Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges


Department of Surgery, City of Hope National Medical Center, Duarte, CA 91010, USA
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(6), 1699; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061699
Submission received: 31 May 2020
/
Revised: 19 June 2020
/
Accepted: 23 June 2020
/
Published: 26 June 2020
(This article belongs to the Special Issue Oncolytic Virus Therapy Against Cancer)
Abstract
:The field of oncolytic virotherapy has seen remarkable advancements in last two decades, leading to approval of the first oncolytic immuno-virotherapy, Talimogene Laherparepvec, for the treatment of melanoma. A plethora of preclinical and clinical studies have demonstrated excellent safety profiles of other oncolytic viruses. While oncolytic viruses show clinical promise in already immunogenic malignancies, response rates are inconsistent. Response rates are even less consistent in immunosuppressed tumor microenvironments like those found in liver, pancreas, and MSI-stable colon cancers. Therefore, the efficacy of oncolytic viruses needs to be improved for more oncolytic viruses to enter mainstream cancer therapy. One approach to increase the therapeutic efficacy of oncolytic viruses is to use them as primers for other immunotherapeutics. The amenability of oncolytic viruses to transgene-arming provides an immense opportunity for investigators to explore different ways of improving the outcome of oncolytic therapy. In this regard, genes encoding immunomodulatory proteins are the most commonly studied genes for arming oncolytic viruses. Other transgenes used to arm oncolytic viruses include those with the potential to favorably modulate tumor stroma, making it possible to image the virus distribution and increase its suitability for combination with other therapeutics. This review will detail the progress made in arming oncolytic viruses with a focus on immune-modulatory transgenes, and will discuss the challenges that need to be addressed for more armed oncolytic viruses to find widespread clinical use.
1. Introduction
Oncolytic viruses (OVs) represent a novel class of therapeutics that use replication-competent, live viruses to treat cancer. OVs are either inherently cancer-selective or are genetically modified to replicate specifically in cancer cells. Cancer cells accrue myriad mutations during the course of evolution which allow them to evade the immune system and grow in an unchecked manner [1]. Interestingly, while preparing themselves for immune evasion, cancer cells often inactivate their antiviral protection mechanisms, such as the interferon pathways, leaving themselves vulnerable to viruses [2]. First-generation OVs utilize these mutated loopholes to specifically replicate in and kill cancer cells while leaving normal cells unharmed. Examples of first-generation OVs include reovirus [3], vesicular stomatitis virus (VSV) [4] and Newcastle disease virus (NDV) [5]. Unlike first-generation OVs which are inherently cancer selective, second-generation OVs are genetically manipulated to selectively replicate in cancer cells. A wide variety of viruses including adenovirus [6], herpes simplex virus (HSV) [7], vaccinia virus (VACV) [8], poliovirus [7], myxomavirus [9] and measles virus [10] have been genetically altered to make them cancer selective. Furthermore, these second-generation oncolytic viruses have also been armed with transgenes to activate antitumor immunity or to aid to the efficacy of OVs in some other way. These armed viruses are often referred to as third generation OVs. This review will discuss armed viruses with a primary focus on immunomodulatory transgenes.
2. Oncolytic Virus and the Immune System
Until one decade ago, the immune system was largely assumed to be one of the biggest hurdles in the success of oncolytic virus. This assumption was based on the premise that antiviral immune response not only causes premature clearance of the virus, but also hampers subsequent rounds of treatment with the virus [11,12,13]. However, the field has evolved to view the immune system not as a hurdle, but as an essential component for the success of OV therapy [14,15,16,17,18,19]. It should be noted that OVs induce not only antiviral immunity, but also antitumor immunity. While antitumor immunity is obviously beneficial in the treatment of cancer, antiviral immunity does not necessarily contravene the efficacy of OVs. In fact, antiviral immunity is thought to be essential for the initial priming of antitumor immunity, as well as for the conversion of immunologically “cold” tumors into immunologically “hot” ones (reviewed by Gujar et al. [19]). Immunologically “cold” tumors display one or more of the following features: low levels of tumor antigen, lack of T cells recognizing tumor antigens, low levels of CD8+ T cells, high levels of immune suppressive cells and/or cytokines. In contrast, immunologically “hot” tumors have high levels of tumor antigens, high levels of effector T cells and low levels of immune suppressive cells and/or cytokines. Furthermore, since OVs preferentially infect cancer cells, immune cells directed against virus antigens will also kill cancer cells [19].
OVs kill cancer cells mostly by inducing immunogenic cell death (ICD) [20,21,22]. This type of cell death, which includes immunogenic apoptosis, pyroptosis, necrosis, necroptosis and autophagy, causes the release of a plethora of damage-associated molecular patters (DAMPs) such as ecto-calreticulin, ATP, heat shock proteins, HMGB1, uric acid as well as pathogen-associated molecular patterns (PAMPs) [20,21]. In addition to DAMPs and PAMPs, a repertoire of tumor-specific antigens (TSAs) and/or tumor-associated antigens (TAAs) is also released from the dying cancer cells [20]. Danger signals provided by DAMPs and PAMPs, together with the TSA/TAA, are required for dendritic cells to evoke an adaptive antitumor immunity [23,24]. Given their role in the activation of antitumor immunity, OVs are now considered a type of immunotherapy [25].
3. Strategies for Arming Oncolytic Viruses
Cancer is a complex disease, and equally complex treatments will be needed to eliminate it. In complex solid tumors, it is almost impossible to infect all cancer cells, no matter how enormous the dose of the virus administered and what route is used to administer the virus. Compared to intratumoral injection, OVs administered systemically are more prone to be cleared by different components of the host immune system, including complement and neutralizing antibodies. Hence, systemically delivered OVs are less efficient at reaching the tumor sites. Investigators have studied different strategies to protect OVs from immune components, including the use of stem cells, immune cells and cancer cells as carriers to deliver OVs. Furthermore, bio-compatible polymers such as polyethylene glycol have also been used to coat OVs in order to protect them from host immune components. While OVs, in theory, can self-amplify within the tumor and possibly infect all cancer cells, this scenario will be possible only if there is nothing to obstruct the replication and spread of the virus. However, in real-life, that is not the case. Clinical trials with OVs have revealed that virus titer rapidly declines within days after injection [26,27]. Therefore, it is highly unlikely that OVs could infect and kill all cancer cells in a tumor. Furthermore, while OVs possess the inherent ability to induce antitumor immunity, the extent of the antitumor activation may not be optimal for maximal tumor destruction. Hence, it is desirable to arm OVs with transgenes coding for proteins that can directly or indirectly aid overall antitumor efficacy. The ability of OVs to specifically infect cancer cells makes them good vectors for the localized expression of transgenes within the tumor microenvironment (TME), thereby reducing the potential toxicities associated with the systemic delivery of the products of the transgenes. Furthermore, the temporal regulation of gene expression in viruses, as well as existence of promoters with different transcriptional strengths, provides flexibility in controlling the timing and extent of transgene expression from oncolytic viruses. For example, gene expression in the poxvirus is programmed into three temporal stages: early, intermediate and late [28,29]. While early and intermediate genes are expressed before DNA replication, late gene expression is initiated only after the replication of the viral genome [28,29]. Therefore, a transgene placed under the control of a late promoter will only be expressed in cells that support replication of the virus. Furthermore, investigators have made synthetic promoters which can further regulate the expression of transgenes [30].
A variety of genes with a wide range of functions including immune-stimulation and stromal-modulation, have been used to arm different types of oncolytic viruses. Some of the most commonly studied oncolytic viruses include adenovirus, herpes simplex virus, poxvirus and reovirus. Table 1 compares and contrasts the properties of these four viruses. While many of the armed viruses are still in the preclinical phase, more than a dozen of them are currently in clinical trials (Table 2); the only OV to have received FDA approval, Talimogene laherparepvec (T-VEC), is also an armed OV.
4. Oncolytic Viruses Armed to Modulate Antitumor Immunity
Generating an adaptive antitumor immune response is a complex, multistep process, starting with the uptake of tumor antigens by antigen presenting cells and ending with the activation of effector T cells that can recognize and kill the tumor cells [34]. However, tumors employ strategies to thwart antitumor immune response by interfering at almost every step of immune activation [35]. Tumor cells, with the help of stromal cells, maintain an immune-suppressive environment within TME [35,36]. While OVs could inherently revert the immune-suppressed tumor microenvironment to some extent [19,37], different immune-modulating genes have been used to arm OVs to further increase the antitumor immune activating property of OVs and ultimately increase antitumor efficacy and reduce the probability of disease relapse. Genes encoding cytokines, chemokines, inhibitors of immune checkpoints, bi-specific T cell engagers, tumor antigens, and targets for chimeric antigen receptor (CAR)-T cells have been used to arm OVs with the purpose of enhancing antitumor immunity.
