Small Molecule Inhibitors of Microenvironmental Wnt/β-Catenin Signaling Enhance the Chemosensitivity of Acute Myeloid Leukemia

 , , , , , , , and
, , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
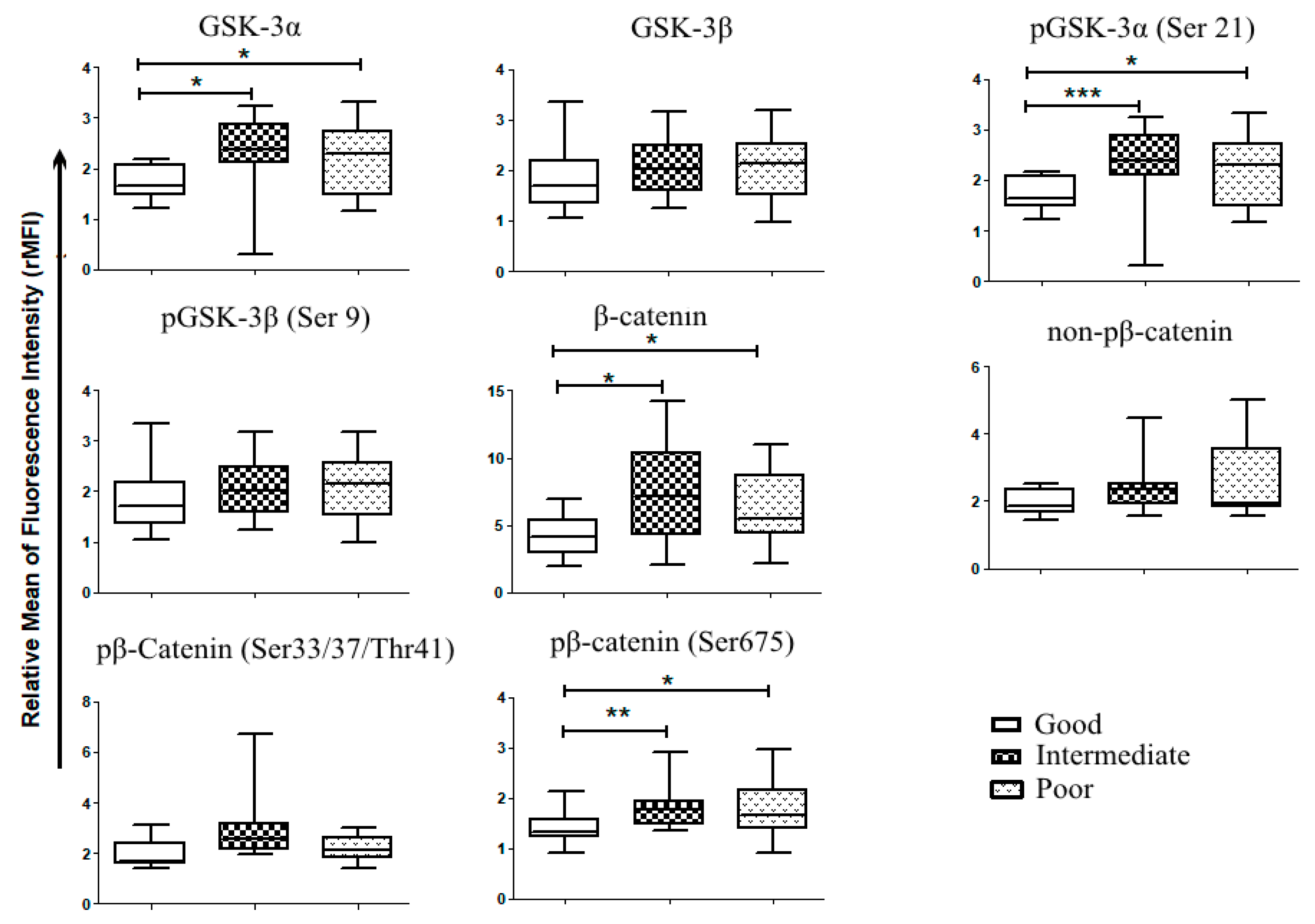
2.2. Patients, Samples and Cell Lines
2.3. Western Blotting
2.4. Cell Proliferation and Apoptosis and Viability Assays
2.5. Xenograft Mouse Model
2.6. Cell Culture and Co-Culture
2.7. Flow Cytometry Analysis of Wnt Molecules
2.8. Gene Reporter
2.9. RNA-seq Analysis
2.10. Statistical Analysis
3. Results
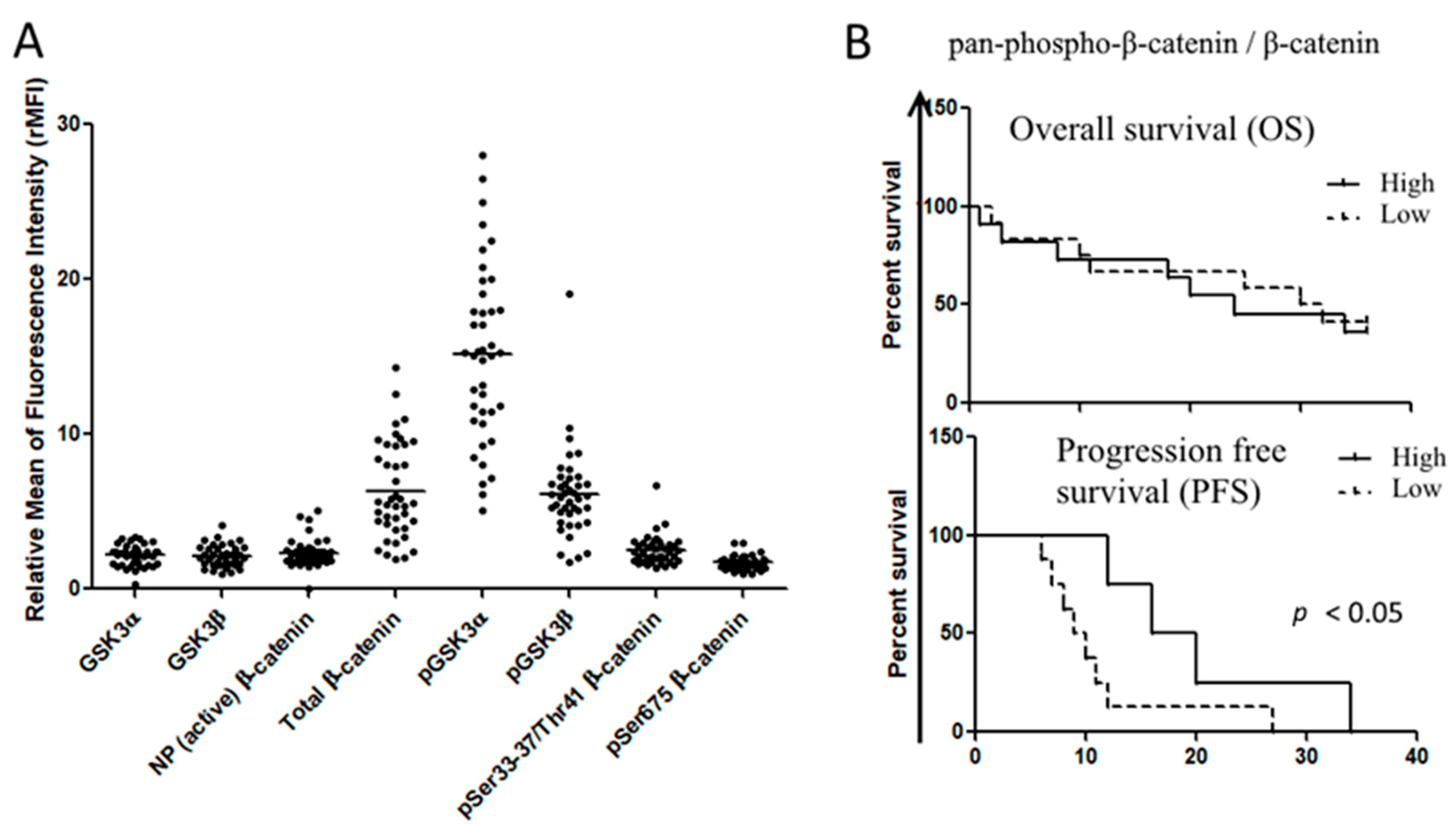
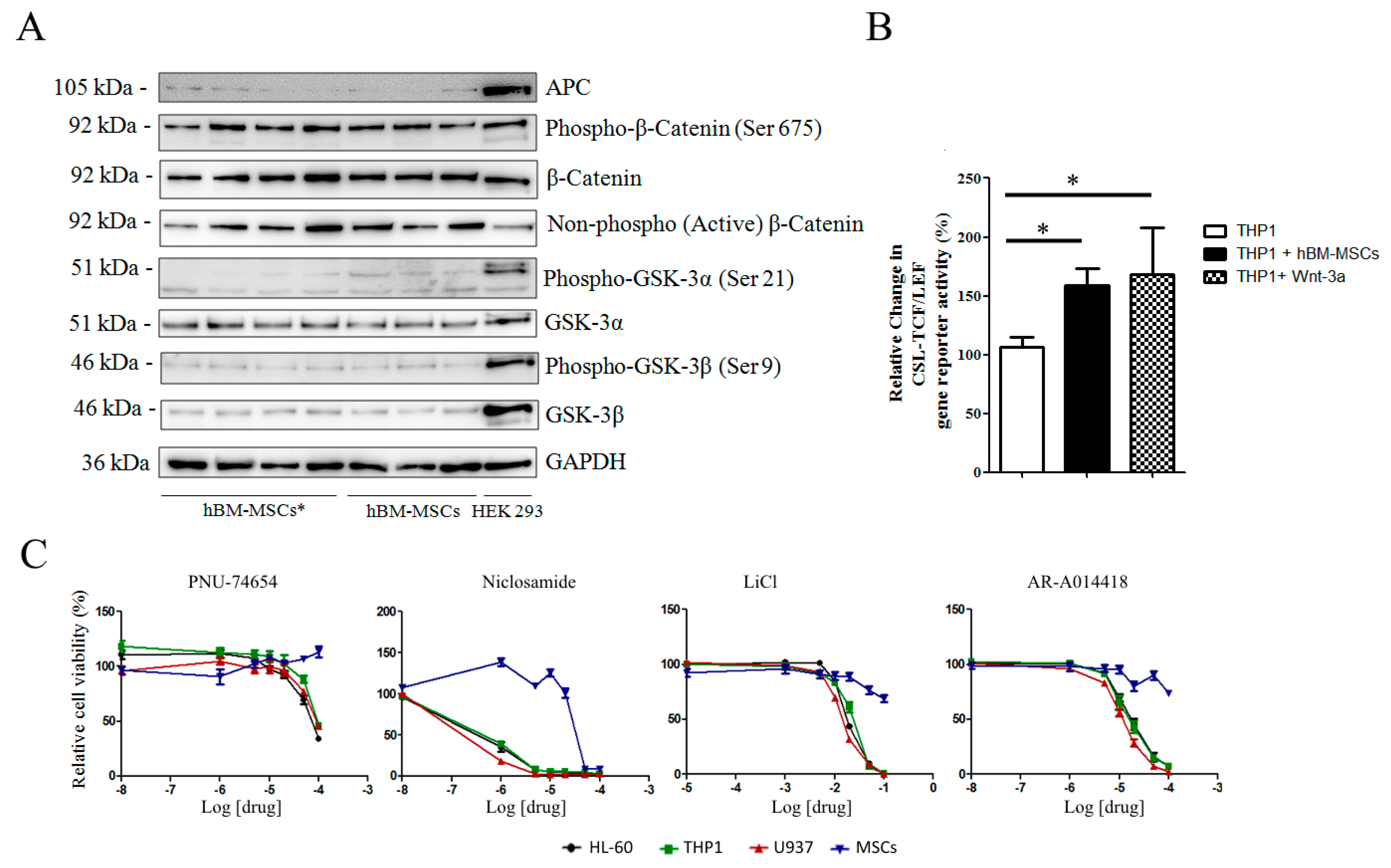
3.1. Wnt/GSK-3 Axis Is Functional in AML Cell Lines
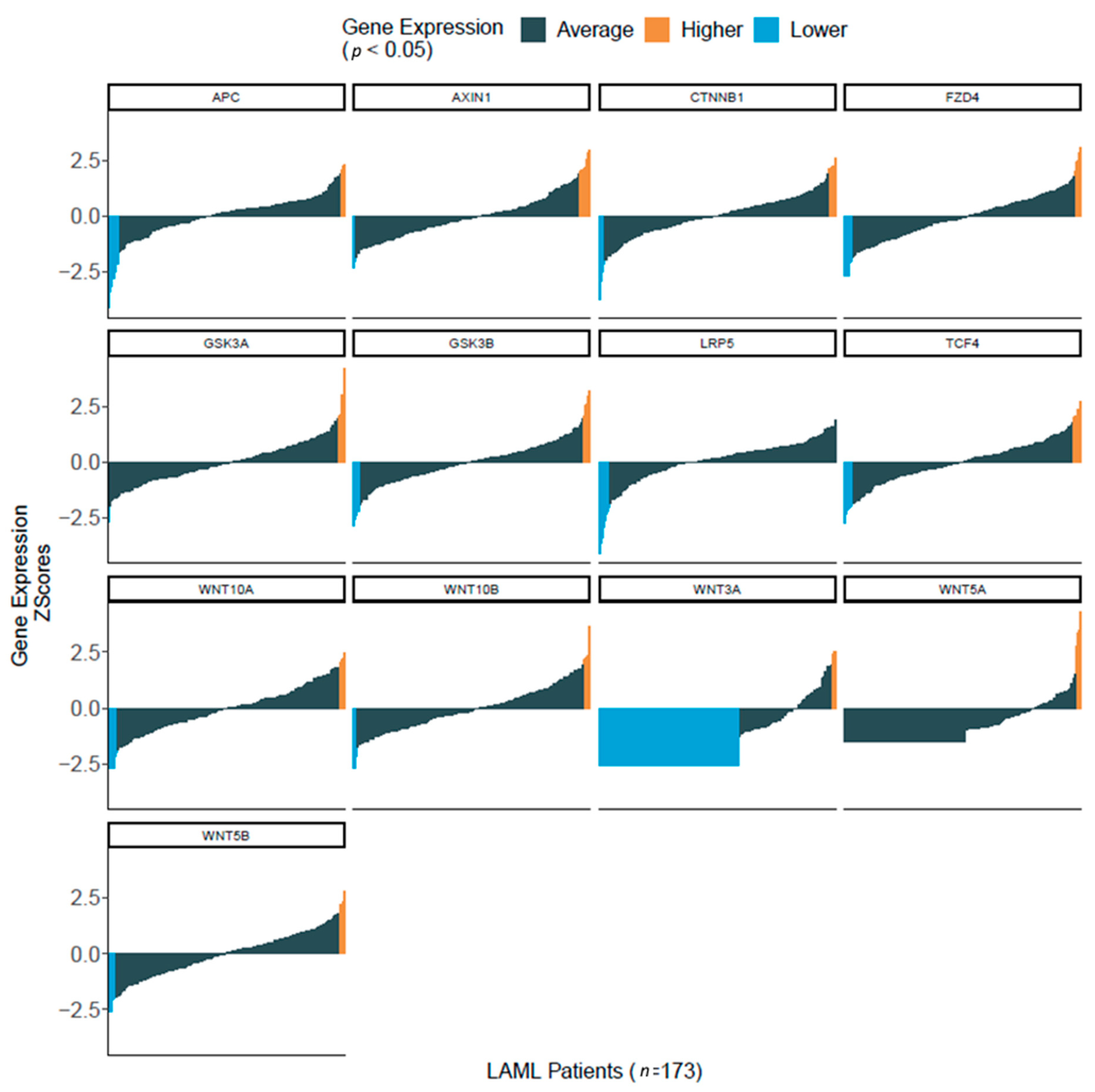
3.2. Wnt Molecules Are Enriched in Patient Samples
3.3. hBM-MSCs Express Wnt Molecules but are Insensitive to Pathway Inhibitors
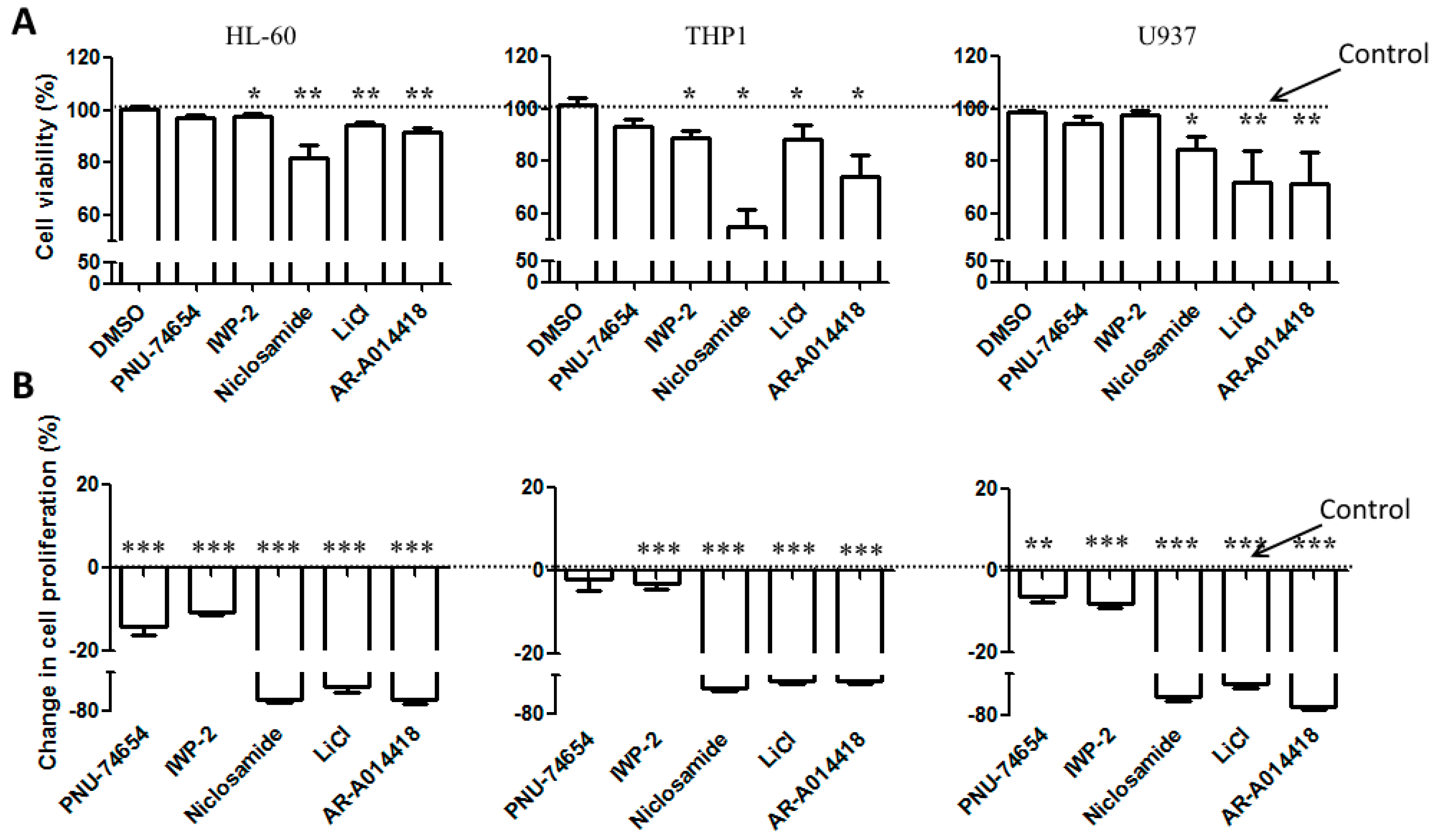
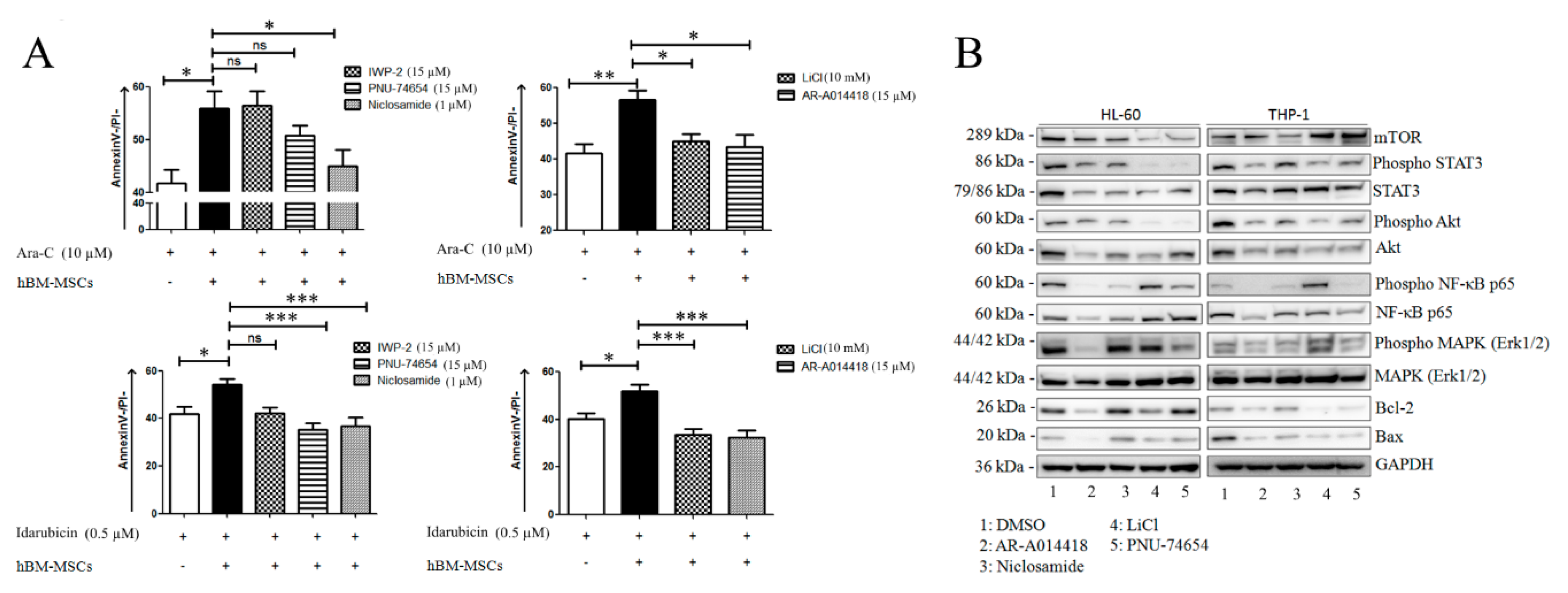
3.4. Wnt Modulators Enhance Chemosensitivity of AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, C.C. Genetic abnormalities and challenges in the treatment of acute myeloid leukemia. Genes Cancer 2011, 2, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcucci, G.; Mrózek, K.; Bloomfield, C.D. Molecular heterogeneity and prognostic biomarkers in adults with acute myeloid leukemia and normal cytogenetics. Curr. Opin. Hematol. 