Molecular Characterization of Astrocytoma Progression Towards Secondary Glioblastomas Utilizing Patient-Matched Tumor Pairs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
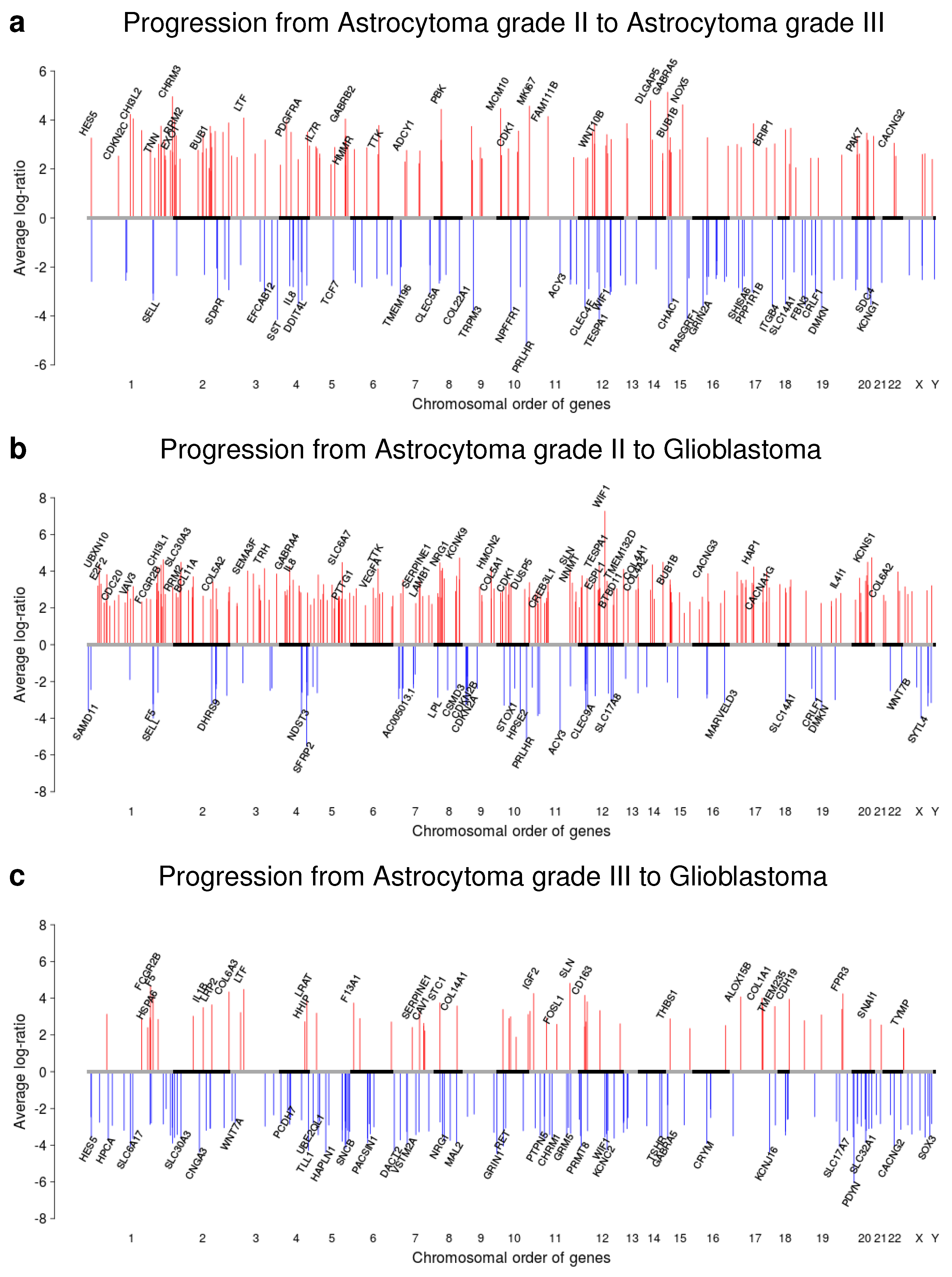
2.1. Gene Copy Number Alterations of Patient-Matched Tumor Pairs
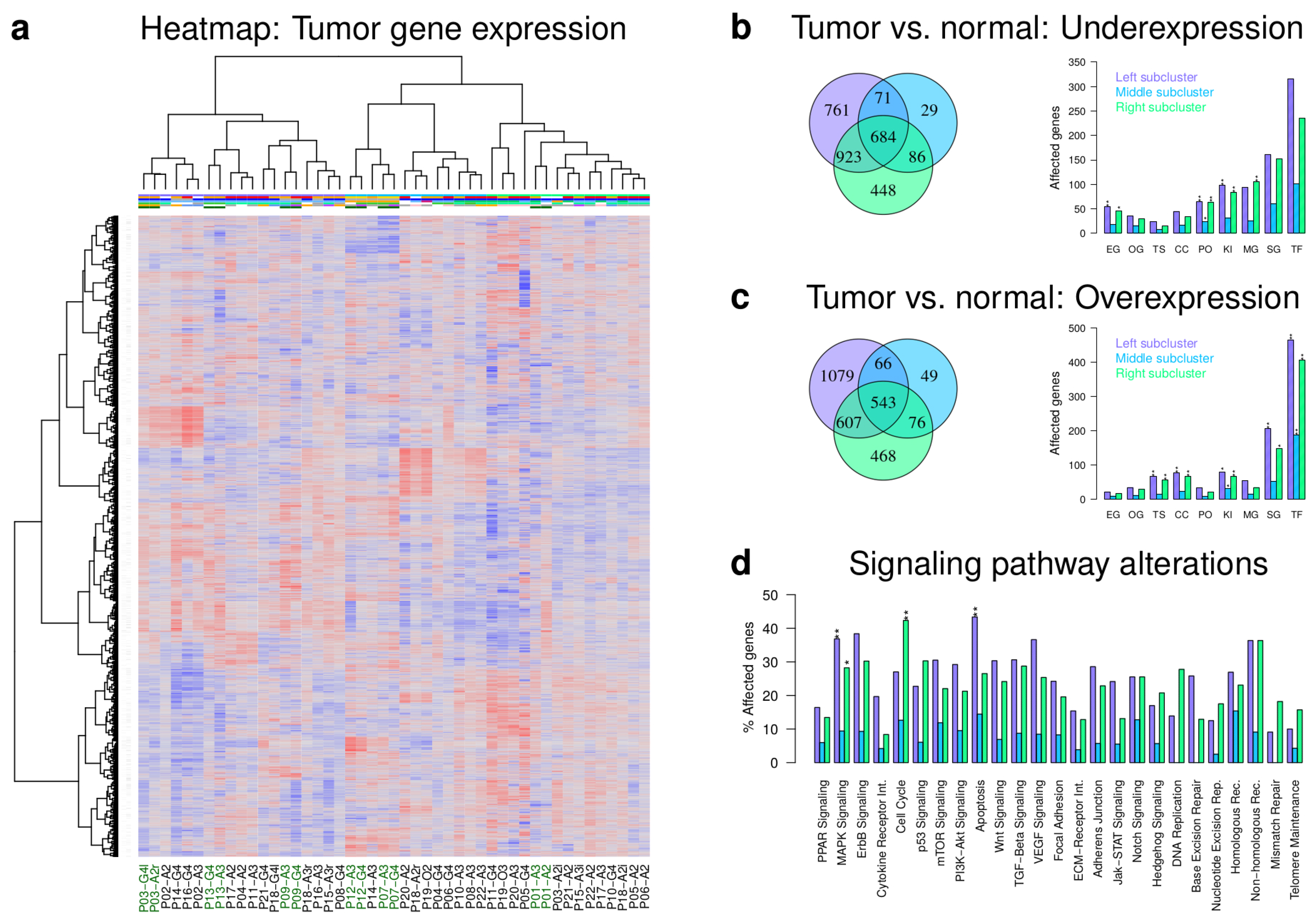
2.2. Majority of Patient-Matched Tumors Have Distinct Expression Profiles
2.3. IDH1 Mutant and Wild Type Tumors Differ in Expression of Cancer Genes and Pathways
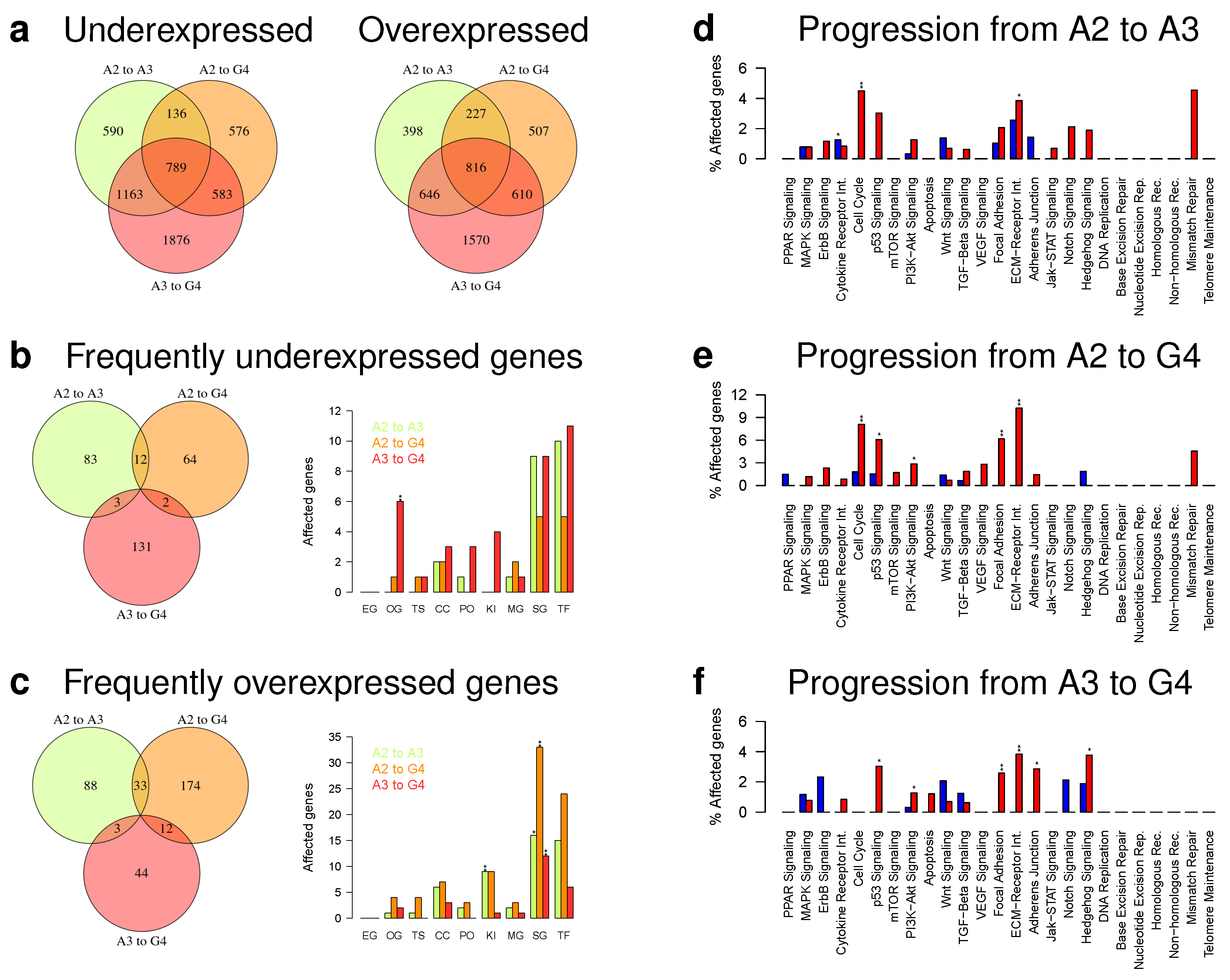
2.4. Initial Tumors Tend to Become Mesenchymal during Progression
2.5. Gene Expression Alterations of Astrocytoma Progression Groups
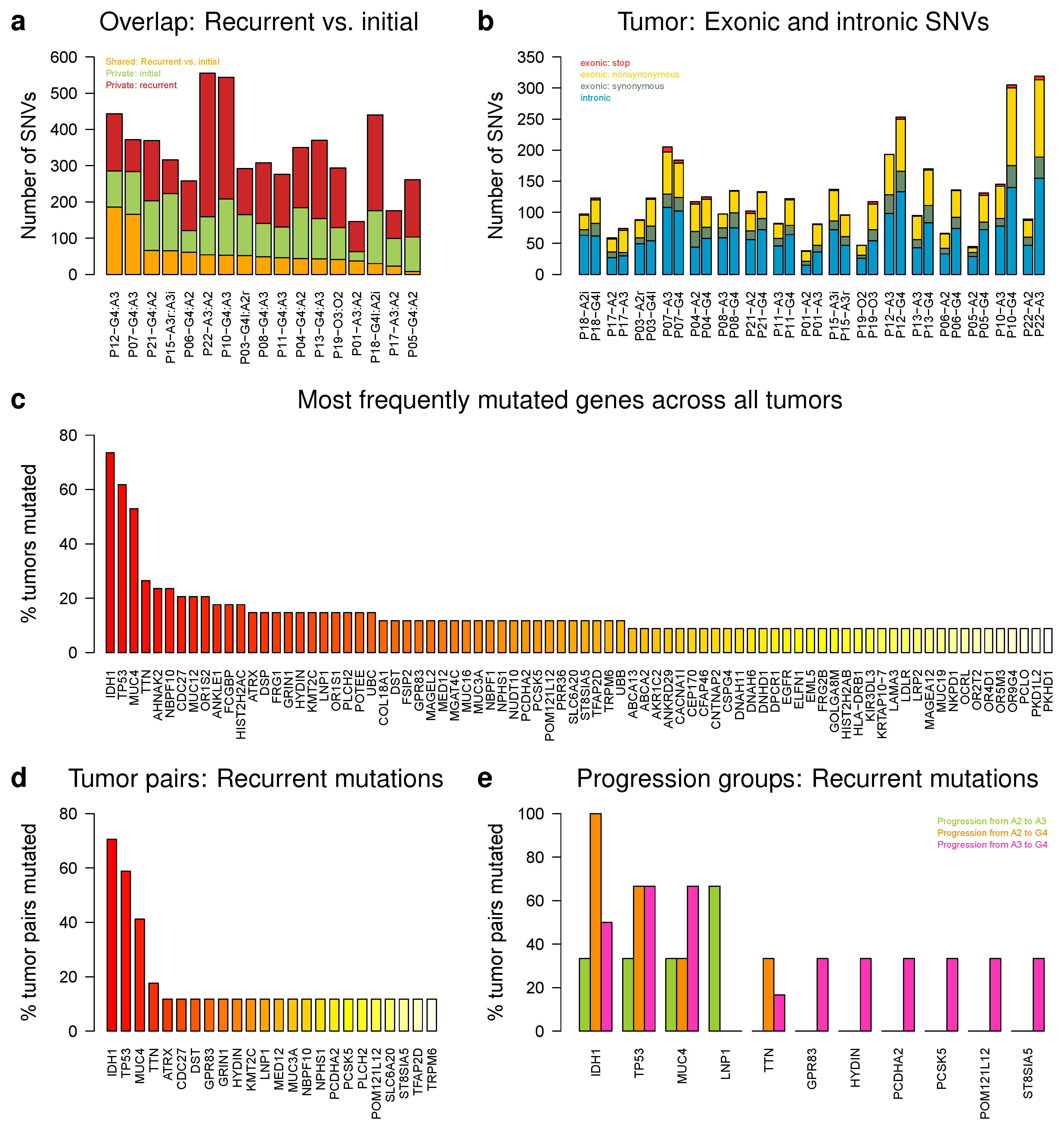
2.6. IDH1, TP53, and MUC4 Are Frequently Affected by Somatic Single Nucleotide Variations
2.7. Genes with Small Insertions or Deletions Were Rare, Except for ATRX
3. Discussion
4. Materials and Methods
4.1. Patients and Tumor Material
4.2. Gene Copy Number Measurements and Data Analysis
4.3. Transcriptome Sequencing and Data Preprocessing
4.4. Gene Expression Heatmap and Differential Expression of Major Tumor Clusters
4.5. G-CIMP and Verhaak Classification
4.6. HMM-Based Analysis of Patient-Matched Tumor Expression Profiles
4.7. Exome Sequencing and Data Preprocessing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ohgaki, H.; Kleihues, P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 2005, 64, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; McKay, R.M.; Parada, L.F. Malignant glioma: Lessons from genomics, mouse models, and stem cells. Cell 2012, 149, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, K.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Gorovets, D.; Kannan, K.; Shen, R.; Kastenhuber, E.R.; Islamdoust, N.; Campos, C.; Pentsova, E.; Heguy, A.; Jhanwar, S.C.; Mellinghoff, I.K.; et al. IDH mutation and neuroglial developmental features define clinically distinct subclasses of lower grade diffuse astrocytic glioma. Clin. Cancer Res. 2012, 18, 2490–2501. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and Adult Glioblastoma: Multiform (Epi)genomic Culprits Emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, A.; Karamchandani, J.; Das, S. Glioblastoma: Molecular Genetics of Secondary Glioblastoma; Codon Publications: Brisbane, Austrlia, 2017; Chapter 2. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N.Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.; Garbe, M.; Friedrich, B.; Mittelbronn, M.; Klink, B. Comparative transcriptomics reveals similarities and differences between astrocytoma grades. BMC Cancer 2015, 15, 952. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wesseling, P.; Verhaak, R.G.W. Reconstructing the molecular life history of gliomas. Acta Neuropathol. 2018, 135, 649–670. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Abou-El-Ardat, K.; Friedrich, B.; Klink, B.; Deutsch, A. Autoregressive Higher-Order Hidden Markov Models: Exploiting Local Chromosomal Dependencies in the Analysis of Tumor Expression Profiles. PLoS ONE 2014, 9, E100295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, M.; Strickert, M.; Schliep, A.; Grosse, I. Exploiting prior knowledge and gene distances in the analysis of tumor expression profiles by extended Hidden Markov Models. Bioinformatics 2011, 27, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E.; et al. Open Targets Platform: New developments and updates two years on. Nucleic Acids Res. 2019, 47, D1056. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aum, D.J.; Kim, D.H.; Beaumont, T.L.; Leuthardt, E.C.; Dunn, G.P.; Kim, A.H. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg. Focus 2014, 37, E11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Boyle, E.A.; Tokita, M.; Mikheev, A.M.; Sanger, M.C.; Girard, E.; Silber, J.R.; Gonzalez-Cuyar, L.F.; Hiatt, J.B.; Adey, A.; et al. Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 2014, 15, 530. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, N.; Gielen, G.H.; Rauschenbach, L.; Kebir, S.; Till, A.; Reinartz, R.; Simon, M.; Niehusmann, P.; Kleinschnitz, C.; Herrlinger, U.; et al. Longitudinal heterogeneity in glioblastoma: Moving targets in recurrent versus primary tumors. J. Transl. Med. 2019, 17, 96. [Google Scholar] [CrossRef] [Green Version]
