes I and IIa HDACs. In addition, butyric acid and phenylbutyric acid inhibit classes I and II HDACS [184]. Another HDACi is romidepsin, which is a cyclic tetrapeptide and a class I inhibitor [200].
HDACis have shown very promising results for the treatment of various neoplasms and several in vitro [201,202,203,204,205] and in vivo [206,207] studies have sought to understand the methods of action and the pathways involved in the anticancer process of these molecules. To date, only vorinostat (SAHA), romidepsin (FK228), panobinostat ((LBH589), belinostat (PXD101) and chidamide (CS055/HBI-8000) have been approved for clinical trials in the United States. These compounds show antineoplastic activity in the treatment of several hematological malignancies [208,209,210,211,212,213,214]. Class III-directed HDACi target NAD+-containing sirtuins and have shown an effect in the treatment of cardiovascular and neurodegenerative diseases and aging [215]. Variable biological effects of HDACis have been observed, which are the result of the individual chemical structure and profile of each inhibitor [199].
Although HDACis are showing important activities, mainly oncological, their adverse effects and cytotoxicity are still serious and do not present selective inhibition among HDACs isoforms [216]. To design an effective HDACi, the molecule must have synergy with other anticancer agents given that the HDACi used as a single agent does not show clinical benefits in nearly all types of solid tumors [217]. The involvement that HDACis have in the levels of epigenetic alteration for which they are responsible explains why they are so involved in altering normal phenotypes in malignant ones. Figure 6 contains the structures of four HDACis but others are currently under development.
7. Alternative Inhibitors
Despite their benefits which were proven in clinical trials, synthetic HDACis still have undesirable adverse effects [200]. One alternative is the search for natural products and their derivatives that are able to inhibit epigenetic changes caused by changes in gene expression, with less risk to the patient [218]. Psammaplin A is a natural product derived from bromotyrosine that is found in marine sponges [219]. This compound has been shown to inhibit the activity of HDACs and DNA methyltransferase, exhibiting low cytotoxicity in in vitro studies [220]. Largazole is a macrocyclic depsipeptide isolated from marine cyanobacteria, which exerts antiproliferative activity by inhibiting HDACs 1, 2, 3 and 6 [221,222]. This compound is a promising pro-drug for the treatment of carcinomas. Several natural products exhibit low HDAC inhibitory activity, but modifications in their chemical structure can produce analogs with high inhibitory activity. FK228 (FR901228), also known as a depsipeptide (a peptide in which one or more of its amide groups are substituted by an ester), is produced by Chromobacterium violaceum and exhibits HDAC inhibitory potential and antitumor activity in vivo [223]. Several natural compounds appear to interfere with most of the molecular mechanisms that involve cell proliferation and death. Natural compounds isolated from plants such as polyphenolic compounds, for example fisetin [224], resveratrol [225], curcumin [226] or flavonoids [227], can induce epigenetic changes, increasing the sensitivity of cancer cells to chemotherapeutic agents, and reduce tumor proliferation. Resveratrol is a natural, biologically active polyphenol that is found in grape seeds and peanut skin and that has therapeutic applications in the treatment of a range of diseases, including cancer [228,229,230]. Resveratrol can reverse the progression of prostate cancer by inhibiting MTA1 that binds to HDAC, forming the MTA1/HDAC complex [231]. This naturally occurring HDACi inhibited the activity of 11 HDACs in hepatoblastoma cells [232], the concentration-dependent histone hyperacetylation in hepatoma cell lines and cytotoxicity, but only at high doses [233]. Combination treatment with resveratrol and other HDACis revealed important antitumor activity in leukemia [234] and ovarian [235] and pancreatic carcinomas [174].
7.1. Chalcones
Chalcones (1,3-diaryl-2-propenones) are intermediates in the biosyntheses of flavonoids and isoflavonoids. They can be synthesized by the condensation between aryl aldehydes and acetophenone [236,237]. Their structure consists of two aromatic rings linked by a three carbon unsaturated keto group [236]. Chalcones are found in medicinal plants, fruits, vegetables, spices and nuts, and have anti-inflammatory [238], antihistaminic [239], antihypertensive [240], antidiabetic [241], antimalarial [242], antiretroviral [243], antioxidant [236] and antitumor [237] properties. In regards to the antitumor properties of chalcones, one should note that they are far less toxic than many current anticancer drugs [244]. In an in vitro and in vivo study, different chalcones synthesized in the laboratory were effective against colon adenocarcinoma, altering epigenetic pathways and inhibiting HDACs [245]. In computer-assisted studies on the activity of chalcones, β-hydroxymethyl chalcone exhibited the best time-dependency (∼24 h) as a broad-spectrum HDACi and β-hydroxymethyl chalcone as a selective inhibitor of HDAC2 [246]. Evaluating the inhibitory activity of 21 natural chalcones, researchers found the significant HDAC inhibitory activity of four, namely isoliquiritigenin, butein, homobutein and marein, against class I, II, and IV HDACs [247]. The 3,2,3’,4-tetrahydroxychalcone inhibited the class III HDAC SIRT1, resulting in tumor suppression [248]. Chalcones exhibited significant anti-proliferative activity against the HDAC inhibitory activity in carcinoma cell lines when compared to the synthetic drug SAHA, which is already used clinically [249]. These data suggest that natural compounds are promising in cancer treatment.
7.2. Curcuminoids
Another potential HDACi is curcumin (diferuloylmethane), a polyphenol and the active component of turmeric (Curcuma longa) [250], which is widely known for its diverse pharmacological activities against various diseases, including cancer [251,252,253,254,255]. Turmeric is composed of 80% curcumin, 17% dimethoxycurcumin and 3% bisdemethoxycurcumin [256]. Curcumin can alter several important molecular signaling pathways that are responsible for cell survival and inflammatory responses and for reducing the expression of genes such as the tumor necrosis factor, adhesion molecules, interleukins (IL-1, IL-6, IL-8), C-X-C chemokine receptor type 4 (CXCR-4), and C-reactive protein [257,258,259]. Curcumin is considered a DNA hypomethylating agent that inhibits DNA methyltransferase and balances the activity of HATs and HDACs (HDAC 1, 3, 4, 5, 8) [260]. This compound was first described as a specific inhibitor of the coactivator p300/CBP. The latter interacts with numerous transcription factors and has been shown to increase the activity of acetyltransferase in cervical cancer cells. Its inhibition induced apoptosis [261]. Several HDACis have been used for the treatment of cancer alone or in combination with chemotherapeutic agents. Curcuminoids have shown deacetylase inhibitory activity that can suppress DNA repair pathways and can be used to increase the efficacy of cancer treatments [262]. A reduction of HDAC1 and HDAC3 activity was found in lymphoblastic cells treated with curcumin [263]. Furthermore, curcumin significantly reduced class I HDAC levels and increased acetylation [264,265]. Calebin A-([(E)-4-(4-hydroxy-3-methoxyphenyl)-2-oxobut-3-enyl]-(E)-3-(4-hydroxy-3-methoxyphenyl)prop-2-enoate) is a curcumin analog that contains a ferulic acid ester bond [266] (see Figure 7).
This compound can be used as an adjuvant in cancer treatment, increasing the efficacy of the chemotherapeutic agents used; however, its bioavailability is low [267]. The inhibition of HDACs promotes cell death and inhibits angiogenesis in different tumor cell lines in in vitro studies [268] (see Table 2).
Curcumin increases the sensitivity to DNA damage, reduces the repair of double-strand breaks and inhibits homologous recombination by inhibiting HDACs and promoting the degradation of recombinases [262]. In another study, in addition to interacting with the active zinc-containing site, curcumin exhibited an excellent inhibition of class 1 and 3 HDACs [275]. The enzymatic activity of HAT was found to be reduced in tumor cells treated with calebin A [276]. However, another study demonstrated that calebin A inhibited HDAC1, similarly to curcumin [277]. Tetrahydrocurcumin is a curcuminoid obtained by the reduction of curcumin. It is synthesized in the laboratory by hydrogenation but can be produced in vivo by metabolism in the liver [278]. Tetrahydrocurcumin did not inhibit HDACs [277], probably because its mechanism of action differed from that of calebin A and curcumin which have similar mechanisms. However, this curcuminoid exhibits anti-inflammatory potential and antioxidant activity which may explain its anticancer activity [279]. Chalcones have been linked to HDACs in an attempt to better understand the mechanisms of action of these deacetylases, with epigenetic importance in the treatment of diseases [247]. A molecular docking study concluded that curcumin does not bind to HDAC8 through the interaction with the zinc ion; this deacetylase is inhibited by the interaction with Arg37, Pro35, Ile34 and Phe152 residues located in the active site of the enzyme [280].
8. Conclusions
This review has outlined the structures and functions of different classes of histone deacetylases (HDACs). A major emphasis has been placed on various inhibitors of HDACs and how they exert their bioactivity. Although neoplasms are currently the main clinical indication for these compounds, future applications may include autoimmune diseases, neurological indications and even parasitic diseases. However, improvements in the therapeutic index of these drugs should be made, as they present high toxicity, inducing symptoms from fatigue, nausea and vomiting to thrombocytopenia, neutropenia and some cardiac toxicity. One path to this improvement may come from more specific inhibitors of individual HDAC isoforms that are critically involved in particular indications. By targeting the most relevant HDAC isoform in a specific indication, it may be possible to significantly improve their efficacy by removing certain toxicities that may be associated with the inhibition of multiple isoforms [281].
Funding
This research was funded by FAPESP, grant number 19/03074-1, 18/50008-1; Maunders McNeil Foundation Inc.; CAPES.