2005, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Kamdje, A.H.N.; Bassi, G.; Pacelli, L.; Malpeli, G.; Amati, E.; Nichele, I.; Pizzolo, G.; Krampera, M. Role of stromal cell-mediated Notch signaling in CLL resistance to chemotherapy. Blood Cancer J. 2012, 2, e73. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Mikesch, J.-H.; Steffen, B.; Berdel, W.E.; Serve, H.; Müller-Tidow, C. The emerging role of Wnt signaling in the pathogenesis of acute myeloid leukemia. Leukemia 2007, 21, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.W.; Ryu, S.; Kim, D.S.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Mesenchymal stem cells in suppression or progression of hematologic malignancy: Current status and challenges. Leukemia 2019, 33, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Scholer-Dahirel, A.; Schlabach, M.R.; Loo, A.; Bagdasarian, L.; Meyer, R.; Guo, R.; Woolfenden, S.; Yu, K.K.; Markovits, J.; Killary, K.; et al. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/β-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 17135–17140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheda, M.L.; Stacker, S.A. Importance of Wnt signaling in the tumor stroma microenvironment. Curr. Cancer Drug Targets 2008, 8, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- Gruszka, A.M.; Valli, D.; Alcalay, M. Wnt Signalling in Acute Myeloid Leukaemia. Cells 2019, 8, 1403. [Google Scholar] [CrossRef] [Green Version]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Vundavalli, M.; Friedman, R.; et al. Leukemogenesis Induced by an Activating β-catenin mutation in Osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Takam Kamga, P.; Collo, G.D.; Resci, F.; Bazzoni, R.; Mercuri, A.; Quaglia, F.M.; Tanasi, I.; Delfino, P.; Visco, C.; Bonifacio, M.; et al. Notch Signaling Molecules as Prognostic Biomarkers for Acute Myeloid Leukemia. Cancers 2019, 11, 1958. [Google Scholar] [CrossRef] [Green Version]
- Takam Kamga, P.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef] [Green Version]
- Kamga, P.T.; Dal Collo, G.; Bassi, G.; Midolo, M.; Delledonne, M.; Chilosi, M.; Bonifacio, M.; Krampera, M. Characterization of a new B-ALL cell line with constitutional defect of the Notch signaling pathway. Oncotarget 2018, 9, 18341–18350. [Google Scholar] [CrossRef] [Green Version]
- Takam Kamga, P.; Dal Collo, G.; Midolo, M.; Adamo, A.; Delfino, P.; Mercuri, A.; Cesaro, S.; Mimiola, E.; Bonifacio, M.; Andreini, A.; et al. Inhibition of Notch Signaling Enhances Chemosensitivity in B-cell Precursor Acute Lymphoblastic Leukemia. Cancer