5. Cytokines
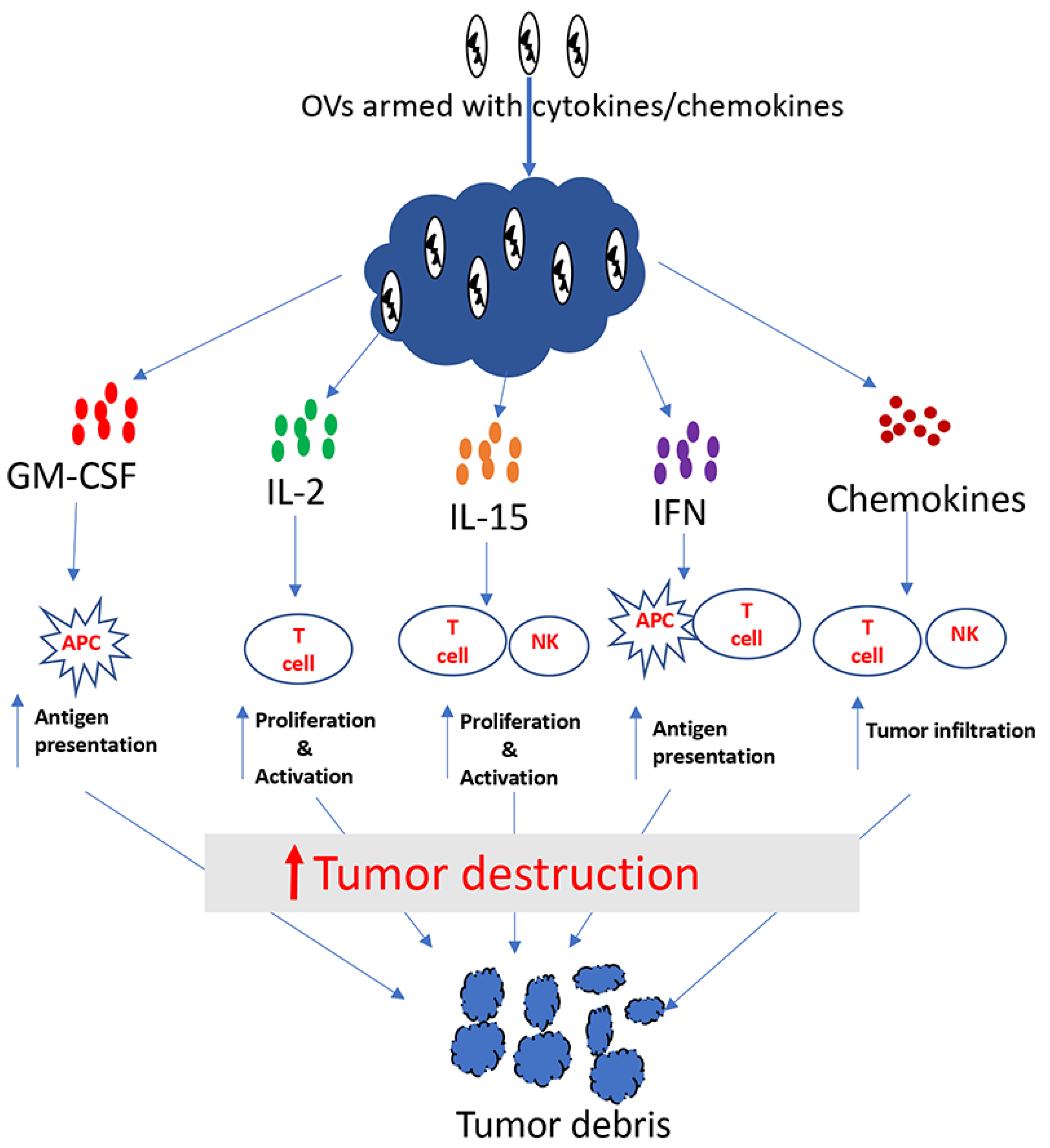
Cytokines are soluble proteins that allow immune cells to communicate. They play an important role in the regulation of both the innate and adaptive immune system. Some crucial aspects of the immune system, including differentiation, proliferation, effector functions and survival of immune cells, are controlled by cytokines [38,39]. Many cytokines including granulocyte macrophage colony stimulating factor (GM-CSF), interleukin (IL)-2, IL-12, IL-15, interferon (IFN)-α, IFN-β and IFN-γ have shown antitumor properties in preclinical studies (Figure 1). Among these cytokines, IFN-α [40] and IL-2 [41] were approved by FDA more than two decades ago for the treatment of some malignancies. Cytokines generally have short half-lives and act over short distances, which is why high doses are used for systemic administration, so that an effective dose can reach the tumor [39,40,42]. Unfortunately, high doses of cytokines often result in severe toxicities. For example, high doses of IL-2 can cause life-threatening side-effects such as vascular leak syndrome [43,44]. Hence, it is logical to surmise that localized expression of cytokines within TME will not only increase their antitumor efficacy, but also reduce systemic toxicity. With this idea, many investigators have used cytokine-encoding genes to arm oncolytic viruses.
5.1. Granulocyte Macrophage Colony Stimulating Factor (GM-CSF)
GM-CSF is one of the most commonly used cytokines for arming oncolytic viruses. GM-CSF is produced from activated T cells, fibroblasts, endothelial cells, macrophages and stromal cells [45]. GM-CSF helps in antigen presentation through recruitment and activation of dendritic cells and macrophages [46]. Several types of viruses, including HSV, VACV and adenovirus, have been armed with GM-CSF and evaluated in preclinical as well as clinical studies in different types of cancer. T-VEC, the only OV that has received FDA approval so far, is armed with GM-CSF. T-VEC is an HSV-1 (JS-1 strain) with both copies of the neurovirulence gene ICP34.5 replaced with two copies of human GM-CSF cDNA [47]. Also, T-VEC is deleted in the ICP47 gene, whose product inhibits the transporter associated with antigen presentation (TAP). The deletion of ICP34.5 genes increases the safety of T-VEC, while the deletion of ICP47 allows for MHC-I-dependent antigen presentation from virus-infected cells; the virus-encoded GM-CSF helps in the recruitment of DC, and promotes their antigen presenting function [48]. Another clinically advanced OV, called Pexa-Vec, is also armed with GM-CSF. Pexa-Vec is a Wyeth strain of the vaccinia virus with the J2R gene deleted, encoding for viral thymidine kinase, and a cDNA encoding human GM-CSF inserted at the J2R locus [49]. Pexa-Vec has completed multiple phase I and II trials with some degree of success in patients with different types of malignancies [50,51]. Likewise, adenoviruses armed with GM-CSF have also been studied in phase I clinical trials and found to be safe with modest antitumor efficacy [52,53].
5.2. Interleukin-2
IL-2 is primarily secreted from CD4+ T cells, but it can also be secreted from CD8+ T cells, NK cells and dendritic cells [54,55]. This cytokine potently induces the activation and proliferation of T cells. In 1992, IL-2 became the first cancer immunotherapy to be approved by the FDA for the treatment of metastatic renal cancer, and in 1998, its use was approved for the treatment of metastatic melanoma [41]. Because high levels of IL-2 delivered systemically can result in severe toxicity, investigators have tried to locally express IL-2 in tumors using different viral vectors [44,56]. Likewise, OVs have also been armed with IL-2. In a recent study, Liu et al. used the IL-2 gene to arm an oncolytic vaccinia virus and studied safety as well as antitumor efficacy of the virus in different murine tumor models [57]. The IL-2 transgene used in this study was modified to contain a glycosylphosphatidylinositol anchor with a rigid peptide linker in order to maintain IL-2 within TME to reduce toxicity. The virus was found to be effective in treating tumors with no side effects.
5.3. Interleukin-12
Many preclinical studies have shown that IL-12 has potent antitumor activity (reviewed by Weiss et al. [58]). In addition to the activation of antitumor immune cells (T cell and NK cells), IL-12 has also been shown to negatively affect tumors through its antiangiogenic activity [59]. Several OVs have been armed with IL-12 and studied for the treatment of different types of cancer. For example, Hellums et al. found higher levels of CD4+, CD8+ and NK cells in syngeneic murine gliomas treated with an IL-12-armed oncolytic HSV-1 compared to tumors treated with an unarmed oncolytic HSV-1 [60]. Consequently, the IL-12-armed oncolytic HSV-1 resulted in better survival compared to the unarmed virus. Ge et al. studied the safety and antitumor efficacy of an oncolytic vaccinia virus encoding a membrane-bound version of IL-12 [61]. The virus was found to convert an immunologically “cold” tumor into a “hot” one, and to increase the survival of mice bearing syngeneic colon tumors. Furthermore, when combined with a PD-1 inhibitor, the virus resulted in complete tumor regression in all mice that had late-stage colon cancer. Other oncolytic viruses, including adenovirus [62,63], VSV [64,65] and Semliki Forest virus [66], have also been armed with IL-12, and shown better efficacy compared to their respective unarmed parental viruses.
5.4. Interleukin-15
The cytokine IL-15 promotes the proliferation, activation and survival of CD8+ T cells, NK cells, NKT and dendritic cells [67]. IL-15 shares two of the three subunits of the IL-2 receptor, and has some overlapping functions [67]. Despite similarities in function, several studies suggest that IL-15 is more potent in controlling tumors and is less toxic than IL-2 [68,69]. A number of oncolytic viruses have been armed with the IL-15 gene and have shown superior antitumor efficacy compared to their unarmed counterparts [70,71,72]. A recent study by Kowalsky et al. armed an oncolytic vaccinia virus with IL-15 superagonist (a fusion protein of IL-15 and IL-15Rα) and found the virus to induce strong antitumor immunity, resulting in better therapeutic benefits in murine tumor models, which were further enhanced when the virus was combined with PD-1 blockade [73].