Acknowledgments
We acknowledge the contribution of Kerstin Markendorf for the English translation and revision.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cheung, W.L.; Briggs, S.D.; Allis, C.D. Acetylation and chromosomal functions. Curr. Opin. Cell Boil. 2000, 12, 326–333. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, G.G.; Worman, H.J. Nuclear positioning. Cell 2013, 152, 1376–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- De La Cruz, X.; Lois, S.; Sánchez-Molina, S.; Martínez-Balbás, M.A. Do protein motifs read the histone code? BioEssays 2005, 27, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Woodcock, C.L.; Skoultchi, A.I.; Fan, Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosom. Res. 2006, 14, 17–25. [Google Scholar] [CrossRef]
- Hansen, J.C. Conformational Dynamics of the Chromatin Fiber in Solution: Determinants, Mechanisms, and Functions. Annu. Rev. Biophys. Biomol. Struct. 2002, 31, 361–392. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacol. 2012, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Allfrey, V.G.; Mirsky, A.E.; Arnon, D.I.; Tsujimoto, H.Y.; McSwain, B.D. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Sciebce 1964, 144, 559. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Hayes, J.J. Chromatin disruption and modification. Nucleic Acids Res. 1999, 27, 711–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, D.G.; Smith, M.M.; Roth, S.Y. Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev. 1996, 10, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Boil. 2013, 20, 259–266. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Manning, L.R.; Russell, J.E.; Popowicz, A.M.; Manning, R.S.; Padovan, J.C.; Manning, J.M. Energetic Differences at the Subunit Interfaces of Normal Human Hemoglobins Correlate with Their Developmental Profile. Biochemistry 2009, 48, 7568–7574. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.-W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Boil. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Y.; Shi, X. Emerging roles of lysine methylation on non-histone proteins. Cell. Mol. Life Sci. 2015, 72, 4257–4272. [Google Scholar] [CrossRef]
- Abbass, S.A.; Hassan, H.A.; Mohamed, M.F.A.; Moustafa, G.A.I.; Abuo-Rahma, G.E.-D.A. Recent Prospectives of Anticancer Histone Deacetylase Inhibitors. J. Adv. Biomed. Pharm. Sci. 2019, 2, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, M.; Smith, M.M. Functional consequences of histone modifications. Curr. Opin. Genet. Dev. 2003, 13, 154–160. [Google Scholar] [CrossRef]
- Berger, S. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Gibbon, S.; Prainsack, B.; Hilgartner, S.; Lamoreaux, J. (Eds.) Routledge Handbook of Genomics, Health and Society; Routledge: Abington, UK, 2018. [Google Scholar]
- Parthun, M.R. Hat1: The emerging cellular roles of a type B histone acetyltransferase. Oncogene 2007, 26, 5319–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, S.; Almouzni, G. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 2005, 220, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vidanes, G.M.; Bonilla, C.Y.; Toczyski, D.P. Complicated Tails: Histone Modifications and the DNA Damage Response. Cell 2005, 121, 973–976. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, B.; Bickmore, W.A. Human diseases with underlying defects in chromatin structure and modification. Hum. Mol. Genet. 2001, 10, 2233–2242. [Google Scholar] [CrossRef] [Green Version]
- Cairns, B.R. Emerging roles for chromatin remodeling in cancer biology. Trends Cell Boil. 2001, 11, S15–S21. [Google Scholar] [CrossRef]
- Hake, S.B.; Xiao, A.; Allis, C.D. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br. J. Cancer 2004, 90, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef]
- Gil, J.; Ramírez-Torres, A.; Guevara, S.E. Lysine acetylation and cancer: A proteomics perspective. J. Proteom. 2017, 150, 297–309. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Dekker, F.J.; Haisma, H.J. Histone acetyl transferases as emerging drug targets. Drug Discov. Today 2009, 14, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced Histone Acetylation and Transcription: A Dynamic Perspective. Mol. Cell 2006, 23, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Paik, W.K.; Borun, T.W. The periodic synthesis of S-adenosylmethionine: Protein methyltransferases during the HeLa S-3 cell cycle. J. Boil. Chem. 1973, 248, 4194–4199. [Google Scholar]
- Wolffe, A.P.; Guschin, D. Review: Chromatin Structural Features and Targets That Regulate Transcription. J. Struct. Boil. 2000, 129, 102–122. [Google Scholar] [CrossRef] [Green Version]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.J.; Zhang, Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Boil. 2013, 20, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; Van Gennip, A.H.; Caron, H.N.; Kemp, S.; Van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Inoue, A.; Fujimoto, D. Enzymatic deacetylation of histone. Biochem. Biophys. Res. Commun. 1969, 36, 146–150. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Boil. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors: Insights into mechanisms of lethality. Expert Opin. Ther. Targets 2005, 9, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Frew, A.J.; Johnstone, R.W.; Bolden, J.E. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009, 280, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.-W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer. Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.A.; Chabner, B.A. Histone Deacetylase Inhibitors in Cancer Therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-Y.; Chen, C.-S.; Lin, S.-P.; Weng, J.-R.; Chen, C.-S. Targeting Histone Deacetylase in Cancer Therapy. Chemin 2006, 37, 397–413. [Google Scholar] [CrossRef]
- Lagger, G.; O’Carroll, D.; Rembold, M.; Khier, H.; Tischler, J.; Weitzer, G.; Schuettengruber, B.; Häuser, C.; Brunmeir, R.; Jenuwein, T.; et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002, 21, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone Deacetylase 4 Controls Chondrocyte Hypertrophy during Skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Arnold, M.; Kim, Y.; Czubryt, M.P.; Phan, D.; McAnally, J.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E. MEF2C Transcription Factor Controls Chondrocyte Hypertrophy and Bone Development. Dev. Cell 2007, 12, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; Von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA Expression in Invasive Carcinoma of the Breast*. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Horiuchi, A.; Kikuchi, N.; Hayashi, T.; Fuseya, C.; Suzuki, A.; Konishi, I.; Shiozawa, T. Type-specific roles of histone deacetylase (HDAC) overexpression in ovarian carcinoma: HDAC1 enhances cell proliferation and HDAC3 stimulates cell migration with downregulation of E-cadherin. Int. J. Cancer 2010, 127, 1332–1346. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzsche, F.R.; Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Scholman, K.; Denkert, C.; Dietel, M.; et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 2008, 8, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyet, C.; Jentsch, B.; Hermanns, T.; Schweckendiek, D.; Seifert, H.-H.; Schmidtpeter, M.; Sulser, T.; Moch, H.; Wild, P.J.; Kristiansen, G. Expression of histone deacetylases 1, 2 and 3 in urothelial bladder cancer. BMC Clin. Pathol. 2014, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.A.; Queiroz, R.D.P.; Valera, E.T.; Silveira, V.S.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. research paper: Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kafeel, M.I.; Avezbakiyev, B.; Chen, C.; Sun, Y.; Rathnasabapathy, C.; Kalavar, M.; He, Z.; Burton, J.; Lichter, S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2012, 81, 325–329. [Google Scholar] [CrossRef]
- Ecker, J.; Oehme, I.; Mazitschek, R.; Korshunov, A.; Kool, M.; Hielscher, T.; Kiss, J.; Selt, F.; Konrad, C.; Lodrini, M.; et al. Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol. Commun. 2015, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566–569. [Google Scholar] [CrossRef]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.-J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer - overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche, P.; Seidler, B.; Schüler, S.; Schnieke, A.; Göttlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [Green Version]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Motoyama, S.; Ogawa, J. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumor Boil. 2010, 31, 533–539. [Google Scholar] [CrossRef]
- Wilmott, J.S.; Colebatch, A.J.; Kakavand, H.; Shang, P.; Carlino, M.S.; Thompson, J.; Long, G.V.; Scolyer, R.A.; Hersey, P. Expression of the class 1 histone deacetylases HDAC8 and 3 are associated with improved survival of patients with metastatic melanoma. Mod. Pathol. 2015, 28, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; Von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Zhu, S.; Wu, C.; Kang, J. Histone Deacetylase (HDAC) 10 Suppresses Cervical Cancer Metastasis through Inhibition of Matrix Metalloproteinase (MMP) 2 and 9 Expression*. J. Boil. Chem. 2013, 288, 28021–28033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Yang, W.-M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.-M.; Yao, Y.-L.; Sun, J.-M.; Davie, J.R.; Seto, E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Boil. Chem. 1997, 272, 28001–28007. [Google Scholar] [CrossRef] [Green Version]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, H.-Y.; Downes, M.; Ordentlich, P.; Evans, R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genome Res. 2000, 14, 55–66. [Google Scholar]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.-F.; Johanson, K.; Sung, C.-M.; Liu, R.; Winkler, J. Cloning and Characterization of a Novel Human Class I Histone Deacetylase That Functions as a Transcription Repressor. J. Boil. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrie, K.; Guidez, F.; Howell, L.; Healy, L.; Waxman, S.; Greaves, M.; Zelent, A. The Histone Deacetylase 9 Gene Encodes Multiple Protein Isoforms. J. Boil. Chem. 2003, 278, 16059–16072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Kao, H.-Y.; Lee, C.-H.; Komarov, A.; Han, C.C.; Evans, R.M. Isolation and Characterization of Mammalian HDAC10, a Novel Histone Deacetylase. J. Boil. Chem. 2001, 277, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Boil. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [Green Version]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1799, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Myers, S.J.; Dingledine, R. Transcriptional repression by REST: Recruitment of Sin3A and histone deacetylase to neuronal genes. Nat. Neurosci. 1999, 2, 867–872. [Google Scholar] [CrossRef]
- Lagger, S.; Meunier, M.; Mikula, M.; Brunmeir, R.; Schlederer, M.; Artaker, M.; Pusch, O.; Egger, G.; Hagelkruys, A.; Mikulits, W.; et al. Crucial function of histone deacetylase 1 for differentiation of teratomas in mice and humans. EMBO J. 2010, 29, 3992–4007. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.L.; Roskams, A.J. Histone deacetylases 1 and 2 are expressed at distinct stages of neuro-glial development. Dev. Dyn. 2008, 237, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leus, N.G.; Zwinderman, M.R.; Dekker, F.J. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-κB-mediated inflammation. Curr. Opin. Chem. Boil. 2016, 33, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänsch, N.; Meyners, C.; Muth, M.; Kopranovic, A.; Witt, O.; Oehme, I.; Meyer-Almes, F.-J. The enzyme activity of histone deacetylase 8 is modulated by a redox-switch. Redox Boil. 2018, 20, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.; Lee, Y.-M.; Goodson, H.V. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Boil. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Tsai, S.-C.; Seto, E. Regulation of Histone Deacetylase 2 by Protein Kinase CK2. J. Boil. Chem. 2002, 277, 31826–31833. [Google Scholar] [CrossRef] [Green Version]