Res. 2019, 79, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Grieselhuber, N.R.; Klco, J.M.; Verdoni, A.M.; Lamprecht, T.; Sarkaria, S.M.; Wartman, L.D.; Ley, T.J. Notch Signaling in Acute Promyelocytic Leukemia. Leukemia 2013, 27, 1548–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estey, E.H. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am. J. Hematol. 2018, 93, 1267–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.; Nowakowski, G.S.; Knox, T.R.; Boysen, J.C.; Maas, M.L.; Schwager, S.M.; Wu, W.; Wellik, L.E.; Dietz, A.B.; Ghosh, A.K.; et al. Bi-directional activation between mesenchymal stem cells and CLL B-cells: Implication for CLL disease progression. Br. J. Haematol. 2009, 147, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Tabe, Y.; Lu, H.; Borthakur, G.; Miida, T.; Kantarjian, H.; Andreeff, M.; Konopleva, M. Mechanisms of apoptosis induction by simultaneous inhibition of PI3K and FLT3-ITD in AML cells in the hypoxic bone marrow microenvironment. Cancer Lett. 2013, 329, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spirli, C.; Locatelli, L.; Morell, C.M.; Fiorotto, R.; Morton, S.D.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. PKA dependent p-Ser-675β-catenin, a novel signaling defect in a mouse model of Congenital Hepatic Fibrosis. Hepatol. Baltim. Md. 2013, 58, 1713–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, L.F.; Bueno, A.C.; Gomes, D.C.; Abduch, R.; de Castro, M.; Antonini, S.R. Inhibition of the Tcf/beta-catenin complex increases apoptosis and impairs adrenocortical tumor cell proliferation and adrenal steroidogenesis. Oncotarget 2015, 6, 43016–43032. [Google Scholar] [CrossRef]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef] [Green Version]
- Staal, F.J.T.; Famili, F.; Garcia Perez, L.; Pike-Overzet, K. Aberrant Wnt Signaling in Leukemia. Cancers 2016, 8, 78. [Google Scholar] [CrossRef]
- Tickenbrock, L.; Hehn, S.; Sargin, B.; Choudhary, C.; Bäumer, N.; Buerger, H.; Schulte, B.; Müller, O.; Berdel, W.E.; Müller-Tidow, C.; et al. Activation of Wnt signalling in acute myeloid leukemia by induction of Frizzled-4. Int. J. Oncol. 2008, 33, 1215–1221. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Pan, W. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, C.; Bienz, M. Inhibition of GSK3 by Wnt signalling--two contrasting models. J. Cell Sci. 2011, 124, 3537–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.-H.; Ding, W.V.; McCormick, F. Wnt Signaling to β-Catenin Involves Two Interactive Components glycogen synthase kinase-3β inhibition and activation of protein kinase C. J. Biol. Chem. 2000, 275, 