5.5. Interferons
The name “interferon” was originally applied to some cytokines due to their ability to interfere with virus replication. Type I interferons, including IFN-α and IFN-β, are potent antiviral cytokines [74]; however, that has not stopped investigators from using these cytokines to arm oncolytic viruses. The rationale for using Type-I IFN as a transgene in oncolytic viruses is that, apart from their antiviral function, they also have antitumor potential due to their role in the maturation of dendritic cells [75] and cytotoxic T cells [76]. An oncolytic adenovirus armed with IFN-α and used in combination with radiation was found to yield a modest increase in antitumor efficacy in murine models of pancreatic cancer [77]. Likewise, some degree of improvement in antitumor efficacy of oncolytic VACV [78], VSV [79], NDV [80] and measles [81] were observed after arming them with IFN-β.
IFN-γ, the only type-II IFN, has also been shown to exert an antitumor effect through a variety of mechanisms [82,83]. First, IFN-γ can inhibit the proliferation of cancer cells and also induce apoptosis [84]. Second, it affects tumors by inhibiting angiogenesis [85]. Lastly, it induces innate and adaptive immunity against cancer [86]. Commensurate with the antitumor role of IFN-γ, an oncolytic VSV engineered to encode IFN-γ exerted superior antitumor effect in multiple types of murine tumor models [87]. Compared to the parental virus, the IFN-γ-encoding virus was found to be more potent in the activation of dendritic cells, and it also resulted in higher levels of proinflammatory cytokines in the treated mice.
6. Chemokines
These are small secreted proteins that act as chemoattractant for leukocytes [88] (Figure 1). Because of their ability to attract immune cells, it may be beneficial to arm OVs with chemokines, especially for the treatment of immunologically “cold” tumors that lack effector immune cells. Indeed, several OVs have been armed with chemokines, resulting in enhancements of the antitumor efficacy. Nishio et al. armed an oncolytic adenovirus with the chemokine RANTES (CCL5) and the cytokine IL-15. They studied the antitumor potency of this virus in combination with CAR-T cells. The authors found that RANTES and IL-15 expressed from the oncolytic virus within the tumor was able to increase tumor infiltration by the CAR-T cells that were delivered systemically [88]. Likewise, Liu et al. used the chemokine CXCL11 to arm an oncolytic VACV, and studied its oncolytic potency in a mesothelioma model [89]. CXCL11 arming was found to attract higher numbers of tumor-specific CD8+ T cells compared to parental virus; consequently, higher therapeutic benefits were achieved with the CXCL11-encoding virus. Another study by Li et al. reported the enhanced safety and antitumor efficacy of an oncolytic VACV armed with CCL5 [90]. Since immunologically “cold” tumors are mostly refractory to immunotherapy, increasing effector immune cells in the tumor through the expression of chemoattractants from OVs seems to be an attractive approach for cancer therapy.
7. Inhibitors of Immunosuppressors
Tumors maintain an immune-tolerant microenvironment through the activation of a plethora of immune-suppressive mechanisms [35]. The immune-suppressed environment within tumors not only inhibits immune cells from mounting antitumor effects, but also reduces the efficacy of immunotherapy. A variety of cellular and secreted factors play crucial roles in maintaining immune suppression. Different strategies have been studied to arm OVs in order to overcome these immune-suppressive factors.
7.1. Inhibition of Secretory Immunosuppressive Factors
Some of the most potent secreted immune-suppressive factors are transforming growth factor-beta (TGF-β), vascular endothelial growth factor (VEGF) and prostaglandin E2 (PGE2) [91]. TGF-β inhibits the activation, proliferation and differentiation of T cells, and hence, negates their cytotoxic ability [92]. Likewise, VEGF has been shown to induce the proliferation of immunosuppressive cells, prevent T-cell recruitment to tumors, and induce exhaustion in T-cells [93]. Similarly, PGE2 exerts its immunosuppressive function through interference with antigen presentation by dendritic cells and by fostering the proliferation of myeloid-derived suppressor cells (MDSCs) [94]. Therefore, it is plausible that expression of proteins from OVs against these immunosuppressive cytokines may boost the antitumor efficacy of OVs. With this rationale, investigators have engineered OVs to encode proteins that can specifically inhibit TGF-β, VEGF or PGE2. For example, Yang et al. created an oncolytic adenovirus that encodes a soluble receptor for TGF-β, which can trap TGF-β and abrogate its immunosuppressive function [95]. The virus was found to increase Th1 cytokine, granzyme and perforin and CD8+ T cells in tumors, and promote the generation of CD4+ memory cells. Conversely, the virus reduced Th2 cytokines, the number of regulatory T (Tregs) cells and bone marrow-derived suppressor cells. Consequently, the virus demonstrated superior antitumor efficacy compared to the parental virus. Likewise, an oncolytic VACV-encoding antibody against VEGF was shown to increase innate immune cells in tumors, resulting in better therapeutic efficacy [96]. In another study, Hou et al. identified PGE2 as a key factor allowing tumors to resist immunotherapies as well as oncolytic virotherapy. In order to inhibit PGE2-mediated resistance, the authors armed an oncolytic VACV with a PGE2-inactivating enzyme, hydroxyprostaglandin dehydrogenase (HPGD) [97]. The HPGD-encoding virus was able to overcome localized immunosuppression and increase tumor infiltration by effector T cells. The armed virus exhibited better antitumor efficacy and sensitized previously resistant tumors to immune-checkpoint inhibitors.
7.2. Inhibition of Cellular Immunosuppressive Factors
T cells express checkpoint receptors such as CTLA-4, PD-1, LAG-3 and TIM-3. When these receptors interact with their ligands expressed on cancer cells, stromal cells or antigen presenting cells, the T cells become dysfunctional [98]. Because the checkpoint receptors need to physically interact with their ligands to inactivate T cells, it is possible to block such interactions using antibodies against the receptors or ligands [99]. Unleashing the antitumor potential of CD8+ T cells through the use of immune checkpoint inhibitors (ICI) can result in a strong antitumor effect. Indeed, ICIs have revolutionized the field of cancer immunotherapy with unprecedented, long-lasting therapeutic efficacies in many types of cancer [100]. However, only a small fraction of patients with immunologically “hot” tumors seem to benefit from ICIs [101]. OVs have been shown to convert immunologically “cold” tumors into “hot” tumors, suggesting that they could sensitize otherwise resistant tumors to ICIs [102,103]. Furthermore, we and others have shown that cancer cells upregulate the expression of checkpoint ligands in response to oncolytic viruses [103,104]. Together, these observations indicate that OVs and ICIs will make an attractive combination with which to fight cancer. Based on this idea, many studies have examined the combination of oncolytic viruses with antibodies against checkpoint ligands or receptors, and based on encouraging preclinical results, numerous clinical trials are currently underway to evaluate the combo therapy in multiple tumor types (Table 2).
At present, both T-VEC and approved ICIs cost well over one hundred thousand each for one course of treatment [105,106]. Given the high cost of ICIs, it will be beneficial to have ICIs encoded from the oncolytic virus itself. Indeed, many OVs have recently been engineered to encode ICIs. An oncolytic VACV encoding PD-1 antibody showed better antitumor efficacy compared to the parental virus, and the efficacy of the armed virus was similar that of the parental virus in combination with systemically administered PD-1 antibodies [107]. In another study, Bartee et al. engineered an oncolytic myxoma virus to encode a soluble form of PD-1 to inhibit the PD1/PD-L1 pathway [108]. Intratumoral injection of this virus in a murine melanoma model resulted in antitumor efficacy that was greater than that of the parental virus combined with the systemic injection of PD-1 antibodies. Similarly, recently, Wang et al. reported the construction and characterization of an oncolytic vaccinia virus encoding a PD-L1 inhibitor and GM-CSF. Using different tumor models, the authors showed that the expression of a PD-L1 inhibitor from the virus increased overall efficacy of the virus [109]. In another study, Dias et al. vectorized a CTLA-4 antibody in an oncolytic adenovirus and observed a modest increase in antitumor efficacy in prostate and lung cancer models [110]. In contrast to all these studies where vectorization of ICIs in OVs enhanced the antitumor efficacy of the viruses, a study by Engeland et al. found no difference between the efficacy of an oncolytic Measles virus encoding anti-PD-L1 or anti-CTLA-4 and the unarmed parental virus in a murine melanoma model [111]. However, systemically administered anti-PD-L1 or anti-CTLA-4 recombinant protein did increase the antitumor efficacy of the parental virus, suggesting that the route of administration for ICIs may determine their antitumor efficacy.
8. Tumor Antigens
Some types of tumors with low mutation burdens may not have adequate levels of neo-antigens; hence, it could be difficult for CD8+ T cells to recognize and eliminate those tumors. Ovarian cancer is an example of cancer with low mutation burden [112]. One way to increase the abundance of tumor antigens would be to use an OV armed with a tumor antigen. A recent study by McGray et al. demonstrated the feasibility of such an approach to boost vaccine priming and increase overall therapeutic response in an ovarian cancer model [113]. The authors used a Maraba virus encoding full-length ovalbumin (OVA) as a tumor antigen to treat a murine ovarian tumor model engineered to express OVA. Mice bearing ovarian tumors were first immunized with OVA admixed with an adjuvant, and then treated with OVA-encoding oncolytic Maraba virus. This prime-boost strategy using tumor antigen-armed OV greatly increased tumor antigen-specific CD8+ T cells, and resulted in improved therapeutic response.
