CRISPR-Mediated Non-Viral Site-Specific Gene Integration and Expression in T Cells: Protocol and Application for T-Cell Therapy


Abstract
:1. Introduction
2. Results
2.1. Gene Knock-In Using Primary T Cells: Overview
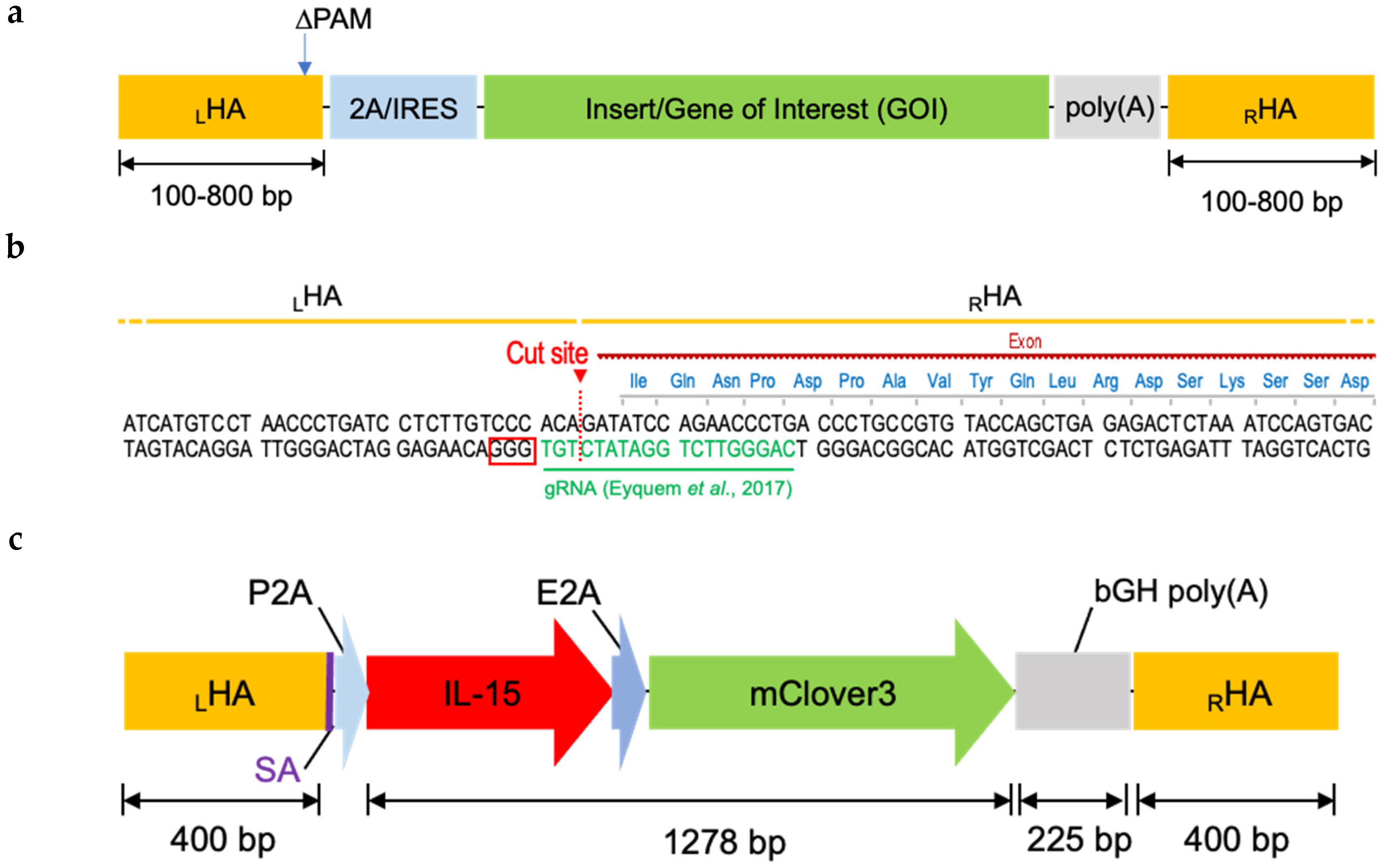
2.2. Designing Donor DNA
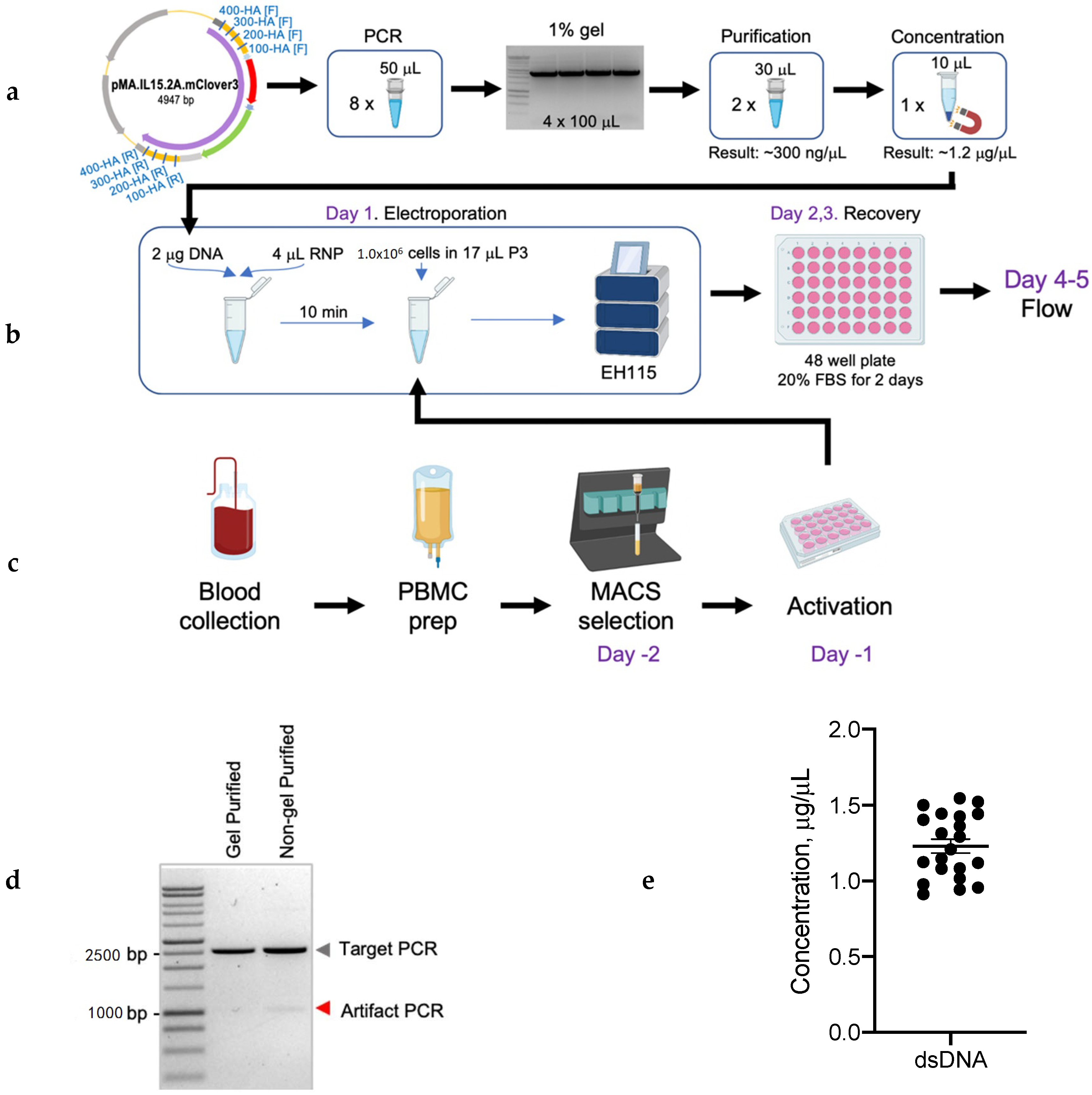
2.3. Donor DNA Amplification, Purification, and Concentration
2.4. Optimizing Transgene Knock-In in Primary Human T Cells
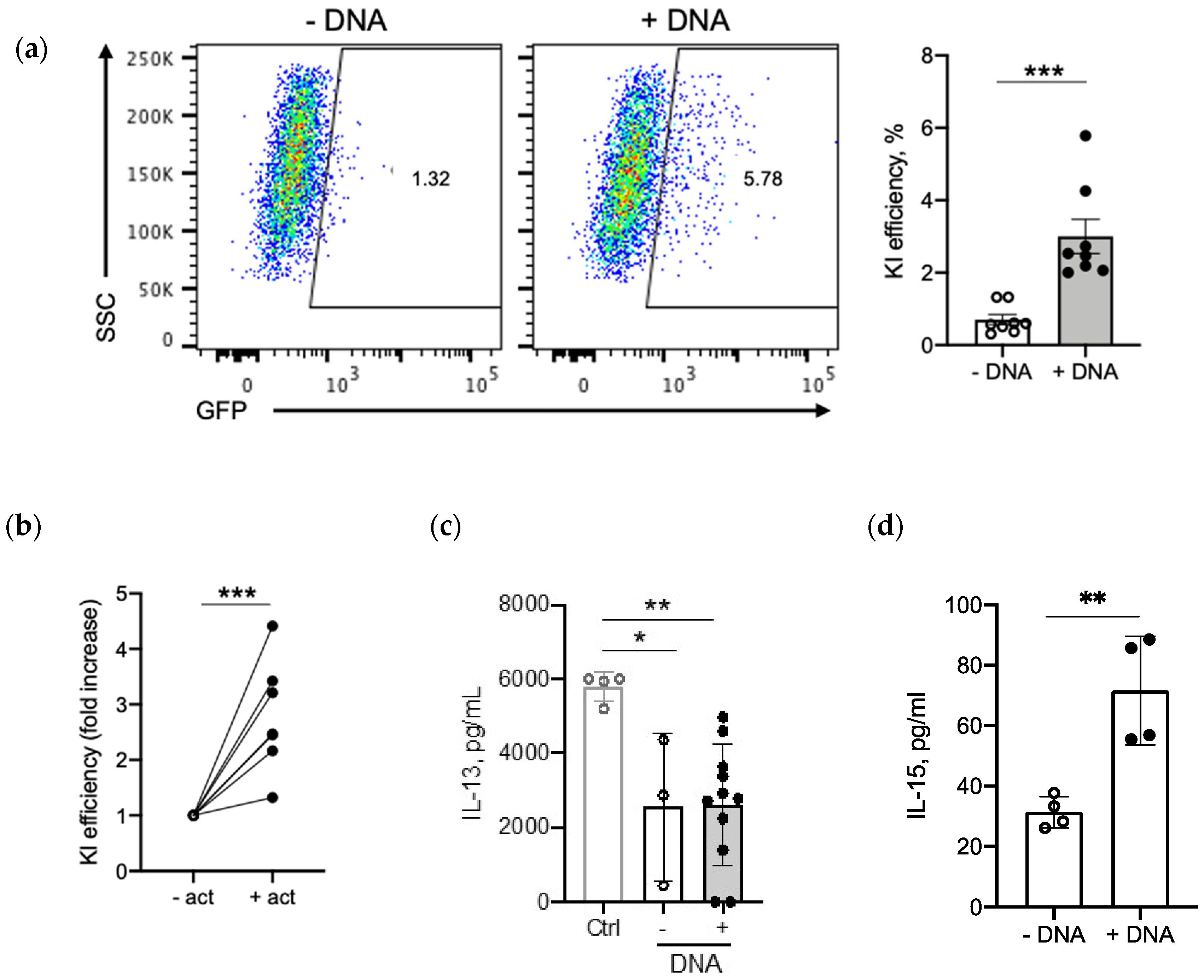
2.4.1. DNA Amount
2.4.2. T-Cell Number
2.4.3. Homology Arms and Recovery Time
2.5. IL-15 Transgene Is Functional
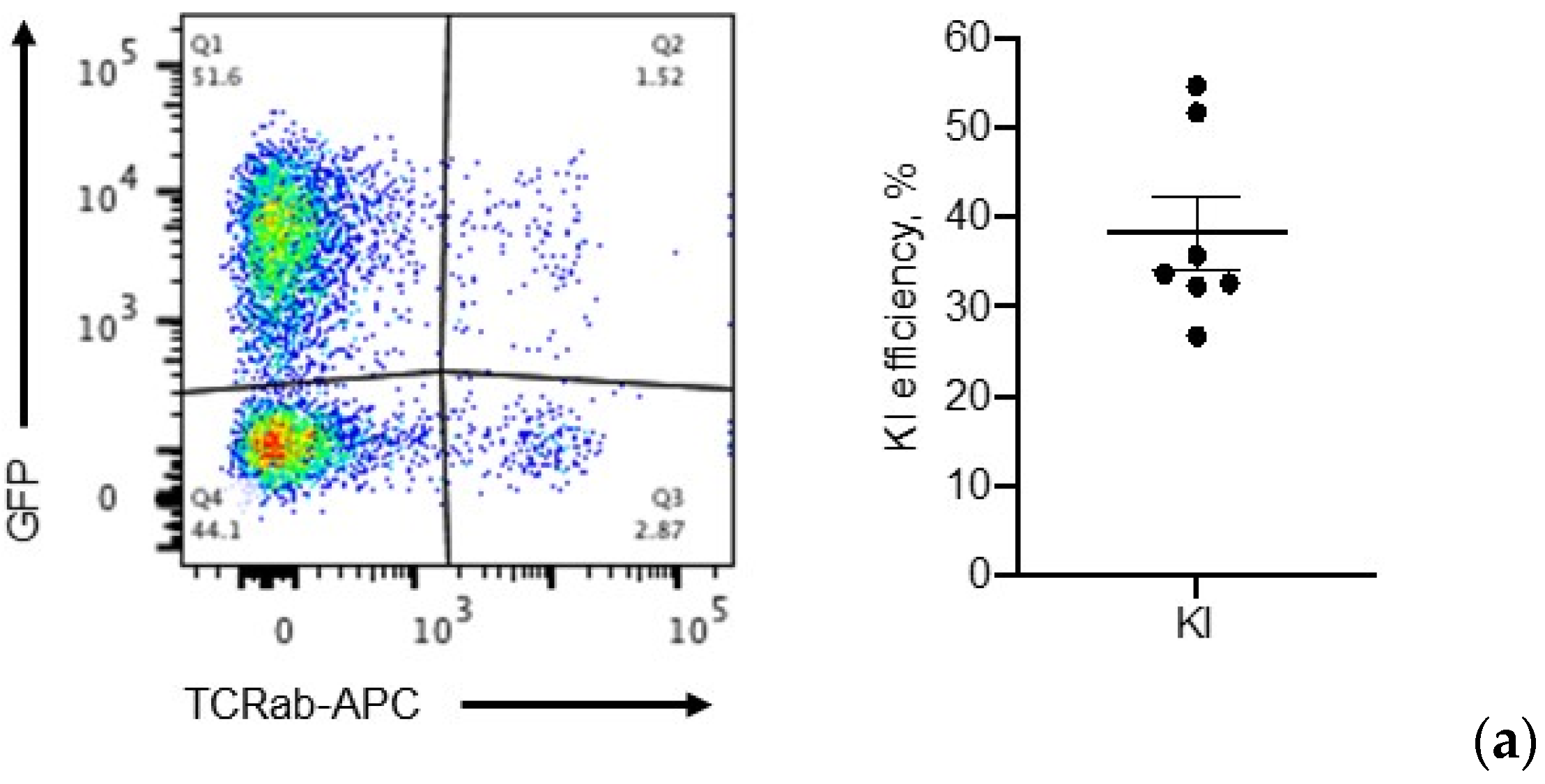
2.6. Application: CAR, BiTE Integration into TRAC Locus and IL-15.E2A.mClover3 Knock-In into IL-13 Locus
3. Discussion
4. Materials and Methods
4.1. Generation of DsDNA Donor Template
4.1.1. Construct Design
4.1.2. PCR Amplification
4.1.3. DNA Purification and Concentration
4.2. Generation of Knock-In T Cells
4.2.1. Primary Human T-Cell Culture
4.2.2. Primary Human T-Cell Electroporation
4.3. Flow Cytometry
4.4. Targeted Deep Sequencing
4.5. Analysis of IL-15 and IL-13 Production
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Riviere, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Nam, C.H.; Rabbitts, T.H. The role of LMO2 in development and in T cell leukemia after chromosomal translocation or retroviral insertion. Mol. Ther. 2006, 13, 15–25. [Google Scholar] [CrossRef]