- Jamaladdin, S.; Kelly, R.D.W.; O’Regan, L.; Dovey, O.M.; Hodson, G.E.; Millard, C.J.; Portolano, N.; Fry, A.M.; Schwabe, J.W.; Cowley, S. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. In Proceedings of the Proceedings of the National Academy of Sciences, Seattle, WA, USA, 3 June 2014; Volume 111, pp. 9840–9845. [Google Scholar]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.S.; Bradley, A.; Cowley, S.; Vasilliou, G. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genome Res. 1999, 13, 1924–1935. [Google Scholar] [CrossRef]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. BioEssays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Burke, L.J.; Baniahmad, A. Co-repressors 2000. FASEB J. 2000, 14, 1876–1888. [Google Scholar] [CrossRef] [Green Version]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.-M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.W.; Chandru, A.; Watson, P.J.; Song, Y.; Blades, M.; Robertson, N.; Jamieson, A.G.; Schwabe, J.W.; Cowley, S. Histone deacetylase (HDAC) 1 and 2 complexes regulate both histone acetylation and crotonylation in vivo. Sci. Rep. 2018, 8, 14690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.D.; Cowley, S. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.A.; Ekwall, K. Sin3: A flexible regulator of global gene expression and genome stability. Curr. Genet. 2004, 47, 1–17. [Google Scholar] [CrossRef]
- Chaubal, A.; Pile, L.A. Same agent, different messages: Insight into transcriptional regulation by SIN3 isoforms. Epigenetics Chromatin 2018, 11, 17. [Google Scholar] [CrossRef]
- Grzenda, A.; Lomberk, G.; Zhang, J.-S.; Urrutia, R. Sin3: Master scaffold and transcriptional corepressor. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1789, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.T.; Martin-Brown, S.A.; Florens, L.; Washburn, M.P.; Workman, J. Deacetylase Inhibitors Dissociate the Histone-Targeting ING2 Subunit from the Sin3 Complex. Chem. Boil. 2010, 17, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, T.C.; Yun, U.J.; Ayer, D. Identification and Characterization of Three New Components of the mSin3A Corepressor Complex. Mol. Cell. Boil. 2003, 23, 3456–3467. [Google Scholar] [CrossRef] [Green Version]
- Binda, O.; Roy, J.-S.; Branton, P.E. RBP1 Family Proteins Exhibit SUMOylation-Dependent Transcriptional Repression and Induce Cell Growth Inhibition Reminiscent of Senescence. Mol. Cell. Boil. 2006, 26, 1917–1931. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, A.Y.; Papanikolaou, N.A.; Li, M.; Qin, J.; Gu, W. Identification of a novel BRMS1-homologue protein p40 as a component of the mSin3A/p33ING1b/HDAC1 deacetylase complex. Biochem. Biophys. Res. Commun. 2004, 323, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Rose, D.W.; Aur, R.; Donohoe, S.; Aebersold, R.; Eisenman, R.N. Identification and Characterization of SAP25, a Novel Component of the mSin3 Corepressor Complex. Mol. Cell. Boil. 2006, 26, 1386–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, K.; Cowley, S.M.; Huang, K.; Loo, L.; Yochum, G.S.; Ayer, D.E.; Eisenman, R.N.; Radhakrishnan, I. Solution structure of the interacting domains of the Mad-Sin3 complex: Implications for recruitment of a chromatin-modifying complex. Cell 2000, 103, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Spronk, A.C.; Tessari, M.; Kaan, A.M.; Jansen, J.F.; Vermeulen, M.; Stunnenberg, H.G.; Vuister, G.W. The Mad1-Sin3B interaction involves a novel helical fold. Nat. Genet. 2000, 7, 1100–1104. [Google Scholar] [CrossRef]
- Sif, S.; Saurin, A.; Imbalzano, A.N.; Kingston, R.E. Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genome Res. 2001, 15, 603–618. [Google Scholar] [CrossRef] [Green Version]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Méndez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef]
- Baltus, G.A.; Kowalski, M.P.; Tutter, A.V.; Kadam, S. A Positive Regulatory Role for the mSin3A-HDAC Complex in Pluripotency through Nanog and Sox2*. J. Boil. Chem. 2009, 284, 6998–7006. [Google Scholar] [CrossRef] [Green Version]
- Gambi, G.; Di Simone, E.; Basso, V.; Ricci, L.; Wang, R.; Verma, A.; Elemento, O.; Ponzoni, M.; Inghirami, G.; Icardi, L.; et al. The Transcriptional Regulator Sin3A Contributes to the Oncogenic Potential of STAT3. Cancer Res. 2019, 79, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Dannenberg, J.-H.; David, G.; Zhong, S.; Van Der Torre, J.; Wong, W.H.; A DePinho, R. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genome Res. 2005, 19, 1581–1595. [Google Scholar] [CrossRef] [Green Version]
- Pile, L.A.; Spellman, P.T.; Katzenberger, R.J.; Wassarman, D.A. The SIN3 deacetylase complex represses genes encoding mitochondrial proteins: Implications for the regulation of energy metabolism. J. Biol. Chem. 2003, 278, 37840–37848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, A.H.; Jansen, P.W.T.C.; Poser, I.; Hyman, A.A.; Vermeulen, M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2012, 41, e28. [Google Scholar] [CrossRef]
- Torchy, M.; Hamiche, A.; Klaholz, B. Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Millard, C.J.; Varma, N.; Saleh, A.; Morris, K.; Watson, P.J.; Bottrill, A.; Fairall, L.R.; Smith, C.J.; Schwabe, J.W. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. eLife 2016, 5, 7285. [Google Scholar] [CrossRef] [PubMed]
- Schmidberger, J.W.; Sharifi Tabar, M.; Torrado, M.; Silva, A.P.; Landsberg, M.J.; Brillault, L.; AlQarni, S.; Zeng, Y.C.; Parker, B.L.; Low, J.K.; et al. The MTA1 subunit of the nucleosome remodeling and deacetylase complex can recruit two copies of RBBP4/7. Protein Sci. 2016, 25, 1472–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denslow, S.A.; Wade, P.A. The human Mi-2/NuRD complex and gene regulation. Oncogene 2007, 26, 5433–5438. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.K.; Hassig, C.A.; Schnitzler, G.R.; Kingston, R.E.; Schreiber, S.L. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 1998, 395, 917–921. [Google Scholar] [CrossRef]
- Le Guezennec, X.; Vermeulen, M.; Brinkman, A.B.; Hoeijmakers, W.A.M.; Cohen, A.; Lasonder, E.; Stunnenberg, H.G. MBD2/NuRD and MBD3/NuRD, Two Distinct Complexes with Different Biochemical and Functional Properties. Mol. Cell. Boil. 2006, 26, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Ishikawa, F. The mCpG-binding Domain of Human MBD3 Does Not Bind to mCpG but Interacts with NuRD/Mi2 Components HDAC1 and MTA2. J. Boil. Chem. 2002, 277, 35434–35439. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, B.; Tweedie, S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003, 19, 269–277. [Google Scholar] [CrossRef]
- Aguilera, C.; Nakagawa, K.; Sancho, R.; Chakraborty, A.; Hendrich, B.; Behrens, A. C-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature 2011, 469, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.L.; Tosti, L.; Radzisheuskaya, A.; Caballero, I.M.; Kaji, K.; Hendrich, B.; Silva, J.C. MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 2014, 15, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.; Han, C.; Park, I.; Lee, B.; Jin, S.; Choi, H.; Kim, H.; Park, Z.Y.; Eddy, E.M.; Cho, C. A Novel Germ Cell-specific Protein, SHIP1, Forms a Complex with Chromatin Remodeling Activity during Spermatogenesis*. J. Boil. Chem. 2008, 283, 35283–35294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaki, S.; Sato, A.; Toyomoto, T.; Hayano, T.; Sugai, M.; Kubota, T.; Koiwai, O. Direct binding of TReP-132 with TdT results in reduction of TdT activity. Genes Cells 2005, 11, 47–57. [Google Scholar] [CrossRef]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic Activity Associated with Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Surdo, P.L.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Dequiedt, F.; Kasler, H.G. Class II histone deacetylases: Versatile regulators. Trends Genet. 2003, 19, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Parra, M.; Verdin, E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr. Opin. Pharmacol. 2010, 10, 454–460. [Google Scholar] [CrossRef]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class IIa histone deacetylases: Conducting development and differentiation. Int. J. Dev. Boil. 2009, 53, 291–301. [Google Scholar] [CrossRef]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class IIa histone deacetylases: Regulating the regulators. Oncogene 2007, 26, 5450–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Methods 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Guardiola, A.R.; Yao, T.-P. Molecular Cloning and Characterization of a Novel Histone Deacetylase HDAC10. J. Boil. Chem. 2001, 277, 3350–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotian, S.; Liyanarachchi, S.; Zelent, A.; Parvin, J.D. Histone Deacetylases 9 and 10 Are Required for Homologous Recombination*. J. Boil. Chem. 2011, 286, 7722–7726. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell. Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.-P.; Vourc’H, C.; Matthias, P.; et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genome Res. 2007, 21, 2172–2181. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell–cell interactions. Trends Cell Boil. 2008, 18, 291–297. [Google Scholar] [CrossRef]
- Yang, W.-M.; Tsai, S.-C.; Wen, Y.-D.; Fejér, G.; Seto, E. Functional Domains of Histone Deacetylase-3. J. Boil. Chem. 2002, 277, 9447–9454. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Maison, C.; Bailly, D.; Peters, A.H.; Quivy, J.-P.; Roche, D.; Taddei, A.; Lachner, M.; Jenuwein, T.; Almouzni, G. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet. 2002, 30, 329–334. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2008, 86, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, Z.-R.; Xu, Y.-F.; Wang, X.-B.; Gong, J.; Liu, Z.-J. Suppression of Histone Deacetylase 11 Promotes Expression of IL-10 in Kupffer Cells and Induces Tolerance Following Orthotopic Liver Transplantation in Rats. J. Surg. Res. 2012, 174, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004, 32, 959–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, B.N. Crafting the Brain—Role of Histone Acetyltransferases in Neural Development and Disease. Cell Tissue Res. 2014, 356, 553–573. [Google Scholar] [CrossRef]
- Gupta, A.; Guerin-Peyrou, T.G.; Sharma, G.G.; Park, C.; Agarwal, M.; Ganju, R.K.; Pandita, S.; Choi, K.; Sukumar, S.; Pandita, R.K.; et al. The Mammalian Ortholog of Drosophila MOF That Acetylates Histone H4 Lysine 16 Is Essential for Embryogenesis and Oncogenesis. Mol. Cell. Boil. 2007, 28, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [Green Version]
- Shivdasanl, R.A.; Mayer, E.L.; Orkin, S.H. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature 1995, 373, 432–434. [Google Scholar] [CrossRef]
- Sebastiano, V.; Dalvai, M.; Gentile, L.; Schubart, K.; Sutter, J.; Wu, G.; Tapia, N.; Esch, D.; Ju, J.-Y.; Hübner, K.; et al. Oct1 regulates trophoblast development during early mouse embryogenesis. Development 2010, 137, 3551–3560. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.J.; Seto, E. Unlocking the mechanisms of transcription factor YY1: Are chromatin modifying enzymes the key? Gene 1999, 236, 197–208. [Google Scholar] [CrossRef]