17894–17899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, M.; Zhao, X.; Huang, Q.; Sun, H.; Sun, C.; Yuan, J.; He, C.; Sun, Y.; Huang, X.; Kong, W.; et al. Activation of Wnt/β-catenin signaling by lithium chloride attenuates d-galactose-induced neurodegeneration in the auditory cortex of a rat model of aging. FEBS Open Bio 2017, 7, 759–776. [Google Scholar] [CrossRef] [PubMed]
- Ding, V.W.; Chen, R.H.; McCormick, F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J. Biol. Chem. 2000, 275, 32475–32481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Wang, L.; Gou, S.-M.; Wang, T.-L.; Zhang, M.; Liu, T.; Wang, C.-Y. ShRNA silencing glycogen synthase kinase-3 beta inhibits tumor growth and angiogenesis in pancreatic cancer. Cancer Lett. 2012, 316, 178–186. [Google Scholar] [CrossRef]
- Dong, J.; Peng, J.; Zhang, H.; Mondesire, W.H.; Jian, W.; Mills, G.B.; Hung, M.-C.; Meric-Bernstam, F. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005, 65, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Dal Col, J.; Dolcetti, R. GSK-3beta inhibition: At the crossroad between Akt and mTOR constitutive activation to enhance cyclin D1 protein stability in mantle cell lymphoma. Cell Cycle Georget. Tex 2008, 7, 2813–2816. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Stefan, T.; Ignatz-Hoover, J.; Moreton, S.; Parizher, G.; Saunthararajah, Y.; Wald, D.N. GSK-3 Inhibition Sensitizes Acute Myeloid Leukemia Cells to 1,25D-Mediated Differentiation. Cancer Res. 2016, 76, 2743–2753. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Gaianigo, N.; Ligorio, F.; Santoro, R.; Merz, V.; Simionato, F.; Zecchetto, C.; Falco, G.; Conti, G.; et al. Adipocytes sustain pancreatic cancer progression through a non-canonical WNT paracrine network inducing ROR2 nuclear shuttling. Int. J. Obes. 2018, 42, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Lin, C.; Roberts, M.J.; Waud, W.R.; Piazza, G.A.; Li, Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS ONE 2011, 6, e29290. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Onono, F.O.; Spielmann, H.P.; Subramanian, T.; Scherr, M.; Venturini, L.; Dallmann, I.; Ganser, A.; Reuter, C.W.M. Modulation of anthracycline-induced cytotoxicity by targeting the prenylated proteome in myeloid leukemia cells. J. Mol. Med. Berl. Ger. 2012, 90, 149–161. [Google Scholar] [CrossRef]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Yokoyama, A. Inhibition of MEK signaling enhances the ability of cytarabine to induce growth arrest and apoptosis of acute myelogenous leukemia cells. Apoptosis Int. J. Program. Cell Death 2009, 14, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Ristic, B.; Bosnjak, M.