- van der Loo, J.C.; Wright, J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2016, 25, R42–R52. [Google Scholar] [CrossRef] [Green Version]
- Merten, O.W.; Charrier, S.; Laroudie, N.; Fauchille, S.; Dugué, C.; Jenny, C.; Audit, M.; Zanta-Boussif, M.A.; Chautard, H.; Radrizzani, M.; et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum. Gene Ther. 2011, 22, 343–356. [Google Scholar] [CrossRef]
- Wright, J.F. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008, 15, 840–848. [Google Scholar] [CrossRef]
- David, R.M.; Doherty, A.T. Viral Vectors: The Road to Reducing Genotoxicity. Toxicol. Sci. 2017, 155, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagarajan, S.; Schmitt, D.; Acker, C.; Rutjens, E. Autologous cryopreserved leukapheresis cellular material for chimeric antigen receptor-T cell manufacture. Cytotherapy 2019, 21, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Webber, B.R.; Lonetree, C.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.A.; et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019, 10, 5222. [Google Scholar] [CrossRef] [Green Version]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Gao, D.; Wang, P.; Chen, J.; Ruan, J.; Xu, J.; Xia, X. Efficient homology-directed gene editing by CRISPR/Cas9 in human stem and primary cells using tube electroporation. Sci. Rep. 2018, 8, 11649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, M.; Lee, B.; Honaker, Y.; Leung, W.