9. Bispecific T Cells Engager (BiTE)
One approach to increasing the T cell-mediated killing of cancer cells is to use a bispecific antibody which can bind to T cells (CD3) and a target antigen on the surface of cancer cells [114]. BiTEs bind to T cells and cancer cells simultaneously and facilitate the lysis of cancer cells, even in the absence of costimulatory signals [115]. So far, one BiTE, called Blinatumomab, has been approved by the FDA for the treatment of B-cell malignancies [116], and many others are currently under clinical trials (clinicaltrials.gov accessed in May 2020). Many investigators have studied the potential of arming oncolytic viruses with BiTEs to achieve higher therapeutic responses. For example, Yu et al. engineered an oncolytic VACV to encode a secretory BiTE that could engage T cells with the tumor cell surface antigen EphA-2 [117]. The virus, in combination with T cells, demonstrated higher antitumor efficacy in a xenograft model of lung cancer. Likewise, Fajardo et al. constructed an oncolytic adenovirus armed with BiTE specific for EGFR and CD3. In an in vitro coculture experiment, oncolysis by BiTE-encoding OV greatly enhanced T-cell activation, proliferation and cytotoxicity [118]. Also, the BiTE-encoding adenovirus increased the persistence and accumulation of T cells in tumors and resulted in higher antitumor efficacy compared to parental virus in xenograft models. Another study using BiTE to arm an OV was recently published by Freedman et al., where they took a different approach from other investigators [119]. In this study, the BiTE was designed to target cancer-associated fibroblasts (CAFs) instead of directly targeting cancer cells. BiTE encoded from the virus was specific for fibroblast activation protein, a protein overexpressed on CAF, and CD3 on T cells. The virus was tested in malignant ascites and prostate biopsies freshly obtained from patients. As expected, BiTE-encoding OV increased T-cell mediated killing of CAFs, which led to the depletion of CAF-mediated immunosuppression, upregulation of proinflammatory cytokine and increased antigen presentation and T cell function in ascites. This study serves as a proof-of-principle that it is possible to gain immune-mediated antitumor responses simply by blocking immune-suppressive stromal cells.
10. Target for CAR-T Cells
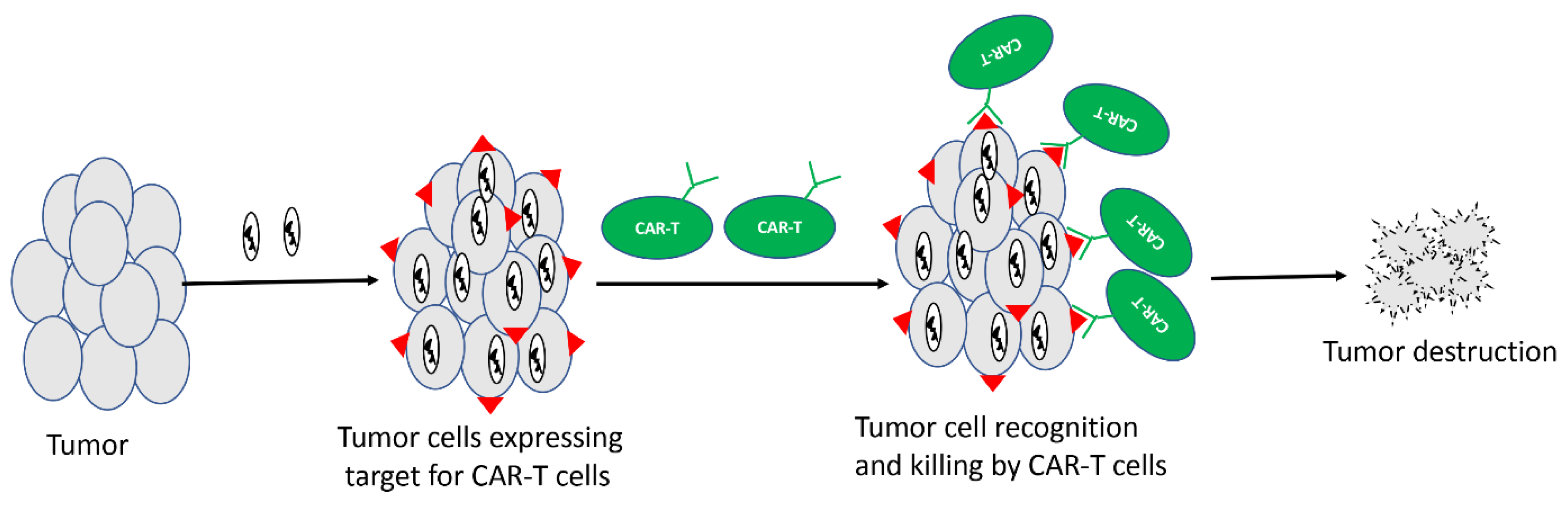
CAR-T cells are T cells redirected against tumors through engineered expression of CARs. These modified T cells are injected into cancer patients where they can specifically kill tumor cells. CAR-T cells directed against CD19 work very well against B cell malignancies, and so far, two CD-19-specific CAR-Ts, namely Kymriah [120] and Yescarta [121], have received FDA approval for the treatment of different types of B cell malignancies. However, unlike in B cell malignancies, CAR-T cells have shown suboptimal efficacy in solid tumors. One of the major reasons for the limited success of CAR-T in solid tumors is the lack of the selective and homogeneous expression of antigens on tumor cells [122]. Given the ability of OVs to selectively infect cancer cells, it may be possible to selectively express a unique antigen from cancer cells by using an antigen-armed OV, followed by treatment with CAR-T cells directed against that antigen (Figure 2). A recent study by Aalipour et al. demonstrated proof-of-principle for such a strategy [123]. In this study, the authors used an oncolytic VACV to selectively deliver CD19 to tumor cells in a murine melanoma model, and used CAR-T cells directed against CD19. The combination of CD-19-directed CAR-T with CD19-encoding OV resulted in improved survival of mice compared to antigen-mismatched combinations.
11. Stromal Modifiers
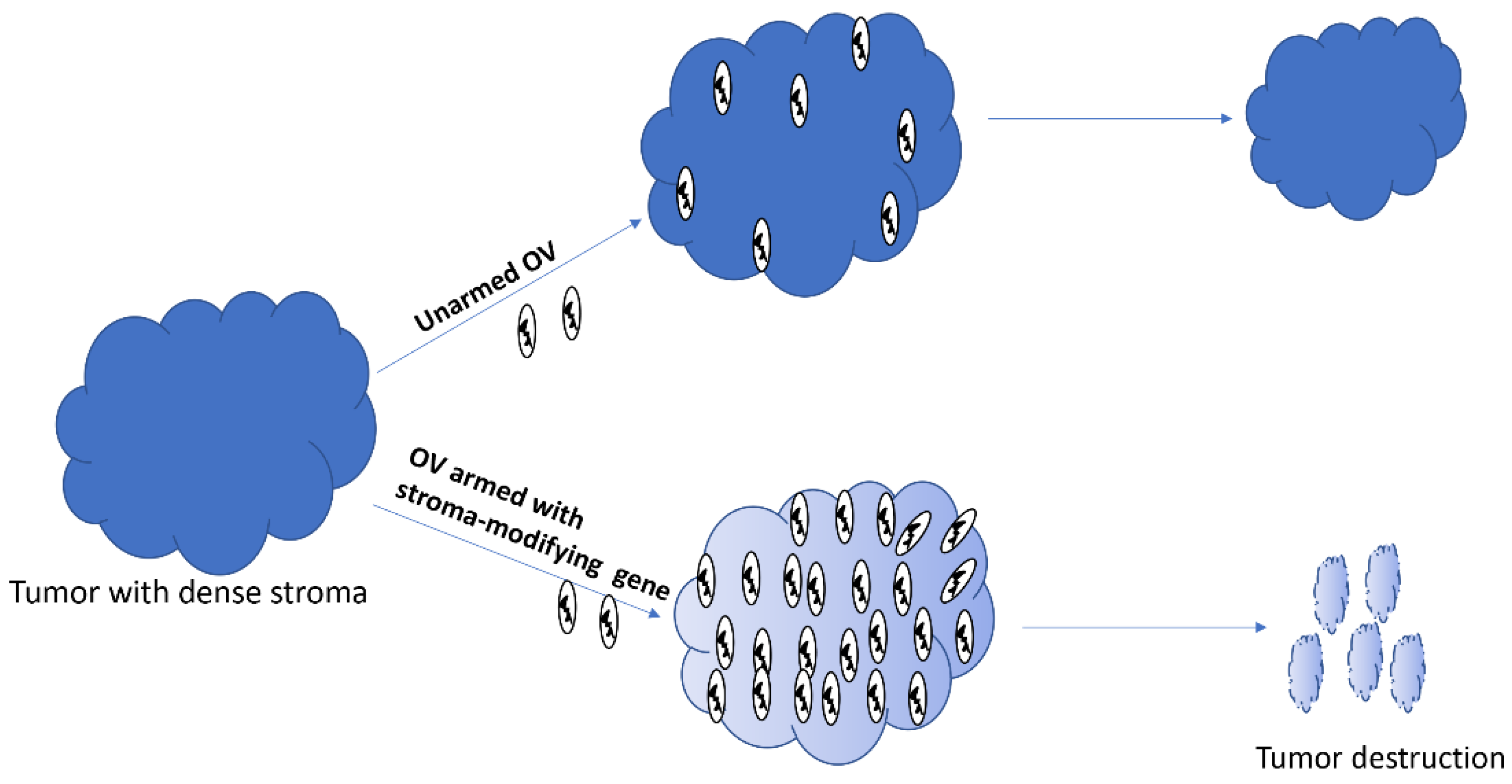
Oncolytic viruses administered intratumorally or through systemic routes can access tumors; however, their ability to spread within the tumors is mired by stromal components, including extracellular matrix (ECM) proteins [124,125]. Collagen and hyaluronic acid are two major components of ECM that increase interstitial fluid pressure and block therapeutics from reaching cancer cells [126]. Therefore, arming OVs with genes whose products can destroy the stromal factors could potentially increase the spread of OVs within dense solid tumors, and hence, improve the overall therapeutic efficacy (Figure 3). Indeed, an OV armed with the proteolytic enzyme hyaluronidase showed better intratumoral spread and higher antitumor efficacy in a xenograft model of melanoma in mice [127]. Likewise, an oncolytic VACV engineered to express metalloproteinase 9 was shown to degrade collagen IV and spread better in prostate cancer xenograft models. Consequently, the armed virus resulted in higher antitumor efficacy compared to the unarmed parental virus. Many other genes with the potential to modify stromal components, including decorin [128] and relaxin [129], have also been used to arm OVs.