- Lee, J.S.; Galvin, K.M.; See, R.H.; Eckner, R.; Livingston, D.; Moran, E.; Shi, Y. Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev. 1995, 9, 1188–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malewicz, M.; Perlmann, T. Function of transcription factors at DNA lesions in DNA repair. Exp. Cell Res. 2014, 329, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, R.M.D.O.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Tsay, Y.-G.; Tan, B.C.; Lo, W.-Y.; Lee, S.-C. Identification and Characterization of a Novel p300-mediated p53 Acetylation Site, Lysine 305. J. Boil. Chem. 2003, 278, 25568–25576. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [Green Version]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. JBIC J. Boil. Inorg. Chem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Zhan, Q.; El-Deiry, W.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- Smith, M.; Chen, I.; Zhan, Q.; Bae, I.; Chen, C.; Gilmer, T.; Kastan, M.; O’Connor, P.; Fornace, A.J. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.-H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.-P. MDM2–HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Beishline, K.; Kelly, C.M.; Olofsson, B.A.; Koduri, S.; Emrich, J.; Greenberg, R.A.; Azizkhan-Clifford, J. Sp1 Facilitates DNA Double-Strand Break Repair through a Nontranscriptional Mechanism. Mol. Cell. Boil. 2012, 32, 3790–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, P.; Pandey, V.; Kumar, V. Stimulation of ribosomal RNA gene promoter by transcription factor Sp1 involves active DNA demethylation by Gadd45-NER pathway. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, J.; Crofts, L.A.; Quinlan, K.G.; Czolij, R.; Perkins, A.; Crossley, M. Human KLF17 is a new member of the Sp/KLF family of transcription factors. Genomics 2006, 87, 474–482. [Google Scholar] [CrossRef] [Green Version]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Solomon, S.S.; Majumdar, G.; Martínez-Hernández, A.; Raghow, R. A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sci. 2008, 83, 305–312. [Google Scholar] [CrossRef]
- Li, L.; Davie, J.R. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat. Anat. Anz. 2010, 192, 275–283. [Google Scholar] [CrossRef]
- Li, G.; Xie, Q.; Yang, Z.; Wang, L.; Zhang, X.; Zuo, B.; Zhang, S.; Yang, A.; Jia, L. Sp1-mediated epigenetic dysregulation dictates HDAC inhibitor susceptibility of HER2-overexpressing breast cancer. Int. J. Cancer 2019, 145, 3285–3298. [Google Scholar] [CrossRef]
- Banerjee, A.; Mahata, B.; Dhir, A.; Mandal, T.K.; Biswas, K. Elevated histone H3 acetylation and loss of the Sp1–HDAC1 complex de-repress the GM2-synthase gene in renal cell carcinoma. J. Boil. Chem. 2018, 294, 1005–1018. [Google Scholar] [CrossRef] [Green Version]
- Juli, G.; Oliverio, M.; Bellizzi, D.; Cantafio, M.E.G.; Grillone, K.; Passarino, G.; Colica, C.; Nardi, M.; Rossi, M.; Procopio, A.; et al. Anti-tumor Activity and Epigenetic Impact of the Polyphenol Oleacein in Multiple Myeloma. Cancers 2019, 11, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Patel, V.K.; Jain, D.K.; Patel, P.; Rajak, H. Panobinostat as Pan-deacetylase Inhibitor for the Treatment of Pancreatic Cancer: Recent Progress and Future Prospects. Oncol. Ther. 2016, 4, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, E.; Montgomery, M.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Ridinger, J.; Koeneke, E.; Kolbinger, F.R.; Koerholz, K.; Mahboobi, S.; Hellweg, L.; Gunkel, N.; Miller, A.K.; Peterziel, H.; Schmezer, P.; et al. Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep. 2018, 8, 10039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilting, R.H.; Yanover, E.; Heideman, M.R.; Jacobs, H.; Horner, J.; Van Der Torre, J.; DePinho, R.A.; Dannenberg, J.-H. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010, 29, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Santoro, F.; Botrugno, O.A.; Zuffo, R.D.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, G.J.A.; Richmond, P.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; Wheeler, G.E.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2017, 46, 1756–1776. [Google Scholar] [CrossRef]
- Yasui, W.; Oue, N.; Ono, S.; Mitani, Y.; Ito, R.; Nakayama, H. Histone acetylation and gastrointestinal carcinogenesis. Ann. New York Acad. Sci. 2003, 983, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Seto, E. Deacetylation of Nonhistone Proteins by HDACs and the Implications in Cancer. Pharmacol. Ther. Cough 2011, 206, 39–56. [Google Scholar] [CrossRef]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, O.; Fotheringham, S.; Wood, V.; Stimson, L.; Zhang, C.; Pezzella, F.; Duvic, M.; Kerr, D.J.; La Thangue, N.B. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 6532–6537. [Google Scholar] [CrossRef] [Green Version]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef]
- Mai, A.; Altucci, L. Epi-drugs to fight cancer: From chemistry to cancer treatment, the road ahead. Int. J. Biochem. Cell Boil. 2009, 41, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, P.V.; Ververis, K.; Hiong, A.; Karagiannis, T.C. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Boil. Targets Ther. 2013, 7, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborová, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.-H.; Hao, C.; Liu, P.; Tian, X.; Wang, L.-H.; Zhao, L.; Zhu, C.-M. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi-1 leukemia cells. Mol. Med. Rep. 2013, 9, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Jiang, S.; Chen, J.; Su, S.B. Suberoylanilide hydroxamic acid suppresses inflammation-induced neovascularization. Can. J. Physiol. Pharmacol. 2014, 92, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Magaña, M.J.R.; Carranza, D.; Ortiz-Ferrón, G.; Ruiz-Ruiz, C. HDAC inhibitors with different gene regulation activities depend on the mitochondrial pathway for the sensitization of leukemic T cells to TRAIL-induced apoptosis. Cancer Lett. 2010, 297, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bolden, E.J.; Shi, W.; Jankowski, K.; Kan, C.-Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, R.G.; Ude, Z.; Docherty, J.R.; Marmion, C.J. Vorinostat and Belinostat, hydroxamate-based anti-cancer agents, are nitric oxide donors. J. Inorg. Biochem. 2020, 206, 110981. [Google Scholar] [CrossRef]
- Kroesen, M.; Gielen, P.R.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Meeran, S.M.; Katiyar, S.K. Epigenetic regulation by selected dietary phytochemicals in cancer chemoprevention. Cancer Lett. 2014, 355, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Sanaei, M.; Kavoosi, F.; Roustazadeh, A.; Shahsavani, H. In Vitro Effect of the Histone Deacetylase Inhibitor Valproic Acid on Viability and Apoptosis of the PLC/PRF5 Human Hepatocellular Carcinoma Cell Line. Asian Pac. J. Cancer Prev. 2018, 19, 2507–2510. [Google Scholar] [PubMed]
- Cortiguera, M.G.; García-Gaipo, L.; Wagner, S.D.; León, J.; Batlle-López, A.; Delgado, M.D. Suppression of BCL6 function by HDAC inhibitor mediated acetylation and chromatin modification enhances BET inhibitor effects in B-cell lymphoma cells. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Chao, M.-W.; Chang, L.-H.; Tu, H.-J.; Chang, C.-D.; Lai, M.-J.; Chen, Y.-Y.; Liou, J.-P.; Teng, C.-M.; Pan, S.-L. Combination treatment strategy for pancreatic cancer involving the novel HDAC inhibitor MPT0E028 with a MEK inhibitor beyond K-Ras status. Clin. Epigenetics 2019, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Bandolik, J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Lyu, J.; Yang, E.J.; Liu, Y.; Wu, C.; Pardeshi, L.; Tan, K.; Chen, Q.; Xu, X.; Deng, C.-X.; et al. Class I histone deacetylase inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells. Acta Pharm. Sin. B 2019, 10, 615–627. [Google Scholar] [CrossRef]
- Groselj, B.; Ruan, J.-L.; Scott, H.; Gorrill, J.; Nicholson, J.; Kelly, J.; Anbalagan, S.; Thompson, J.; Stratford, M.R.; Jevons, S.J.; et al. Radiosensitization In Vivo by Histone Deacetylase Inhibition with No Increase in Early Normal Tissue Radiation Toxicity. Mol. Cancer Ther. 2017, 17, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Paillas, S.; Then, C.K.; Kilgas, S.; Ruan, J.-L.; Thompson, J.; Elliott, A.; Smart, S.; Kiltie, A.E. The Histone Deacetylase Inhibitor Romidepsin Spares Normal Tissues While Acting as an Effective Radiosensitizer in Bladder Tumors in Vivo. Int. J. Radiat. Oncol. 2020, 107, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Finkler, N.J.; Dizon, D.S.; Braly, P.; Micha, J.; Lassen, U.; Celano, P.; Glasspool, R.; Crowley, E.; Buhl-Jensen, P.; Penson, R.T. Phase II multicenter trial of the histone deactylase inhibitor (HDACi) belinostat, carboplatin and paclitaxel (BelCaP) in patients (pts) with relapsed epithelial ovarian cancer (EOC). J. Clin. Oncol. 2008, 26, 5519. [Google Scholar] [CrossRef]
- Gao, S.; Li, X.; Zang, J.; Xu, W.; Zhang, Y. Preclinical and Clinical Studies of Chidamide (CS055/HBI-8000), An Orally Available Subtype-selective HDAC Inhibitor for Cancer Therapy. Anti-Cancer Agents Med. Chem. 2017, 17, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [Green Version]
- Piekarz, R.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II Multi-Institutional Trial of the Histone Deacetylase Inhibitor Romidepsin As Monotherapy for Patients With Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niesvizky, R.; Ely, S.; Mark, T.; Aggarwal, S.; Gabrilove, J.L.; Wright, J.J.; Chen-Kiang, S.; Sparano, J.A. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 2010, 117, 336–342. [Google Scholar] [CrossRef] [Green Version]
- San-Miguel, J.; Richardson, P.G.; Günther, A.; Sezer, O.; Siegel, D.; Bladé, J.; Leblanc, R.; Sutherland, H.; Sopala, M.; Mishra, K.K.; et al. Phase Ib Study of Panobinostat and Bortezomib in Relapsed or Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2013, 31, 3696–3703. [Google Scholar] [CrossRef] [PubMed]
- Yazbeck, V.; Shafer, D.; Perkins, E.B.; Coppola, M.; Sokol, L.; Richards, K.L.; Shea, T.; Ruan, J.; Parekh, S.; Strair, R.; et al. A Phase II Trial of Bortezomib and Vorinostat in Mantle Cell Lymphoma and Diffuse Large B-cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 569–575.e1. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Boil. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, M. HDAC inhibitors still need a home run, despite recent approval. Nat. Rev. Drug Discov. 2015, 14, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.H. Health-Promoting Components of Fruits and Vegetables in the Diet12. Adv. Nutr. 2013, 4, 384S–392S. [Google Scholar] [CrossRef]
- Thomas, T.R.A.; Kavlekar, D.P.; LokaBharathi, P.A. Marine Drugs from Sponge-Microbe Association—A Review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar] [CrossRef] [Green Version]
- Piña, I.C.; Gautschi , J.T.; Wang, G.Y.; Sanders, M.L.; Schmitz, F.J.; France, D.; Cornell-Kennon, S.; Sambucetti, L.C.; Remiszewski, S.W.; Perez, L.B.; et al. Psammaplins from the Sponge Pseudoceratina p urpurea: Inhibition of Both Histone Deacetylase and DNA Methyltransferase. J. Org. Chem. 2007, 68, 3866–3873. [Google Scholar]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef]
- Ying, Y.; Taori, K.; Kim, H.; Hong, J.; Luesch, H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008, 130, 8455–8459. [Google Scholar] [CrossRef]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot (Tokyo) 2012, 47, 301–310. [Google Scholar]
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Boil. 2016, 40–41, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Boil. 2016, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Duvoix, A.; Blasius, R.; Delhalle, S.; Schnekenburger, M.; Morceau, F.; Henry, E.; Dicato, M.; Diederich, M. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005, 223, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Menezes, J.C.J.M.D.S.; Orlíková, B.; Morceau, F.; Diederich, M. Natural and Synthetic Flavonoids: Structure–Activity Relationship and Chemotherapeutic Potential for the Treatment of Leukemia. Crit. Rev. Food Sci. Nutr. 2015, 56, S4–S28. [Google Scholar] [CrossRef]