-H.; Grier, A.E.; Jacobs, H.M.; Sommer, K.; Sahni, J.; Jackson, S.W.; Scharenberg, A.M.; et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajar, B.T.; Wang, E.S.; Lam, A.J.; Kim, B.B.; Jacobs, C.L.; Howe, E.S.; Davidson, M.W.; Lin, M.Z.; Chu, J. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci. Rep. 2016, 6, 20889. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Roth, T.L.; Li, P.J.; Chen, P.A.; Apathy, R.; Mamedov, M.R.; Vo, L.T.; Tobin, V.R.; Goodman, D.; Shifrut, E.; et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020, 38, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Kan, Y.; Ruis, B.; Takasugi, T.; Hendrickson, E.A. Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 2017, 27, 1099–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2018, 9, 691. [Google Scholar] [CrossRef]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef]
- Odak, A.; Yuan, H.; Mansilla-Soto, J.; Eyquem, J.; Vedantam, P.; Leslie, C.; Sadelain, M. ASGCT Abstract Book. Abstract 941: Novel Genomic Safe Harbors for Effective CAR T Cell Engineering. Mol. Ther. 2019, 27, 1. [Google Scholar]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems for Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenciute, G.; Krebs, S.; Torres, D.; Wu, M.; Liu, H.; Dotti, G.; Li, X.