- Pollack, J.R.; Sorlie, T.; Perou, C.M.; Rees, C.A.; Jeffrey, S.S.; Lonning, P.E.; Tibshirani, R.; Botstein, D.; Borresen-Dale, A.L.; Brown, P.O. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 12963–12968. [Google Scholar] [CrossRef] [Green Version]
- de Tayrac, M.; Etcheverry, A.; Aubry, M.; Saikali, S.; Hamlat, A.; Quillien, V.; Treut, A.L.; Galibert, M.D.; Mosser, J. Integrative genome-wide analysis reveals a robust genomic glioblastoma signature associated with copy number driving changes in gene expression. Genes Chromosom. Cancer 2009, 48, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, H.; Reis, R.M.; Nakamura, M.; Colella, S.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Lab. Investig. 2000, 80, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Zlatescu, M.C.; Betensky, R.A.; Ino, Y.; Cairncross, J.G.; Louis, D.N. PTEN is a target of chromosome 10q loss in anaplastic oligodendrogliomas and PTEN alterations are associated with poor prognosis. Am. J. Pathol. 2001, 159, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Wemmert, S.; Ketter, R.; Rahnenführer, J.; Beerenwinkel, N.; Strowitzki, M.; Feiden, W.; Hartmann, C.; Lengauer, t.; Stockhammer, F.; Zang, K.D.; et al. Patients with high-grade gliomas harboring deletions of chromosomes 9p and 10q benefit from temozolomide treatment. Neoplasia 2005, 7, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Mollgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek Jr, J.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Y.; Qi, X.; Gordon, R.E.; O’Brien, J.A.; Yuan, H.; Zhang, J.; Wang, Z.; Zhang, M.; Song, Y.; et al. EZH2 Phosphorylation Promotes Self-Renewal of Glioma Stem-Like Cells Through NF-kB Methylation. Front. Oncol. 2019, 9, 641. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.J.; Koty, Z.; Nalbantoglu, J. Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 2009, 69, 5433–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Han, G.; Li, Y.; Yue, Z.; Wang, L.; Liu, J. FOXO1 associated with sensitivity to chemotherapy drugs and glial-mesenchymal transition in glioma. J. Cell Biochem. 2019, 120, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef]