- Ko, J.-H.; Sethi, G.; Um, J.-Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.-H.; Ko, J.-H.; Lee, H.; Jung, J.; Kong, M.; Lee, J.-W.; Lee, J.; Chinnathambi, A.; Zayed, M.; Alharbi, S.A.; et al. Resveratrol inhibits STAT3 signaling pathway through the induction of SOCS-1: Role in apoptosis induction and radiosensitization in head and neck tumor cells. Phytomedicine 2016, 23, 566–577. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Sethi, G.; Diagaradjane, P.; Anand, P.; Pandey, M.K.; Gelovani, J.; Krishnan, S.; Guha, S.; Aggarwal, B.B. Resveratrol, a multitargeted agent, can enhance antitumor activity of gemcitabinein vitroand in orthotopic mouse model of human pancreatic cancer. Int. J. Cancer 2010, 127, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.; Kumar, A.; Li, K.; Tzivion, G.; Levenson, A.S. Resveratrol regulates PTEN/Akt pathway through inhibition of MTA1/HDAC unit of the NuRD complex in prostate cancer. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Venturelli, S.; Berger, A.; Böcker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a Pan-HDAC Inhibitor Alters the Acetylation Status of Jistone Proteins in Human-Derived Hepatoblastoma Cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bots, M.; Verbrugge, I.; Martin, B.P.; Salmon, J.M.; Ghisi, M.; Baker, A.; Stanley, K.; Shortt, J.; Ossenkoppele, G.J.; Zuber, J.; et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014, 123, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Helland, Ø.; Popa, M.; Bischof, K.; Gjertsen, B.T.; Mc Cormack, E.; Bjørge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLOS ONE 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, L.; Sinha, R.; Itokawa, H.; Bastow, K.F.; Tachibana, Y.; Nakanishi, Y.; Kilgore, N.; Lee, K.H. Anti-HIV and cytotoxic activities of Ru(II)/Ru(III) polypyridyl complexes containing 2,6-(2′-benzimidazolyl)-pyridine/chalcone as co-ligand. Bioorganic Med. Chem. 2001, 9, 1667–1671. [Google Scholar] [CrossRef]
- Zhang, E.-H.; Wang, R.-F.; Guo, S.; Liu, B. An Update on Antitumor Activity of Naturally Occurring Chalcones. Evidence-Based Complement. Altern. Med. 2013, 2013, 1–22. [Google Scholar] [CrossRef]
- Jeon, J.-H.; Kim, S.-J.; Kim, C.-G.; Kim, J.-K.; Jun, J.-G. Synthesis of Biologically Active Chalcones and their Anti-inflammatory Effects. Bull. Korean Chem. Soc. 2012, 33, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Kantevari, S.; Addla, D.; Bagul, P.K.; Sridhar, B.; Banerjee, S.K. Synthesis and evaluation of novel 2-butyl-4-chloro-1-methylimidazole embedded chalcones and pyrazoles as angiotensin converting enzyme (ACE) inhibitors. Bioorganic Med. Chem. 2011, 19, 4772–4781. [Google Scholar] [CrossRef]
- Detsi, A.; Majdalani, M.; Kontogiorgis, C.A.; Hadjipavlou-Litina, D.; Kefalas, P. Natural and synthetic 2′-hydroxy-chalcones and aurones: Synthesis, characterization and evaluation of the antioxidant and soybean lipoxygenase inhibitory activity. Bioorganic Med. Chem. 2009, 17, 8073–8085. [Google Scholar] [CrossRef] [PubMed]
- Romagnolo, D.F.; Selmin, O.I. Flavonoids and Cancer Prevention: A Review of the Evidence. J. Nutr. Gerontol. Geriatr. 2012, 31, 206–238. [Google Scholar] [CrossRef]
- Israf, D.; Khaizurin, T.; Syahida, A.; Lajis, N.; Khozirah, S. Cardamonin inhibits COX and iNOS expression via inhibition of p65NF-κB nuclear translocation and Iκ-B phosphorylation in RAW 264.7 macrophage cells. Mol. Immunol. 2007, 44, 673–679. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. ChemInform Abstract: Chalcones and Their Therapeutic Targets for the Management of Diabetes: Structural and Pharmacological Perspectives. Cheminform 2015, 46, 839–865. [Google Scholar] [CrossRef]
- Syam, S.; Abdelwahab, S.; Al-Mamary, M.A.; Mohan, S. Synthesis of Chalcones with Anticancer Activities. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef]
- Pande, A.N.; Biswas, S.; Reddy, N.D.; Jayashree, B.S.; Kumar, N.; Rao, C.M. In vitro and in vivo anticancer studies of 2′-hydroxy chalcone derivatives exhibit apoptosis in colon cancer cells by HDAC inhibition and cell cycle arrest. EXCLI J. 2017, 16, 448. [Google Scholar]
- Zhou, J.; Li, M.; Chen, N.; Wang, S.; Luo, H.-B.; Zhang, Y.; Wu, R. Computational Design of a Time-Dependent Histone Deacetylase 2 Selective Inhibitor. ACS Chem. Boil. 2015, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Schnekenburger, M.; Zloh, M.; Golais, F.; Diederich, M.; Tasdemir, D. Natural chalcones as dual inhibitors of HDACs and NF-κB. Oncol. Rep. 2012, 28, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahyo, T.; Ichikawa, S.; Hatanaka, T.; Yamada, M.; Setou, M. A novel chalcone polyphenol inhibits the deacetylase activity of SIRT1 and cell growth in HEK293T cells. J. Pharmacol. Sci. 2008, 108, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, M.F.; Shaykoon, M.S.; Abdelrahman, M.H.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G.E.-D.A. Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors. Bioorganic Chem. 2017, 72, 32–41. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sung, B. Pharmacological basis for the role of curcumin in chronic diseases: An age-old spice with modern targets. Trends Pharmacol. Sci. 2009, 30, 85–94. [Google Scholar] [CrossRef]
- Shehzad, A.; Shahzad, R.; Lee, Y.S. Curcumin: A Potent Modulator of Multiple Enzymes in Multiple Cancers. Enzymes 2014, 36, 149–174. [Google Scholar]
- Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Anand, P.; Guha, S.; Aggarwal, B.B. Curcumin circumvents chemoresistance in vitro and potentiates the effect of thalidomide and bortezomib against human multiple myeloma in nude mice model. Mol. Cancer Ther. 2009, 8, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Kamat, A.M.; Sethi, G.; Aggarwal, B.B. Curcumin potentiates the apoptotic effects of chemotherapeutic agents and cytokines through down-regulation of nuclear factor- B and nuclear factor- B-regulated gene products in IFN- -sensitive and IFN- -resistant human bladder cancer cells. Mol. Cancer Ther. 2007, 6, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishodia, S.; Sethi, G.; Aggarwal, B.B. Curcumin: Getting Back to the Roots. Ann. New York Acad. Sci. 2005, 1056, 206–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Ahn, K.S.; Murakami, A.; Sethi, G.; Limtrakul, P.; Badmaev, V.; Aggarwal, B.B. Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory and anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 2007, 28, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Lao, C.D.; Ruffin, M.; Normolle, D.P.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.-J.; Chun, K.-S.; Cha, H.-H.; Han, S.S.; Keum, Y.-S.; Park, K.-K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat. Res. Mol. Mech. Mutagen. 2001, 480, 243–268. [Google Scholar] [CrossRef]
- Skommer, J.; Wlodkowic, D.; Pelkonen, J. Gene-expression profiling during curcumin-induced apoptosis reveals downregulation of CXCR4. Exp. Hematol. 2007, 35, 84–95. [Google Scholar] [CrossRef]
- Teiten, M.-H.; Dicato, M.; Diederich, M. Curcumin as a regulator of epigenetic events. Mol. Nutr. Food Res. 2013, 57, 1619–1629. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Wang, S.-H.; Lin, P.-Y.; Chiu, Y.-C.; Huang, J.-S.; Kuo, Y.-T.; Wu, J.-C.; Chen, C.-C. Curcumin-Mediated HDAC Inhibition Suppresses the DNA Damage Response and Contributes to Increased DNA Damage Sensitivity. PLOS ONE 2015, 10, e0134110. [Google Scholar] [CrossRef]
- Zeng, L.-L.; Shu, W.; Chen, W.; Wu, Q.; Liu, H.; Cui, G. Curcumin, both Histone Deacetylase and p300/CBP-Specific Inhibitor, Represses the Activity of Nuclear Factor Kappa B and Notch 1 in Raji Cells. Basic Clin. Pharmacol. Toxicol. 2007, 101, 427–433. [Google Scholar] [CrossRef]
- Liu, H.-L.; Chen, Y.; Cui, G.-H.; Zhou, J.-F. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol. Sin. 2005, 26, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Q.; Yu, K.; Yan, Q.X.; Xing, C.Y.; Chen, Y.; Yan, Z.; Shi, Y.F.; Zhao, K.W.; Gao, S.M. Pure curcumin increases the expression of SOCS1 and SOCS3 in myeloproliferative neoplasms through suppressing class I histone deacetylases. Carcinogenesis 2013, 34, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, W.-S.; Lin, C.; Lee, P.-S.; Kalyanam, N.; Ho, C.-T.; Pan, M.-H. Calebin-A induces cell cycle arrest in human colon cancer cells and xenografts in nude mice. J. Funct. Foods 2016, 26, 781–791. [Google Scholar] [CrossRef]
- Oliveira, A.L.D.P.; Martinez, S.E.; Nagabushnam, K.; Majeed, M.; Alrushaid, S.; Sayre, C.; Davies, N.M.; Oliveir, A.L.D.P.; Martine, S.E.; Nagabushna, K.; et al. Calebin A: Analytical Development for Pharmacokinetics Study, Elucidation of Pharmacological Activities and Content Analysis of Natural Health Products. J. Pharm. Pharm. Sci. 2015, 18, 494. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [Green Version]
- Qing, W.; Yan, C.; Xinggang, L. HDAC1 expression and effect of curcumin on proliferation of Raji cells. Acta Acad. Med. Wuhan 2006, 26, 199–201. [Google Scholar] [CrossRef]
- Sarkar, R.; Mukherjee, A.; Mukherjee, S.; Biswas, R.; Biswas, J.; Roy, M. Curcumin augments the efficacy of antitumor drugs used in leukemia by modulation of heat shock proteins via HDAC6. J. Environ. Pathol. Toxicol. Oncol. 2014, 33, 247–263. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Jha, A.; Rupasinghe, H.V. Novel carbocyclic curcumin analog CUR3d modulates genes involved in multiple apoptosis pathways in human hepatocellular carcinoma cells. Chem. Interactions 2015, 242, 107–122. [Google Scholar] [CrossRef]
- Shu, L.; Khor, T.O.; Lee, J.H.; Boyanapalli, S.S.S.; Huang, Y.; Wu, T.-Y.; Saw, C.; Cheung, K.-L.; Kong, A.-N. Epigenetic CpG Demethylation of the Promoter and Reactivation of the Expression of Neurog1 by Curcumin in Prostate LNCaP Cells. AAPS J. 2011, 13, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Shu, L.; Zhang, C.; Su, Z.-Y.; Kong, A.-N. Curcumin inhibits anchorage-independent growth of HT29 human colon cancer cells by targeting epigenetic restoration of the tumor suppressor gene DLEC1. Biochem. Pharmacol. 2015, 94, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soflaei, S.S.; Momtazi-Borojeni, A.A.; Majeed, M.; DeRosa, G.; Maffioli, P.; Sahebkar, A. Curcumin: A Natural Pan-HDAC Inhibitor in Cancer. Curr. Pharm. Desn 2018, 24, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.R.; Sangeetha, S.; Ranjitha, S.; Murugan, K. Breast Cancer Specific Histone Deacetylase Inhibitors and Lead Discovery using Molecular Docking and Descriptor Study. Trends Bioinform. 2013, 6, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-J.; Tsai, Y.-J.; Lin, M.-Y.; You, H.-L.; Kalyanam, N.; Ho, C.-T.; Pan, M.-H. Calebin-A induced death of malignant peripheral nerve sheath tumor cells by activation of histone acetyltransferase. Phytomedicine 2019, 57, 377–384. [Google Scholar] [CrossRef]
- Novaes, J.T.; Lillico, R.; Sayre, C.; Nagabhushanam, K.; Majeed, M.; Chen, Y.; Ho, E.; Oliveira, A.L.D.P.; Martinez, S.E.; Alrushaid, S.; et al. Disposition, Metabolism and Histone Deacetylase and Acetyltransferase Inhibition Activity of Tetrahydrocurcumin and Other Curcuminoids. Pharmaceutics 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pari, L.; Amali, D.R. Protective role of tetrahydrocurcumin (THC) an active principle of turmeric on chloroquine induced hepatotoxicity in rats. J. Pharm. Pharm. Sci. 2005, 8, 115–123. [Google Scholar] [PubMed]
- Aggarwal, B.B.; Deb, L.; Prasad, S. Curcumin Differs from Tetrahydrocurcumin for Molecular Targets, Signaling Pathways and Cellular Responses. Molecules 2014, 20, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Bora-Tatar, G.; Dayangac-Erden, D.; Demir, A.S.; Dalkara, S.; Yelekçi, K.; Yurter, H.E. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorganic Med. Chem. 2009, 17, 5219–5228. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef]
Figure 1.
Modifications in the structure of histones mediated by histone acetyltransferase (HAT) and histone deacetylase (HDAC). The figure shows the lysine residue undergoing acetylation (HAT) and deacetylation (HDAC).
Figure 1.
Modifications in the structure of histones mediated by histone acetyltransferase (HAT) and histone deacetylase (HDAC). The figure shows the lysine residue undergoing acetylation (HAT) and deacetylation (HDAC).
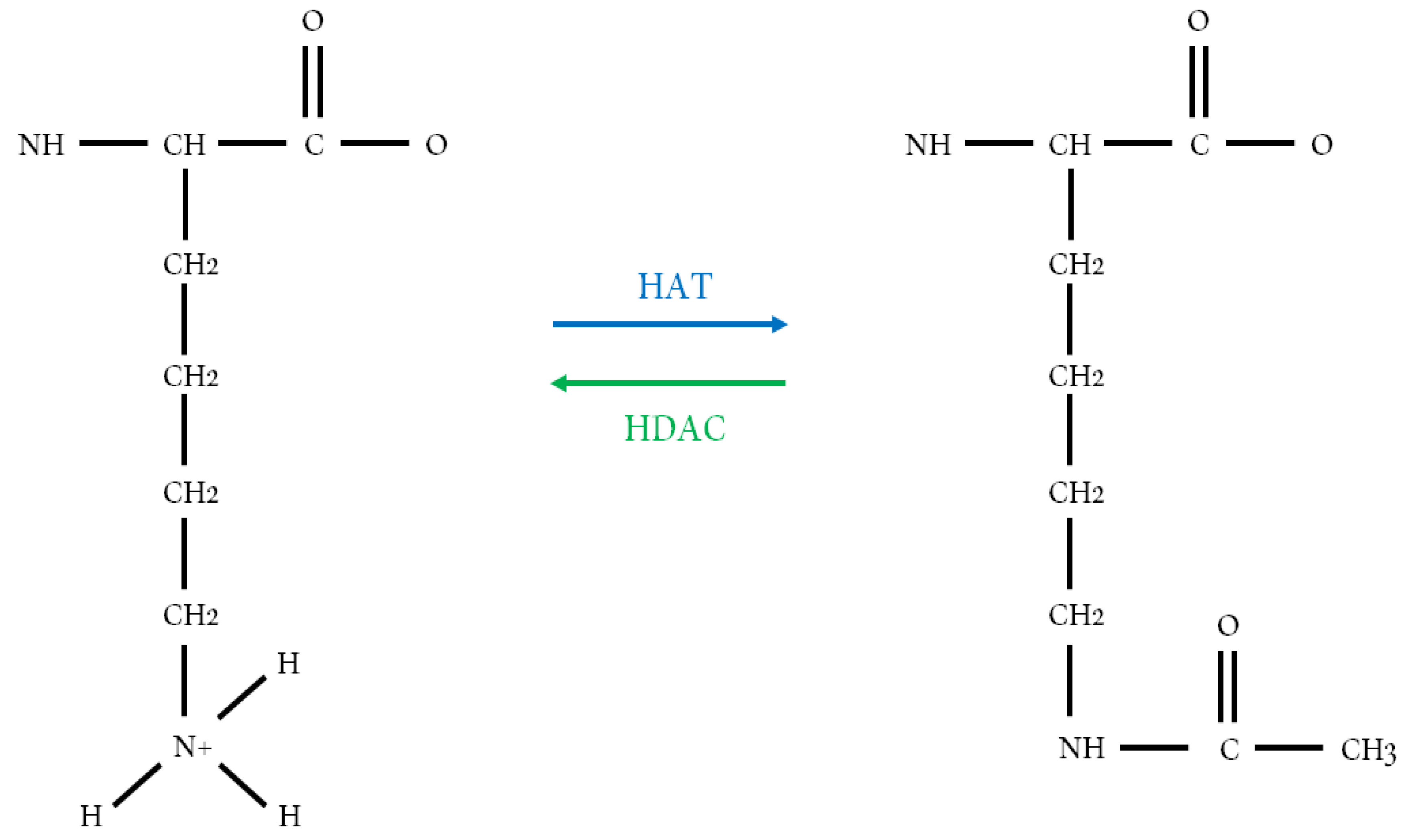
Figure 2.
Schematic organization of the HDAC classes I, II and IV showing the Zn2+-dependent catalytic domains in blue, the NAD+-dependent catalytic domains in pink, the nuclear localization in yellow and the ubiquitin-binding domain of HDAC6 in orange.
Figure 2.
Schematic organization of the HDAC classes I, II and IV showing the Zn2+-dependent catalytic domains in blue, the NAD+-dependent catalytic domains in pink, the nuclear localization in yellow and the ubiquitin-binding domain of HDAC6 in orange.