12. Transgenes for Imaging
The ability to noninvasively track OVs using molecular imaging allows researchers to monitor the safety and efficacy of the virus in real time. Two types of molecular imaging exist: optical and deep-tissue. Many oncolytic viruses have been armed with transgenes to facilitate such imaging. While fluorescence- or luminescence-based optical imaging works well for preclinical studies, it may not be adequate for clinical purposes. Therefore, it is preferable to arm viruses with transgenes that will facilitate deep tissue imaging using MRI, PET or SPECT scanners (reviewed by Haddad et al. [130]). For optical imaging, transgenes encoding florescent proteins such as green fluorescent protein (GFP) and red fluorescent protein (RFP) are commonly used, whereas Firefly luciferase, Renilla luciferase and Gaussia luciferase are commonly used as transgenes for the bioluminescence-based optical imaging of viruses [130,131,132]. For deep tissue imaging, oncolytic viruses are armed with genes whose products work as prodrug converting enzymes or receptors or symporter/transporter. For example, the thymidine kinase gene from HSV-1 (HSV-tk) is used as a prodrug converting enzyme that phosphorylates the membrane permeable radiolabeled prodrug ganciclovir and traps it inside the cells, enabling PET imaging of virus-infected tissues/tumors [133]. Another approach to facilitate deep tissue imaging is the use of the human somatostatin receptor 2 (hSSR2) gene in oncolytic viruses. The expression of hSSR2 on the surface of infected cells binds to the radiolabeled, high-affinity, synthetic peptide pentetreotide and allows SPECT-based imaging to track virus infection [134,135]. The third strategy for deep tissue imaging uses a sodium iodide symporter (NIS) gene to arm oncolytic viruses. Virus-encoded NIS protein allows the uptake of radioisotopes such as 124I, 131I and rhenium by the infected cells. The NIS protein allows the uptake of most radioisotopes that have been approved for use in humans. Several studies have shown the potential of the NIS gene for imaging oncolytic viruses [136,137]. In our lab, we generated a chimeric oncolytic poxvirus and armed it with the human NIS gene under the control of a synthetic early/late promoter [136]. We used 124I to successfully image the virus infection and spread in murine models, and 131I to increase the therapeutic efficacy of the virus.
13. Discussion and Future Directions
Oncolytic viruses are multi-mechanistic bio-therapeutics against cancer. In addition to the direct lysis of cancer cells, OVs also activate antitumor immunity and damage tumor vasculature [138]. Most OVs are amenable to the incorporation of transgenes in their genome. This aspect confers upon OVs an unlimited possibility for modifications with the goal of increasing safety and antitumor efficacy. It is possible to modify OVs to encode one or more transgenes, each of which can be a therapeutic on its own. Not surprisingly, a wide range of therapeutic genes have been used to arm OVs. Most of the transgenes used to arm OVs possess immune-stimulatory function. One of the reasons why immunotherapy is so appealing is that antitumor response to immunotherapy can be long-lasting, due to the generation and maintenance of tumor-specific memory T cells [101]. Furthermore, the use of OVs to express immune-modulatory therapeutics has several advantages over the combination of OV with exogenous immune-therapeutics. First, immune-therapeutics such as cytokines have short half-lives and act over short distances, which is why repeated injections of high doses are required to achieve meaningful antitumor effects. However, OV-encoded cytokines can be restricted to TME and, at least in theory, will be made continuously as long as the virus persists in the tumors. Thus, the cytokine or other immune-modulatory therapeutics encoded by OVs within the tumor milieu will be more effective and less toxic. Second, arming oncolytic viruses with immune-therapeutics is cost-effective. Immune-therapeutics such as cytokines and ICIs are very expensive, as is T-VEC, the only OV approved for clinical use. Therefore, instead of treating patients with a combination of two expensive drugs, it would be more cost-effective to use one drug that has the combined effect of both. Lastly, OVs provide a versatile platform for the expression of immuno-therapeutics. Through the use of temporally regulated viral promoters of varying transcriptional strength, it is possible to have multiple transgenes encoded from an OV with some transgenes expressed early and at high levels, and other expressed late at low levels, or vice-versa.
Although it seems logical and beneficial to arm OVs with immune-therapeutic genes instead of simply combining OVs with exogenous immune-therapeutics, the arming approach may not be applicable for all immune-therapeutics. For example, regulation of T cell function through CTLA-4 takes place at the site of T cell priming, i.e., in secondary lymphoid organs [139]. Therefore, systemic delivery of CTLA-4 inhibitors will be more effective than intratumoral delivery since systemic delivery allows more of the therapeutics to reach secondary lymphoid organs. Indeed, a study by Engeland et al. using anti-CTLA-4-armed OV found no benefit of anti-CTLA-4 expressed from virus in the tumors, whereas systemically delivered anti-CTLA-4 was able to enhance the antitumor efficacy of the parental virus. Also, a large number of preclinical studies have been published showing the enhanced efficacy of oncolytic viruses armed with PD/PD-L1 inhibitors. However, somewhat surprisingly, not one of them has entered clinical testing, whereas a plethora of studies are currently in clinical trial, testing combinations of oncolytic viruses with systemic delivery of PD-1/PD-L1 inhibitors. This again suggests that not all the immune-therapeutics may be used to arm OVs.
The efficacy of OV against solid tumors is often limited by their inability to spread within tumors due to presence of dense stroma. Several studies have tried to address this issue by arming oncolytic viruses with stromal-degrading enzymes including collagenase, hyaluronidase and decorin. While these armed viruses have shown promising results in preclinical models, it remains to be seen if such a strategy will succeed in clinical settings. Other strategies in which arming of oncolytic viruses could be taken advantage of includes the delivery of de novo targets for CAR-T cells which could make it possible to harness the benefits of CAR-T cells in the treatment of solid tumors. Furthermore, oncolytic viruses could also be armed with tumor antigens which would work as in situ vaccinations and increase the overall efficacy of the OVs. In short, the sky is the limit for what OVs can be armed with to achieve better treatments of cancer.
Future studies should focus on the clinical aspects of OVs, such as the dosing and route of administration. For example, preclinical studies have used many different dosing strategies, most of which have resulted in antitumor benefits to some extent. Given the cost associated with clinical trials, it may not be feasible to test all dosing strategies in clinical settings. Therefore, preclinical studies should reach a consensus on the dosing strategy that is most likely to result in optimal therapeutic efficacy in clinical studies. Furthermore, most preclinical studies have used the intratumoral (i.t.) route for administration; however, this may not always be feasible, especially in cases of inaccessible tumors. One reason for intratumoral delivery is to avoid virus neutralization by circulating antibodies, especially if multiple dosing is to be used. One strategy to avoid virus neutralization would be to use antigenically different strains of the virus for each dosing. In summary, more preclinical studies are needed to determine the best dosing strategy, improve virus delivery and enhance immunogenicity.
Author Contributions
Conceptualization, S.C. and S.G.W.; writing—original draft preparation, S.C.; review and editing, S.G.W. and Y.F. All authors have read and agreed to the published version of the manuscript.
Funding
This review article received no external funding.