- Annibali, D.; Whitfield, J.R.; Favuzzi, E.; Jauset, T.; Serrano, E.; Cuartas, I.; Redondo-Campos, S.; Folch, G.; Gonzalez-Junca, A.; Sodir, N.M.; et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 4632. [Google Scholar] [CrossRef]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dube, C.; Gibert, M.J.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT Promoter Methylation in Malignant Gliomas: Ready for Personalized Medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandith, A.A.; Qasim, I.; Zahoor, W.; Shah, P.; Bhat, A.R.; Sanadhya, D.; Shah, Z.A.; Naikoo, N.A. Concordant Association Validates MGMT Methylation and Protein Expression as Favorable Prognostic Factors in Glioma Patients on Alkylating Chemotherapy (Temozolomide). Sci. Rep. 2018, 8, 6704. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Yu, J.; Tu, L.; Huang, N.; Li, H.; Luo, Y. Isocitrate Dehydrogenase Mutations in Glioma: From Basic Discovery to Therapeutics Development. Front. Oncol. 2019, 9, 506. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, I.; Zinn, P.; Lai, M.; Rajakannu, P.; Hamou, M.F.; Hegi, M.E. WIF1 re-expression in glioblastoma inhibits migration through attenuation of non-canonical WNT signaling by downregulating the lncRNA MALAT1. Oncogene 2016, 35, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Lambiv, W.L.; Vassallo, I.; Delorenzi, M.; Shay, T.; Diserens, A.C.; Misra, A.; Feuerstein, B.; Murat, A.; Migliavacca, E.; Hamou, M.; et al. The Wnt inhibitory factor 1 (WIF1) is targeted in glioblastoma and has a tumor suppressing function potentially by induction of senescence. Neuro Oncol. 2011, 13, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.G.; Pezuk, J.A.; Brassesco, M.S.; de Oliveira, J.C.; de Paula Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; de Oliveira, H.F.; Scrideli, C.A.; et al. BUB1 and BUBR1 inhibition decreases proliferation and colony formation, and enhances radiation sensitivity in pediatric glioblastoma cells. Childs Nerv. Syst. 2013, 29, 2241–2248. [Google Scholar] [CrossRef]