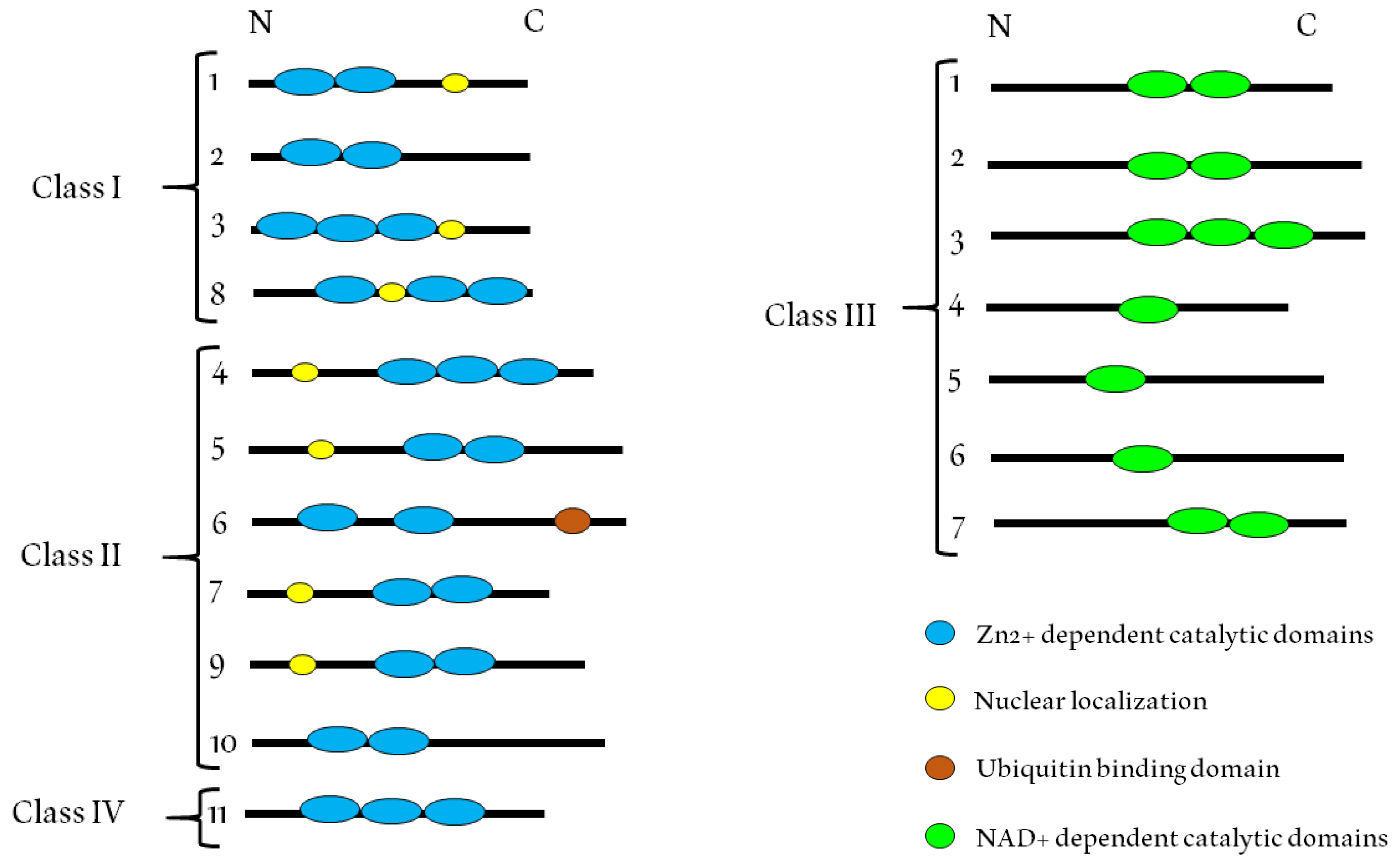
Figure 3.
Structure of class I HDAC co-repressor Complexes. HDAC1 and HDAC2 are recruited to the (A) SIN3 and the (B) NuRD. Adapted from [96,100,115].
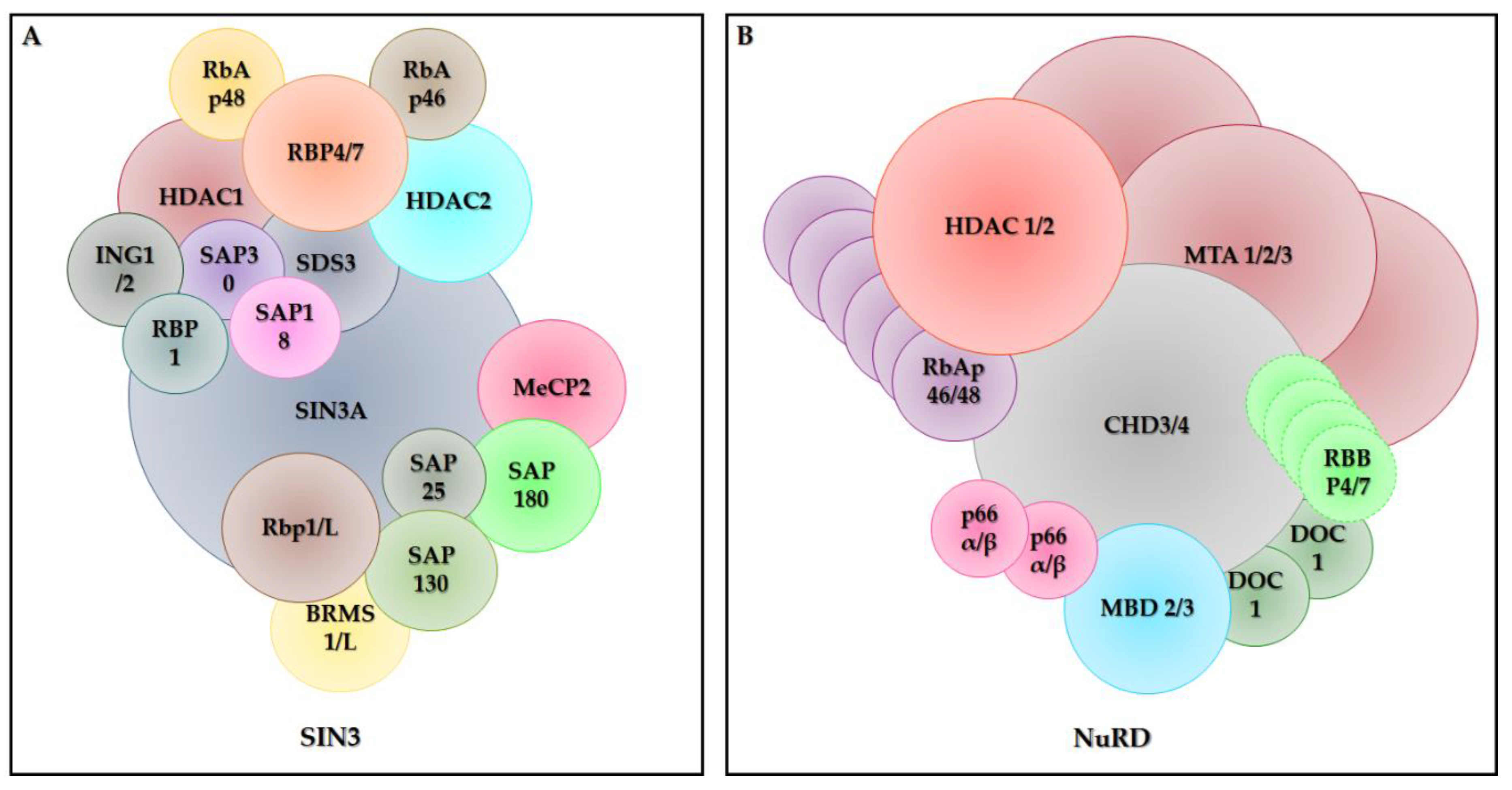
Figure 4.
Some non-histone proteins affected by histone deacetylases (HDACs) and the changes caused by these interactions.
Figure 4.
Some non-histone proteins affected by histone deacetylases (HDACs) and the changes caused by these interactions.

Figure 5.
Some pathways that are altered by the activity of histone deacetylases. Acetylation and deacetylation of histones alter the chromatin activity, causing important epigenetic changes. In addition, the activity of non-histone proteins is altered, including transcription factors, chaperones and structural proteins, influencing the activity of the different pathways involved in the cell cycle control, apoptosis, differentiation, angiogenesis and cell invasion.
Figure 5.
Some pathways that are altered by the activity of histone deacetylases. Acetylation and deacetylation of histones alter the chromatin activity, causing important epigenetic changes. In addition, the activity of non-histone proteins is altered, including transcription factors, chaperones and structural proteins, influencing the activity of the different pathways involved in the cell cycle control, apoptosis, differentiation, angiogenesis and cell invasion.
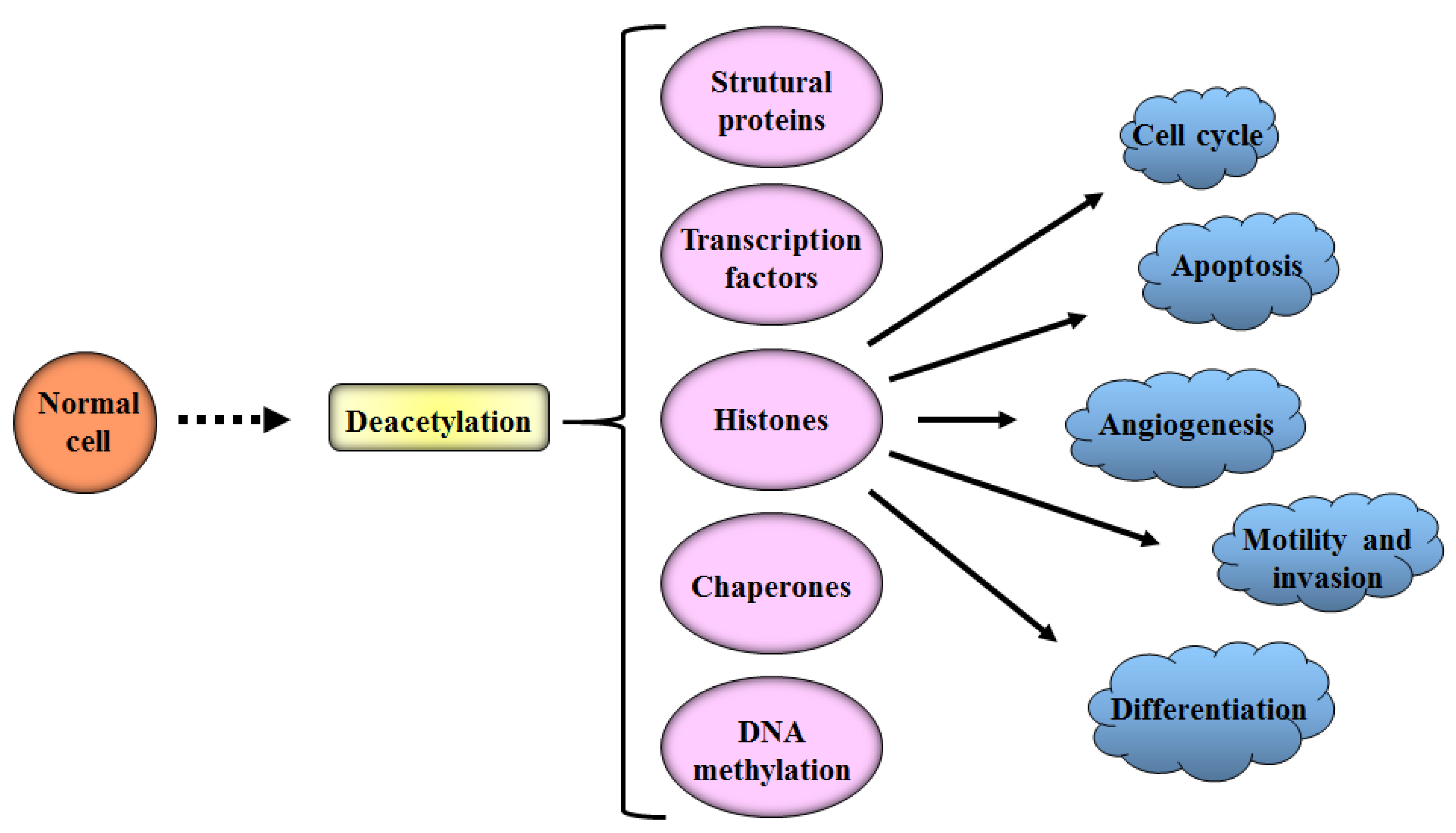
Figure 6.
Structures of vorinostat (i), trichostatin A (ii), entinostat (iii) and valproic acid (iv).
Figure 6.
Structures of vorinostat (i), trichostatin A (ii), entinostat (iii) and valproic acid (iv).