-N.; Lesniak, M.S.; Balyasnikova, I.V.; Gottschalk, S. Characterization and Functional Analysis of scFv-based Chimeric Antigen Receptors to Redirect T Cells to IL13Ralpha2-positive Glioma. Mol. Ther. 2016, 24, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, A.; BethHilton, M.; Seaman, S.; Haines, D.C.; Stevenson, S.; Lemotte, P.K.; Tschantz, W.R.; Zhang, X.M.; Saha, S.; Fleming, T.; et al. TEM8/ANTXR1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell 2012, 21, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.; Lundqvist, A.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Sanz, L.M.; Liang, S.; Murray, S.; et al. 31st Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer (SITC 2016): P51 T cells redirected to TEM8 have antitumor activity but induce ‘on target/off cancer toxicity’ in preclinical models. In Journal for Immunotherapy of Cancer; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Iwahori, K.; Kakarla, S.; Velasquez, M.P.; Yu, F.; Yi, Z.; Gerken, C.; Song, X.-T.; Gottschalk, S. Engager T cells: A new class of antigen-specific T cells that redirect bystander T cells. Mol. Ther. 2015, 23, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Bohmer, R.M.; Bandala-Sanchez, E.; Harrison, L.C. Forward light scatter is a simple measure of T-cell activation and proliferation but is not universally suited for doublet discrimination. Cytometry A 2011, 79, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connelly, J.P.; Pruett-Miller, S.M. CRIS.py: A Versatile and High-throughput Analysis Program for CRISPR-based Genome Editing. Sci. Rep. 2019, 9, 4194. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sgRNA | 3 µL [60 µM] |