Acknowledgments
Shyambabu Chaurasiya and Susanne G. Warner are supported through the generosity of Natalie & David Roberts. These authors wish to thank them for their philanthropy.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashiro, G.; Loh, P.C.; Yau, J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977, 54, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Sinkovics, J.G.; Horvath, J.C. Newcastle disease virus (NDV): Brief history of its oncolytic strains. J. Clin. Virol. 2000, 16, 1–15. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Alemany, R.; Medrano, D.; Alonso, M.; Bekele, B.N.; Lin, E.; Conrad, C.C.; Yung, W.K.; Fueyo, J. Comparative effect of oncolytic adenoviruses with E1A-55 kDa or E1B-55 kDa deletions in malignant gliomas. Neoplasia 2005, 7, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.J.; Wong, J.; Fong, Y. Herpes simplex virus NV1020 as a novel and promising therapy for hepatic malignancy. Expert Opin. Investig. Drugs 2008, 17, 1105–1113. [Google Scholar] [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar]
- Lun, X.; Yang, W.; Alain, T.; Shi, Z.Q.; Muzik, H.; Barrett, J.W.; McFadden, G.; Bell, J.; Hamilton, M.G.; Senger, D.L.; et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005, 65, 9982–9990. [Google Scholar] [CrossRef] [Green Version]
- Grote, D.; Russell, S.J.; Cornu, T.I.; Cattaneo, R.; Vile, R.; Poland, G.A.; Fielding, A.K. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood 2001, 97, 3746–3754. [Google Scholar] [CrossRef] [Green Version]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos de Matos, A.; Franco, L.S.; McFadden, G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Mol. Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Filley, A.C.; Dey, M. Immune System, Friend or Foe of Oncolytic Virotherapy? Front. Oncol 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Chen, N.G.; Fong, Y. Oncolytic viruses and immunity. Curr. Opin. Immunol. 2018, 51, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [Green Version]
- Kleijn, A.; Kloezeman, J.; Treffers-Westerlaken, E.; Fulci, G.; Leenstra, S.; Dirven, C.; Debets, R.; Lamfers, M. The therapeutic efficacy of the oncolytic virus Delta24-RGD in a murine glioma model depends primarily on antitumor immunity. Oncoimmunology 2014, 3, e955697. [Google Scholar] [CrossRef]
- Miller, C.G.; Fraser, N.W. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol. Ther. 2003, 7, 741–747. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol 2014, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasiya, S.; Chen, N.G.; Lu, J.; Martin, N.; Shen, Y.; Kim, S.I.; Warner, S.G.; Woo, Y.; Fong, Y. A chimeric poxvirus with J2R (thymidine kinase) deletion shows safety and anti-tumor activity in lung cancer models. Cancer GeneTher. 2020, 27, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway, C.A., Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-man study of western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity. Mol. Ther. 2015, 23, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Bommareddy, P.K. Two roads for oncolytic immunotherapy development. J. Immunother. Cancer 2019, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Maruri-Avidal, L.; Sisler, J.; Stuart, C.A.; Moss, B. Cascade regulation of vaccinia virus gene expression is modulated by multistage promoters. Virology 2013, 447, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Wittek, R. Organization and expression of the poxvirus genome. Experientia 1982, 38, 285–297. [Google Scholar] [CrossRef]
- Hammond, J.M.; Oke, P.G.; Coupar, B.E. A synthetic vaccinia virus promoter with enhanced early and late activity. J. Virol. Methods 1997, 66, 135–138. [Google Scholar] [CrossRef]
- Small, E.J.; Carducci, M.A.; Burke, J.M.; Rodriguez, R.; Fong, L.; van Ummersen, L.; Yu, D.C.; Aimi, J.; Ando, D.; Working, P.; et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol. Ther. 2006, 14, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.; Ball, M.; Tanay, M.; Nutting, C.; Newbold, K.; Gore, M.E.; Larkin, J.; Syrigos, K.N.; et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012, 18, 2080–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125 (Suppl. 2), S3–S23. [Google Scholar] [CrossRef]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [Green Version]
- Pure, E.; Lo, A. Can Targeting Stroma Pave the Way to Enhanced Antitumor Immunity and Immunotherapy of Solid Tumors? Cancer Immunol. Res. 2016, 4, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Achard, C.; Surendran, A.; Wedge, M.E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers (Basel) 2011, 3, 3856–3893. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castanon, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: A side effect of immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Hew, P.; Crosley, P.; Sharon, D.; Potts, K.; Agopsowicz, K.; Long, M.; Shi, C.; Hitt, M.M. Breast cancer gene therapy using an adenovirus encoding human IL-2 under control of mammaglobin promoter/enhancer sequences. Cancer Gene Ther. 2016, 23, 178–187. [Google Scholar] [CrossRef]
- Shiomi, A.; Usui, T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediat. Inflamm. 2015, 2015, 568543. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Cook, A.D.; Hamilton, J.A. Functions of granulocyte-macrophage colony-stimulating factor. Crit. Rev. Immunol. 2005, 25, 405–428. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Kohlhapp, F.J.; Kaufman, H.L. Molecular Pathways: Mechanism of Action for Talimogene Laherparepvec, a New Oncolytic Virus Immunotherapy. Clin. Cancer Res. 2016, 22, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Breitbach, C.J.; Moon, A.; Burke, J.; Hwang, T.H.; Kirn, D.H. A Phase 2, Open-Label, Randomized Study of Pexa-Vec (JX-594) Administered by Intratumoral Injection in Patients with Unresectable Primary Hepatocellular Carcinoma. Methods Mol. Biol. 2015, 1317, 343–357. [Google Scholar]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Parviainen, S.; Juhila, J.; Turkki, R.; Linder, N.; Lundin, J.; Kankainen, M.; Ristimaki, A.; Koski, A.; Liikanen, I.; et al. Immunological data from cancer patients treated with Ad5/3-E2F-Delta24-GMCSF suggests utility for tumor immunotherapy. Oncotarget 2015, 6, 4467–4481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Sarkioja, M.; et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas-Ramirez, N.; Woytschak, J.; Boyman, O. Interleukin-2: Biology, Design and Application. Trends Immunol. 2015, 36, 763–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelante, T.; Fric, J.; Wong, A.Y.; Ricciardi-Castagnoli, P. Interleukin-2 production by dendritic cells and its immuno-regulatory functions. Front. Immunol. 2012, 3, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobol, R.E.; Shawler, D.L.; Carson, C.; Van Beveren, C.; Mercola, D.; Fakhrai, H.; Garrett, M.A.; Barone, R.; Goldfarb, P.; Bartholomew, R.M.; et al. Interleukin 2 gene therapy of colorectal carcinoma with autologous irradiated tumor cells and genetically engineered fibroblasts: A Phase I study. Clin. Cancer Res. 1999, 5, 2359–2365. [Google Scholar] [PubMed]
- Liu, Z.; Ge, Y.; Wang, H.; Ma, C.; Feist, M.; Ju, S.; Guo, Z.S.; Bartlett, D.L. Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat. Commun. 2018, 9, 4682. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.M.; Subleski, J.J.; Wigginton, J.M.; Wiltrout, R.H. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin. Biol. Ther. 2007, 7, 1705–1721. [Google Scholar] [CrossRef]
- Voest, E.E.; Kenyon, B.M.; O’Reilly, M.S.; Truitt, G.; D’Amato, R.J.; Folkman, J. Inhibition of angiogenesis in vivo by interleukin 12. J. Natl. Cancer Inst. 1995, 87, 581–586. [Google Scholar] [CrossRef]
- Hellums, E.K.; Markert, J.M.; Parker, J.N.; He, B.; Perbal, B.; Roizman, B.; Whitley, R.J.; Langford, C.P.; Bharara, S.; Gillespie, G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-oncology 2005, 7, 213–224. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, H.; Ren, J.; Liu, W.; Chen, L.; Chen, H.; Ye, J.; Dai, E.; Ma, C.; Ju, S.; et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J. Immunother. Cancer 2020, 8, e000710. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.H.; Choi, K.J.; Choi, I.K.; Kim, H.; Cho, S.; Cho, B.C.; Yun, C.O. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006, 12, 5859–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.H.; Zhang, S.N.; Choi, K.J.; Choi, I.K.; Kim, J.H.; Lee, M.G.; Kim, H.; Yun, C.O. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol. Ther. 2010, 18, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.K.; Choi, B.; Wanna, G.; Genden, E.M.; Woo, S.L.; Shin, E.J. Combined VSV oncolytic virus and chemotherapy for squamous cell carcinoma. Laryngoscope 2008, 118, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Shin, E.J.; Wanna, G.B.; Choi, B.; Aguila, D., 3rd; Ebert, O.; Genden, E.M.; Woo, S.L. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope 2007, 117, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Quetglas, J.I.; Labiano, S.; Aznar, M.A.; Bolanos, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sanchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehniger, T.A.; Cooper, M.A.; Caligiuri, M.A. Interleukin-2 and interleukin-15: Immunotherapy for cancer. Cytokine Growth Factor Rev. 2002, 13, 169–183. [Google Scholar] [CrossRef]
- Waldmann, T.A. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat. Rev. Immunol. 2006, 6, 595–601. [Google Scholar] [CrossRef]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef]
- Niu, Z.; Bai, F.; Sun, T.; Tian, H.; Yu, D.; Yin, J.; Li, S.; Li, T.; Cao, H.; Yu, Q.; et al. Recombinant Newcastle Disease virus Expressing IL15 Demonstrates Promising Antitumor Efficiency in Melanoma Model. Technol Cancer Res. Treat. 2015, 14, 607–615. [Google Scholar] [CrossRef]
- Yan, Y.; Li, S.; Jia, T.; Du, X.; Xu, Y.; Zhao, Y.; Li, L.; Liang, K.; Liang, W.; Sun, H.; et al. Combined therapy with CTL cells and oncolytic adenovirus expressing IL-15-induced enhanced antitumor activity. Tumour Biol. 2015, 36, 4535–4543. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.B.; Barra, N.G.; Davies, E.; Ashkar, A.A.; Lichty, B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012, 19, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, A.K.; Chen, G.; Zheng, D.; Tang, H.; Cheng, G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. [Google Scholar] [CrossRef] [Green Version]