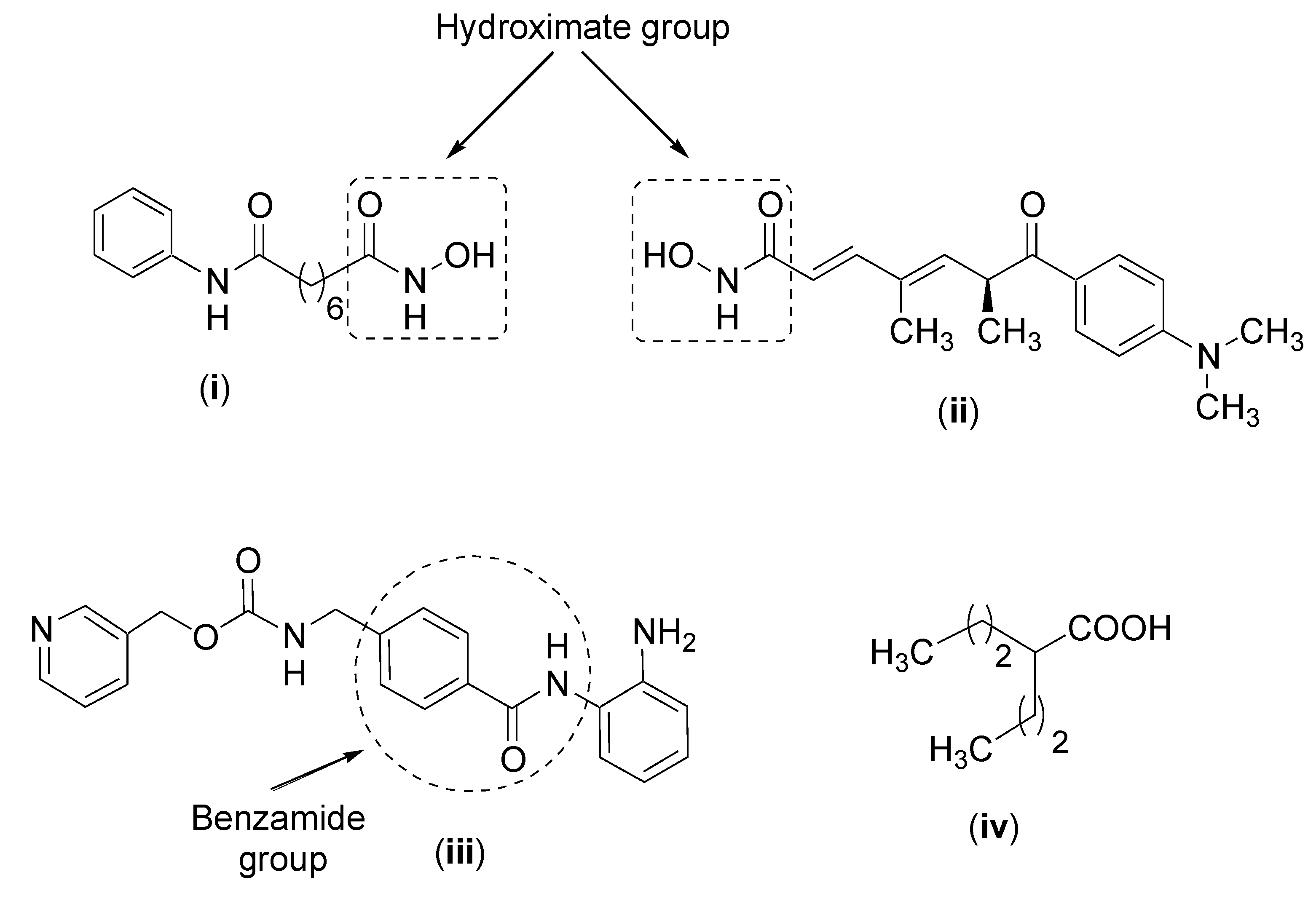
Figure 7.
Structures of calebin A (i) and ferulic acid (ii).
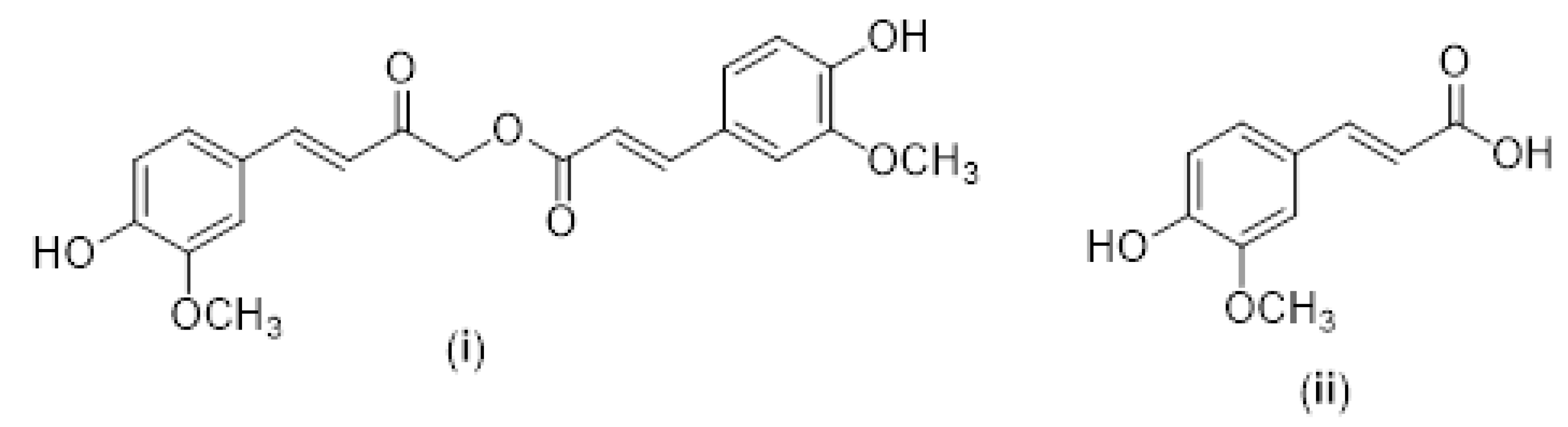

{kind=link}
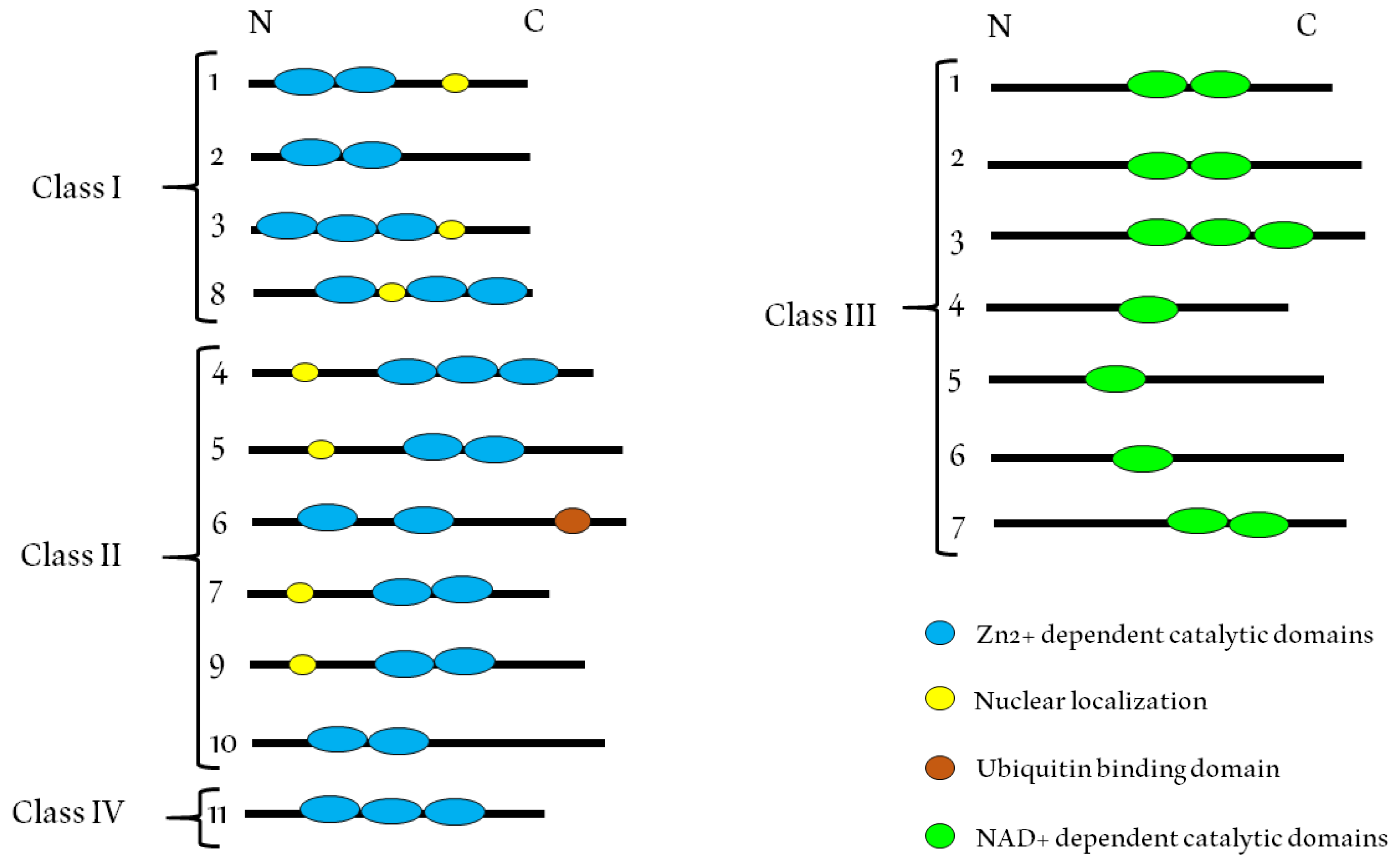{kind=link}
{kind=link}
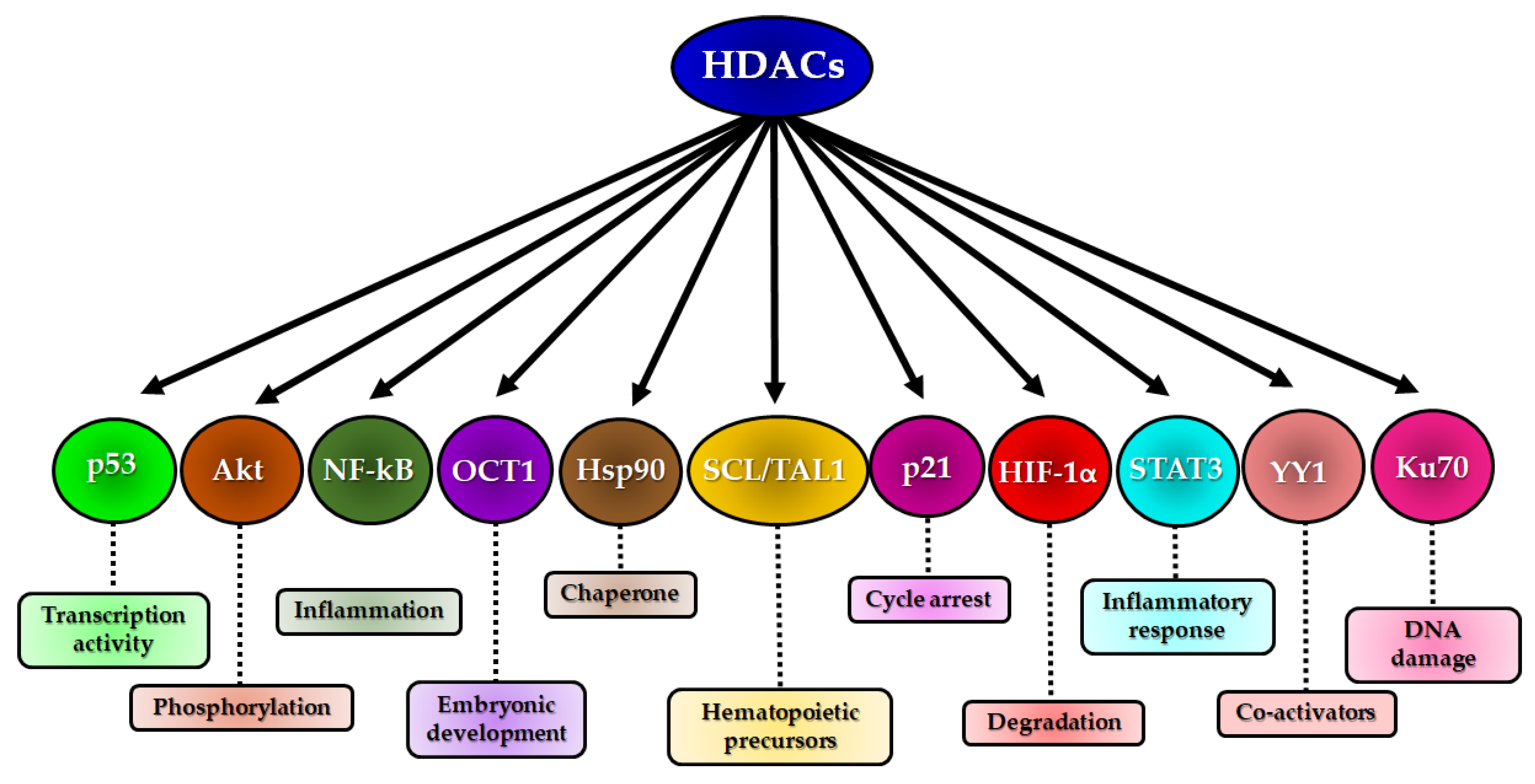{kind=link}
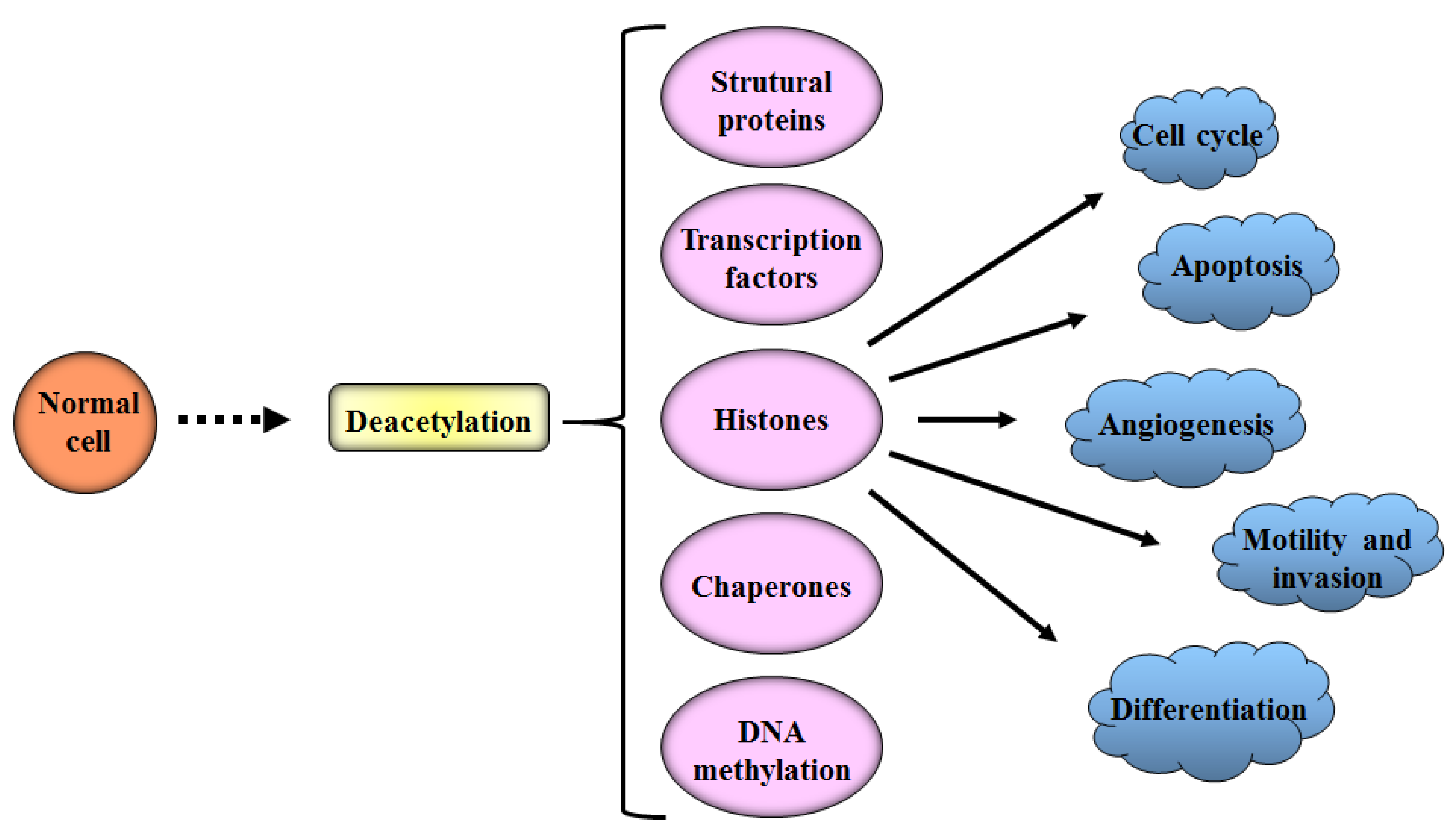{kind=link}
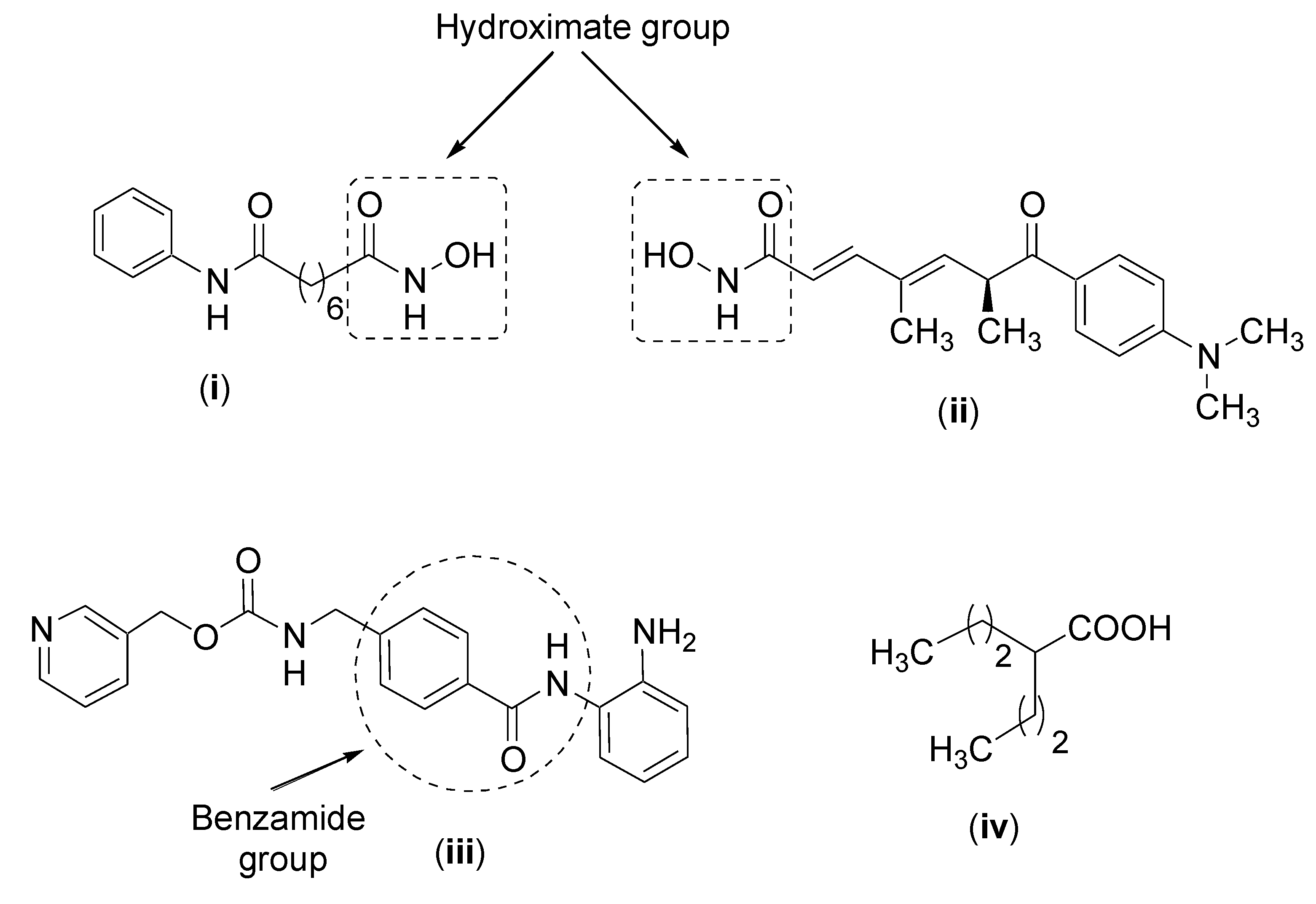{kind=link}
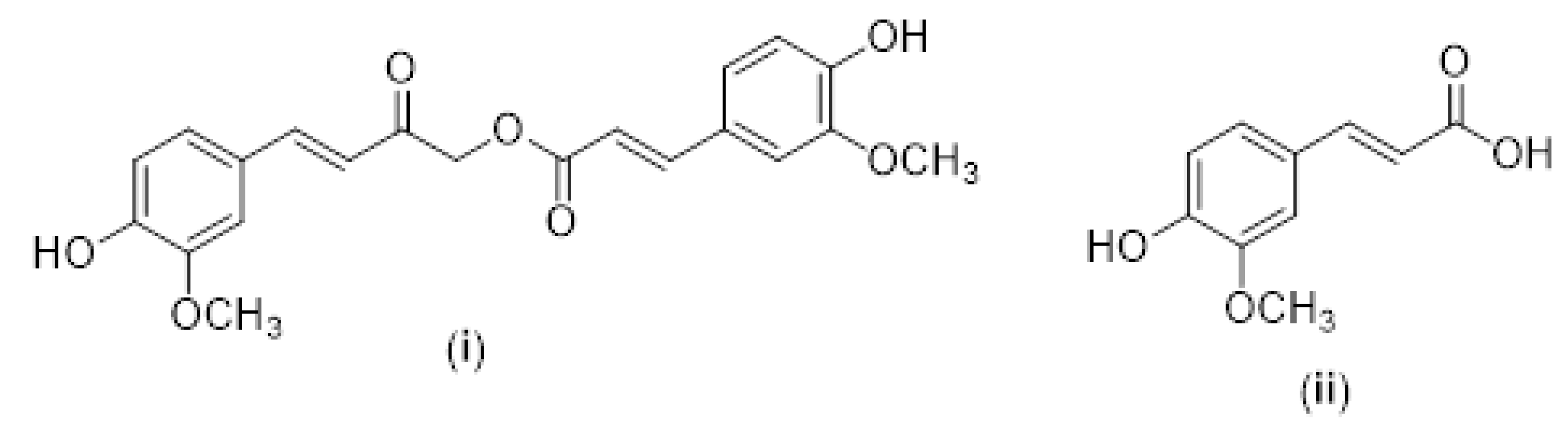{kind=link}
Table 1.
Type, class, protein size and localization of histone deacetylases (HDACs).
| HDAC Type | HDAC Class | Amino Acids | Cellular Localization | Cancer Type | Referencers |
|---|---|---|---|---|---|
| HDAC1 | I | 482 | Nucleus | Lung, gastric, liver, breast, ovarian, prostate, renal, bladder, hematological tumors | [52,53,54,55,56,57,58,59] |
| HDAC2 | I | 488 | Nucleus | Medulloblastoma, gastric, pancreatic, colorectal, breast, ovarian, prostate, renal, bladder, hematological tumors | [54,55,56,57,58,60,61,62,63] |
| HDAC3 | I | 428 | Nucleus and cytoplasm | Lung, gastric, breast, ovarian, prostate, bladder, melanoma, Hematological tumors | [54,55,57,58,62,64,65] |
| HDAC4 | IIa | 1084 | Nucleus and cytoplasm | Hematological tumors | [58] |
| HDAC5 | IIa | 1122 | Nucleus and cytoplasm | Hematological tumors | [58] |
| HDAC6 | IIb | 1215 | cytoplasm | Hematological tumors | [58] |
| HDAC7 | IIa | 912 | Nucleus and cytoplasm | Hematological tumors | [58] |
| HDAC8 | I | 377 | Nucleus, Mitochondria and cytoplasm | Neuroblastoma, melanoma, hematological tumors | [58,65,66] |
| HDAC9 | IIa | 1011 | Nucleus and cytoplasm | Hematological tumors | [58] |