|---|---|
| Cas9 | 1 µL [40 µM] |
| sgRNA:Cas9 (molar ratio) | 4.5:1 |
| RNP incubation | 10 min, RT |
| RNP volume | 4 µL |
| Cells | 1 × 106/17 µL |
| Electroporation solution | P3 + S1 supplement (Lonza) |
| Template | 2 µg dsDNA in 3 µL |
| Homology arm length | 200–400 bp |
| Vol for electroporation | 23 µL |
| Format/program Neon | Strip/EH-115 |
| Incubation after electroporation | 30 min at 37 °C (in 80 µL of RPMI+20%FBS+IL7/15) |
| Transfer | 48-well plate (650 µL of RPMI+20%FBS+IL7/15) |
| Reactions per well after electroporation | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Odé, Z.; Condori, J.; Peterson, N.; Zhou, S.; Krenciute, G. CRISPR-Mediated Non-Viral Site-Specific Gene Integration and Expression in T Cells: Protocol and Application for T-Cell Therapy. Cancers 2020, 12, 1704. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061704
Odé Z, Condori J, Peterson N, Zhou S, Krenciute G. CRISPR-Mediated Non-Viral Site-Specific Gene Integration and Expression in T Cells: Protocol and Application for T-Cell Therapy. Cancers. 2020; 12(6):1704. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061704
Chicago/Turabian StyleOdé, Zelda, Jose Condori, Nicolas Peterson, Sheng Zhou, and Giedre Krenciute. 2020. "CRISPR-Mediated Non-Viral Site-Specific Gene Integration and Expression in T Cells: Protocol and Application for T-Cell Therapy" Cancers 12, no. 6: 1704. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061704