- Pantel, A.; Teixeira, A.; Haddad, E.; Wood, E.G.; Steinman, R.M.; Longhi, M.P. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014, 12, e1001759. [Google Scholar] [CrossRef]
- von Hoegen, P. Synergistic role of type I interferons in the induction of protective cytotoxic T lymphocytes. Immunol. Lett. 1995, 47, 157–162. [Google Scholar] [CrossRef]
- Salzwedel, A.O.; Han, J.; LaRocca, C.J.; Shanley, R.; Yamamoto, M.; Davydova, J. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget 2018, 9, 18041–18052. [Google Scholar] [CrossRef] [Green Version]
- Kirn, D.H.; Wang, Y.; Le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007, 4, e353. [Google Scholar] [CrossRef]
- Patel, M.R.; Jacobson, B.A.; Ji, Y.; Drees, J.; Tang, S.; Xiong, K.; Wang, H.; Prigge, J.E.; Dash, A.S.; Kratzke, A.K.; et al. Vesicular stomatitis virus expressing interferon-beta is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget 2015, 6, 33165–33177. [Google Scholar] [CrossRef] [Green Version]
- Buijs, P.; van Nieuwkoop, S.; Vaes, V.; Fouchier, R.; van Eijck, C.; van den Hoogen, B. Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma. Viruses 2015, 7, 2980–2998. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Peng, K.W.; Dingli, D.; Kratzke, R.A.; Russell, S.J. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010, 17, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-gamma in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, C.; Uekusa, Y.; Iwasaki, M.; Yamaguchi, N.; Mukai, T.; Gao, P.; Tomura, M.; Ono, S.; Tsujimura, T.; Fujiwara, H.; et al. A role of interferon-gamma (IFN-gamma) in tumor immunity: T cells with the capacity to reject tumor cells are generated but fail to migrate to tumor sites in IFN-gamma-deficient mice. Cancer Res. 2001, 61, 3399–3405. [Google Scholar] [PubMed]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Yang, Y.; Luo, X.; Cheng, Y.; Zhang, M.; Wang, K.; Ge, C. Inhibition of tumor angiogenesis by interferon-gamma by suppression of tumor-associated macrophage differentiation. Oncol. Res. 2014, 21, 227–235. [Google Scholar] [CrossRef]
- Ni, L.; Lu, J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Falls, T.; Twumasi-Boateng, K.; St-Germain, L.E.; Marguerie, M.; Garcia, V.; Selman, M.; Jennings, V.A.; Pettigrew, J.; et al. Oncolytic vesicular stomatitis virus expressing interferon-gamma has enhanced therapeutic activity. Mol. Ther. Oncolytics 2016, 3, 16001. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Li, J.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology 2016, 5, e1091554. [Google Scholar] [CrossRef]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.S.; Kalinski, P.; Thorne, S.H.; Bartlett, D.L. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011, 19, 650–657. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of tumor-derived cytokines and growth factors on differentiation and immune suppressive features of myeloid cells in cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 295–342. [Google Scholar] [PubMed]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Xu, W.; Peng, D.; Wang, H.; Zhang, X.; Wang, H.; Xiao, F.; Zhu, Y.; Ji, Y.; Gulukota, K.; et al. An Oncolytic Adenovirus Targeting Transforming Growth Factor beta Inhibits Protumorigenic Signals and Produces Immune Activation: A Novel Approach to Enhance Anti-PD-1 and Anti-CTLA-4 Therapy. Hum. Gene Ther. 2019, 30, 1117–1132. [Google Scholar] [CrossRef]
- Patil, S.S.; Gentschev, I.; Adelfinger, M.; Donat, U.; Hess, M.; Weibel, S.; Nolte, I.; Frentzen, A.; Szalay, A.A. Virotherapy of canine tumors with oncolytic vaccinia virus GLV-1h109 expressing an anti-VEGF single-chain antibody. PLoS ONE 2012, 7, e47472. [Google Scholar] [CrossRef]
- Hou, W.; Sampath, P.; Rojas, J.J.; Thorne, S.H. Oncolytic Virus-Mediated Targeting of PGE2 in the Tumor Alters the Immune Status and Sensitizes Established and Resistant Tumors to Immunotherapy. Cancer Cell 2016, 30, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Sledzinska, A.; Menger, L.; Bergerhoff, K.; Peggs, K.S.; Quezada, S.A. Negative immune checkpoints on T lymphocytes and their relevance to cancer immunotherapy. Mol. Oncol. 2015, 9, 1936–1965. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasiya, S.; Yang, A.; Kang, S.; Lu, J.; Kim, S.I.; Park, A.K.; Sivanandam, V.; Zhang, Z.; Woo, Y.; Warner, S.G.; et al. Oncolytic poxvirus CF33-hNIS-DeltaF14.5 favorably modulates tumor immune microenvironment and works synergistically with anti-PD-L1 antibody in a triple-negative breast cancer model. Oncoimmunology 2020, 9, 1729300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, A. Treating with Checkpoint Inhibitors-Figure $1 Million per Patient. Am. Health Drug Benefits 2015, 8, 9. [Google Scholar]
- Orloff, M. Spotlight on talimogene laherparepvec for the treatment of melanoma lesions in the skin and lymph nodes. Oncolytic Virother. 2016, 5, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinpeter, P.; Fend, L.; Thioudellet, C.; Geist, M.; Sfrontato, N.; Koerper, V.; Fahrner, C.; Schmitt, D.; Gantzer, M.; Remy-Ziller, C.; et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death -1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology 2016, 5, e1220467. [Google Scholar] [CrossRef]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Tumor-Localized Secretion of Soluble PD1 Enhances Oncolytic Virotherapy. Cancer Res. 2017, 77, 2952–2963. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Loskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef] [PubMed]
- McGray, A.J.R.; Huang, R.Y.; Battaglia, S.; Eppolito, C.; Miliotto, A.; Stephenson, K.B.; Lugade, A.A.; Webster, G.; Lichty, B.D.; Seshadri, M.; et al. Oncolytic Maraba virus armed with tumor antigen boosts vaccine priming and reveals diverse therapeutic response patterns when combined with checkpoint blockade in ovarian cancer. J. Immunother. Cancer 2019, 7, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef] [Green Version]
- Suryadevara, C.M.; Gedeon, P.C.; Sanchez-Perez, L.; Verla, T.; Alvarez-Breckenridge, C.; Choi, B.D.; Fecci, P.E.; Sampson, J.H. Are BiTEs the “missing link” in cancer therapy? Oncoimmunology 2015, 4, e1008339. [Google Scholar] [CrossRef] [Green Version]
- Jen, E.Y.; Xu, Q.; Schetter, A.; Przepiorka, D.; Shen, Y.L.; Roscoe, D.; Sridhara, R.; Deisseroth, A.; Philip, R.; Farrell, A.T.; et al. FDA Approval: Blinatumomab for Patients with B-cell Precursor Acute Lymphoblastic Leukemia in Morphologic Remission with Minimal Residual Disease. Clin. Cancer Res. 2019, 25, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, C.A.; Guedan, S.; Rojas, L.A.; Moreno, R.; Arias-Badia, M.; de Sostoa, J.; June, C.H.; Alemany, R. Oncolytic Adenoviral Delivery of an EGFR-Targeting T-cell Engager Improves Antitumor Efficacy. Cancer Res. 2017, 77, 2052–2063. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Gottschalk, S. CAR T cells for solid tumors: Armed and ready to go? Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aalipour, A.; Le Boeuf, F.; Tang, M.; Murty, S.; Simonetta, F.; Lozano, A.X.; Shaffer, T.M.; Bell, J.C.; Gambhir, S.S. Viral Delivery of CAR Targets to Solid Tumors Enables Effective Cell Therapy. Mol. Ther. Oncolytics 2020, 17, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Yaacov, B.; Lazar, I.; Tayeb, S.; Frank, S.; Izhar, U.; Lotem, M.; Perlman, R.; Ben-Yehuda, D.; Zakay-Rones, Z.; Panet, A. Extracellular matrix constituents interfere with Newcastle disease virus spread in solid tissue and diminish its potential oncolytic activity. J. Gen. Virol. 2012, 93 Pt 8, 1664–1672. [Google Scholar] [CrossRef] [Green Version]
- Wojton, J.; Kaur, B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010, 21, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Rojas, J.J.; Gros, A.; Mercade, E.; Cascallo, M.; Alemany, R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. 2010, 18, 1275–1283. [Google Scholar] [CrossRef]
- Xu, W.; Neill, T.; Yang, Y.; Hu, Z.; Cleveland, E.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.R.; Shevrin, D.; et al. The systemic delivery of an oncolytic adenovirus expressing decorin inhibits bone metastasis in a mouse model of human prostate cancer. Gene Ther. 2015, 22, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Park, H.R.; Rhee, J.; Park, Y.M.; Kim, S.H. Therapeutic effect of oncolytic adenovirus expressing relaxin in radioresistant oral squamous cell carcinoma. Oncol. Res. 2013, 20, 419–425. [Google Scholar] [CrossRef]
- Haddad, D.; Fong, Y. Molecular imaging of oncolytic viral therapy. Mol. Ther. Oncolytics 2015, 1, 14007. [Google Scholar] [CrossRef]
- Shah, K.; Jacobs, A.; Breakefield, X.O.; Weissleder, R. Molecular imaging of gene therapy for cancer. Gene Ther. 2004, 11, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.M.; McGregor, A. A bright future for bioluminescent imaging in viral research. Future Virol. 2015, 10, 169–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Alvarez, K.A.; Altomonte, J.; Laitinen, I.; Ziegler, S.; Steiger, K.; Esposito, I.; Schmid, R.M.; Ebert, O. PET imaging of oncolytic VSV expressing the mutant HSV-1 thymidine kinase transgene in a preclinical HCC rat model. Mol. Ther. 2015, 23, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCart, J.A.; Mehta, N.; Scollard, D.; Reilly, R.M.; Carrasquillo, J.A.; Tang, N.; Deng, H.; Miller, M.; Xu, H.; Libutti, S.K.; et al. Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: Molecular imaging after systemic delivery using 111In-pentetreotide. Mol. Ther. 2004, 10, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.E.; Parry, J.J.; Andrews, R.; Cordopatis, P.; Nock, B.A.; Maina, T. MicroPET imaging of gene transfer with a somatostatin receptor-based reporter gene and (94m)Tc-Demotate 1. J. Nucl. Med. 2005, 46, 1889–1897. [Google Scholar]
- Warner, S.G.; Kim, S.I.; Chaurasiya, S.; O’Leary, M.P.; Lu, J.; Sivanandam, V.; Woo, Y.; Chen, N.G.; Fong, Y. A Novel Chimeric Poxvirus Encoding hNIS Is Tumor-Tropic, Imageable, and Synergistic with Radioiodine to Sustain Colon Cancer Regression. Mol. Ther. Oncolytics 2019, 13, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Jun, K.H.; Gholami, S.; Song, T.J.; Au, J.; Haddad, D.; Carson, J.; Chen, C.H.; Mojica, K.; Zanzonico, P.; Chen, N.G.; et al. A novel oncolytic viral therapy and imaging technique for gastric cancer using a genetically engineered vaccinia virus carrying the human sodium iodide symporter. J. Exp. Clin. Cancer Res. 2014, 33, 2. [Google Scholar] [CrossRef] [Green Version]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Tumor destruction by cytokine/chemokine-armed oncolytic viruses.