| HDAC10 | IIb | 669 | Cytoplasm | Cervical, chronic lymphocytic leukemia | [59,67] |
| HDAC11 | IV | 347 | Nucleus | Lung | [68] |
Table 2.
In vitro studies on the effects of curcumin on histone deacetylases (HDACs).
| Cell Line | Curcumin | Molecular Effects | Result | References |
|---|---|---|---|---|
| Lymphoma Raji cells | 1.6–50 μM | Cell proliferation | ↓HDAC1 mRNA and protein expression | [269] |
| Human cervical cancer cell lines | 0.5–50 μM | Augments the efficacy of antitumor drugs | ↓HDAC activity, ↓HDAC1and 2 expression | [270] |
| HepG2 cell line | 100 μM of different curcumin analogues | Modulates genes | ↓HDAC1, 2, 4, 6, 8, 11 expression | [271] |
| K562, HEL, and MPN cell lines | 5–40 μM | Increases the expression of suppressor of cytokine signaling 1 and 3 | ↓HDAC activity ↓HDAC1, 3, 8 expression | [265] |
| LNCaP cells | 5 μM | CpG demethylation | ↓HDAC activity ↑HDAC1, 4, 5 and 8 and ↓HDAC3 protein expression | [272] |
| HT29 cell line | 2.5 and 5 μM | Inhibitory effect on cell proliferation | ↓HDAC4, 5, 6 and 8 expression | [273] |
Adapted from [274].
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061664
AMA Style
Verza FA, Das U, Fachin AL, Dimmock JR, Marins M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers. 2020; 12(6):1664. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061664
Chicago/Turabian StyleVerza, Flávia Alves, Umashankar Das, Ana Lúcia Fachin, Jonathan R. Dimmock, and Mozart Marins. 2020. "Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy" Cancers 12, no. 6: 1664. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061664
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.