- Takashima, S.; Oka, Y.; Fujiki, F.; Morimoto, S.; Nakajima, H.; Nakae, Y.; Nakata, J.; Nishida, S.; Hosen, N.; Tatsumi, N.; et al. Syndecan-4 as a biomarker to predict clinical outcome for glioblastoma multiforme treated with WT1 peptide vaccine. Future Sci. OA 2016, 2, FSO96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, G.F.; Pekmezci, M.; Hansen, H.M.; Rice, T.; Marshall, R.E.; Molinaro, A.M.; Phillips, J.J.; Vogel, H.; Wiencke, J.K.; Wrensch, M.R.; et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J. Neuropathol. Exp. Neurol. 2015, 74, 442–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appay, R.; Dehais, C.; Maurage, C.A.; Alentorn, A.; Carpentier, C.; Colin, C.; Ducray, F.; Escande, F.; Idbaih, A.; Kamoun, A.; et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019, 21, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xie, X.; Ye, Y.; Wang, L.; Che, F. BCL11A: A potential diagnostic biomarker and therapeutic target in human diseases. Biosci. Rep. 2019, 39, BSR20190604. [Google Scholar] [CrossRef] [Green Version]
- Balbous, A.; Cortes, U.; Guilloteau, K.; Villalva, C.; Flamant, S.; Gaillard, A.; Milin, S.; Wager, M.; Sorel, N.; Guilhot, J.; et al. A mesenchymal glioma stem cell profile is related to clinical outcome. Oncogenesis 2014, 3, 91. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef] [Green Version]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Stein, T.I.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database 2010, 2010, baq020. [Google Scholar] [CrossRef]
- Moniaux, N.; Chaturvedi, P.; Van, S.I.; Porchet, N.; Singh, A.P.; Batra, S.K. MUC4 (mucin 4, cell surface associated). Atlas Genet. Cytogenet. Oncol. Haematol. 2007, 11, 201–206. [Google Scholar] [CrossRef]
- Li, W.; Wu, C.; Yao, Y.; Dong, B.; Wei, Z.; Lv, X.; Zhang, J.; Xu, Y. MUC4 modulates human glioblastoma cell proliferation and invasion by upregulating EGFR expression. Neurosci. Lett. 2014, 566, 82–87. [Google Scholar] [CrossRef]