Figure 2.
Oncolytic viruses delivering target for CAR-T cells. Unique antigens can be delivered to tumor cells using oncolytic virus, and CAR-T cells specific for that unique antigen can be used in combination to destroy tumors.
Figure 2.
Oncolytic viruses delivering target for CAR-T cells. Unique antigens can be delivered to tumor cells using oncolytic virus, and CAR-T cells specific for that unique antigen can be used in combination to destroy tumors.

Figure 3.
Oncolytic viruses armed with stroma-modifying genes can spread better within the tumor, and subsequently destroy it.
Figure 3.
Oncolytic viruses armed with stroma-modifying genes can spread better within the tumor, and subsequently destroy it.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of oncolytic virus vectors.
| Adenovirus | Herpes Simplex Virus | Vaccinia Virus | Reovirus | |
|---|---|---|---|---|
| Genome | dsDNA | dsDNA | dsDNA | Segmented dsRNA |
| Capsid | Icosahedral | Icosahedral | Complex | icosahedral |
| Virion diameter | 70–90 nm | 150–200 nm | 170–200 nm | 60–100 nm |
| Genome size | 36–40 kb | 150–200 kb | 130–280 kb | 0.2–3 kb |
| Replication site | Nucleus | Nucleus | Cytoplasm | Cytoplasm |
| Life cycle | 24 h | 12 h | 8 h | 18 h |
| Immunogenicity | High | Moderate | High | Low |
| Transgene expression | Transient | Potentially long-term | Transient | Transgene expression is not common |
| Ease of manipulation | Easy | Difficult | Easy | Very difficult |
| Cloning capacity | 8 kb | >30 kb | 25 kb | n/a |
| Easily achievable titers | 1012 PFU/mL | 1010 PFU/mL | 109 PFU/mL | 109 PFU/mL |
| Maximum Dose used in human | 6 × 1012 VP [31] | 4 × 108 PFU [32] | 3 × 109 PFU [26] | 3 × 1010 TCID50 [33] |
| Virulence of WT virus | Yes | Mild | Mild | No |
dsDNA, Double stranded DNA; dsRNA, Double stranded RNA; kb, kilobase; PFU, plaque forming unit; VP, virus particle; TCID50, median tissue culture infectious dose.
Table 2.
Armed OVs currently in clinical trials (source: clinicaltrials.gov; accessed May 2020).
Table 2.
Armed OVs currently in clinical trials (source: clinicaltrials.gov; accessed May 2020).
| Virus | Transgene | Function of Transgene | Combination | Tumor type | Phase | References |
|---|---|---|---|---|---|---|
| TG6002 (Vaccinia virus) | FCU1 | Conversion of 5-FC to 5-FU | 5-FC | Glioblastoma | Phase 1 and 2 | NCT03294486 |
| Pexa-Vec (Vaccinia virus) | GM-CSF | Activation of APC | αPD-L1 αCTLA-4 | Colorectal cancer | Phase 1 and 2 | NCT03206073 |
| RP1 (HSV-1) | GM-CSF GALV-GP | Activation of APC; fusion of cells | None | Cutaneous squamous cell carcinoma | Phase 1b | NCT04349436 |
| OH2 (HSV-2) | GM-CSF | Activation of APC | αPD-1 | Gastrointestinal and other solid tumors | Phase 1 | NCT03866525 |
| T-Vec (HSV-1) | GM-CSF | Activation of APC | αPD-L1 αCTLA-4 | Breast cancer | Phase 1 | NCT04185311 |
| TILT123 (Adenovirus) | TNFα and IL-2 | Activation of T cells | Tumor infiltrating lymphocytes | Melanoma | Phase 1 | NCT04217473 |
| TBio-6517 (Vaccinia virus) | FLT3 ligand, IL-12 and αCTLA-4 | Immune activation | αPD-1 | Solid tumors, TNBC, microsatellite stable colorectal cancer | Phase 1 and 2 | NCT04301011 |
| MG1-MAGEA3 (Maraba virus) | MAGEA3 | Tumor antigen for melanoma | αPD-1 Cyclophosphamide Ad-MAGEA3 | Metastatic melanoma, Squamous cell skin carcinoma | Phase 1b | NCT03773744 |
| GL-ONC1 (Vaccinia virus) | Luc-GFP fusion, β-galactosidase, β-glucuronidase | Imaging | Chemotherapy or bevacizumab | Ovarian cancer, peritoneal carcinomatosis and fallopian tube cancer | Phase 1 and 2 | NCT02759588 |
| M032 (HSV-1) | IL-12 | Immune-stimulation and anti-angiogenesis | None | Glioblastoma Astrocytoma Gliosarcoma | Phase 1 | NCT02062827 |
| C134 (HSV-1) | IRS1 | PKR evasion | None | Glioblastoma Astrocytoma Gliosarcoma | Phase 1 | NCT03657576 |
| TMV-018 (Measles virus) | Cytosine deaminase | Conversion of 5-FC to 5-FU | 5-FC | Gastrointestinal cancer | Phase 1 and 2 | NCT04195373 |
| LOAd703 (Adenovirus) | CD40L, 41BBL | Immune stimulation | None | PDAC, ovarian cancer, biliary carcinoma and colorectal cancer | Phase 1 and 2 | NCT03225989 |
| MV-NIS (Measles virus) | NIS | Imaging | Mesenchymal stem cells transplantation | Solid tumors | Phase 1 and 2 | NCT02068794 |
| ONCR-177 (HSV-1) | IL-12, CCL4, FLT3L, αPD-L1, αCTLA-4 | Immune stimulation | αPD-1 | Solid tumors | Phase 1 | NCT04348916 |
| NG-350A (Adenovirus) | Anti-CD40 | Immune stimulation | None | Epithelial tumors | Phase 1 | NCT03852511 |
| NG-641 (Adenovirus) | BiTE (FAP-TAc), CXCL9, CXCL10, IFNα | Killing cancer associated fibroblast; Immune stimulation | None | Epithelial tumors | Phase 1 | NCT04053283 |
FCU1, cytosine deaminase and uracil phosphoribosyltransferase; 5-FC, 5-fluorocytosine; 5-FU, 5-fluorouracil; GM-CSF, granulocyte macrophage colony stimulating factor; GALV-GP, gibbon-ape leukemia virus glycoprotein; APC, antigen presenting cell, HSV, herpes simplex virus; TNFα, tumor necrosis factor α; IL-2, interleukin-2; IL-12, interleukin-12; FLT3, fms-like tyrosine kinase; PD-1, programmed death-1; PD-L1, programmed death ligand-1; CTLA-4, cytotoxic T lymphocyte antigen-4; MAGEA3, melanoma antigen gene A3; Luc-GFP, fusion of luciferase and GFP genes; NIS, sodium-iodide symporter; BiTE, bispecific T cell engager; FAP-Tac, fibroblast activation protein-T cell activator; IFNα, interferon α; PKR, protein kinase R.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chaurasiya, S.; Fong, Y.; Warner, S.G. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers 2020, 12, 1699. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061699
AMA Style
Chaurasiya S, Fong Y, Warner SG. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers. 2020; 12(6):1699. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061699
Chicago/Turabian StyleChaurasiya, Shyambabu, Yuman Fong, and Susanne G. Warner. 2020. "Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges" Cancers 12, no. 6: 1699. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061699
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.