- Singh, A.P.; Moniaux, N.; Chauhan, S.C.; Meza, J.L.; Batra, S.K. Inhibition of MUC4 expression suppresses pancreatic tumor cell growth and metastasis. Cancer Res. 2004, 64, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Gautam, S.K.; Kumar, S.; Cannon, A.; Hall, B.; Bhatia, R.; Nasser, M.W.; Mahapatra, S.; Batra, S.K.; Jain, M. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin. Ther. Targets 2017, 21, 657–669. [Google Scholar] [CrossRef]
- Rowson-Hodel, A.R.; Wald, J.H.; Hatakeyama, J.; O’Neal, W.K.; Stonebraker, J.R.; VanderVorst, K.; Saldana, M.J.; Borowsky, A.D.; Sweeney, C.; Carraway 3rd, K.L. Membrane Mucin Muc4 promotes blood cell association with tumor cells and mediates efficient metastasis in a mouse model of breast cancer. Oncogene 2018, 37, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Arora, S.; Wang, B.; Grizzle, W.E.; Singh, A.P. MicroRNA-150 directly targets MUC4 and suppresses growth and malignant behavior of pancreatic cancer cells. Carcinogenesis 2011, 32, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Yang, Z.; Hu, M.; Liu, D.; Hu, Y.; Qian, L.; Zhang, W.; Chen, H.; Guo, L.; Yu, M.; et al. Catecholamine-Induced beta2-adrenergic receptor activation mediates desensitization of gastric cancer cells to trastuzumab by upregulating MUC4 expression. J. Immunol. 2013, 190, 5600–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiestler, B.; Capper, D.; Holland-Letz, T.; Korshunov, A.; von Deimling, A.; Pfister, S.M.; Platten, M.; Weller, M.; Wick, W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013, 126, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Núñez, F.J.; Méndez, F.M.; Núñez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef]
- Abou-El-Ardat, K.; Seifert, M.; Becker, K.; Eisenreich, S.; Lehmann, M.; Hackmann, K.; Rump, A.; Meijer, G.; Carvalho, B.; Temme, A.; et al. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro Oncol. 2017, 19, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Venkatraman, E.S.; Olshen, A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 2007, 23, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Friedrich, B.; Beyer, A. Importance of rare gene copy number alterations for personalized tumor characterization and survival analysis. Genome Biol. 2016, 17, 204. [Google Scholar] [CrossRef] [Green Version]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 12, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC. Babraham Institute, 2012. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 9 December 2019).
- Krueger, F. Trim Galore: A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files; Babraham Institute: Cambridge, UK, 2012; Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 9 December 2019).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Picard Toolkit. 2019. Available online: http://broadinstitute.github.io/picard/ (accessed on 9 December 2019).
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Lauber, C.; Klink, B.; Seifert, M. Comparative analysis of histologically classified oligodendrogliomas reveals characteristic molecular differences between subgroups. BMC Cancer 2018, 18, 399. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. Technical Report. 2013. Available online: https://arxiv.org/abs/1303.3997 (accessed on 9 December 2019).
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019, 861054. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seifert, M.; Schackert, G.; Temme, A.; Schröck, E.; Deutsch, A.; Klink, B. Molecular Characterization of Astrocytoma Progression Towards Secondary Glioblastomas Utilizing Patient-Matched Tumor Pairs. Cancers 2020, 12, 1696. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061696
Seifert M, Schackert G, Temme A, Schröck E, Deutsch A, Klink B. Molecular Characterization of Astrocytoma Progression Towards Secondary Glioblastomas Utilizing Patient-Matched Tumor Pairs. Cancers. 2020; 12(6):1696. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061696
Chicago/Turabian StyleSeifert, Michael, Gabriele Schackert, Achim Temme, Evelin Schröck, Andreas Deutsch, and Barbara Klink. 2020. "Molecular Characterization of Astrocytoma Progression Towards Secondary Glioblastomas Utilizing Patient-Matched Tumor Pairs" Cancers 12, no. 6: 1696. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061696