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Bogdanovic, A.; Perovic, V.; Martinovic, T.; Kravic-Stevovic, T.; Bumbasirevic, V.; et al. Idarubicin induces mTOR-dependent cytotoxic autophagy in leukemic cells. Exp. Cell Res. 2014, 326, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Lazzaroni, F.; Giacco, L.D.; Biasci, D.; Turrini, M.; Prosperi, L.; Brusamolino, R.; Cairoli, R.; Beghini, A. Intronless WNT10B-short variant underlies new recurrent allele-specific rearrangement in acute myeloid leukaemia. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Parmar, A.; Marz, S.; Rushton, S.; Holzwarth, C.; Lind, K.; Kayser, S.; Döhner, K.; Peschel, C.; Oostendorp, R.A.J.; Götze, K.S. Stromal niche cells protect early leukemic FLT3-ITD+ progenitor cells against first-generation FLT3 tyrosine kinase inhibitors. Cancer Res. 2011, 71, 4696–4706. [Google Scholar] [CrossRef] [Green Version]
- Behrmann, L.; Wellbrock, J.; Fiedler, W. Acute Myeloid Leukemia and the Bone Marrow Niche—Take a Closer Look. Front. Oncol. 2018, 8, 444. [Google Scholar] [CrossRef] [Green Version]
- Tabe, Y.; Konopleva, M. Role of Microenvironment in Resistance to Therapy in AML. Curr. Hematol. Malig. Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef]
- Toni, F.D.; Racaud-Sultan, C.; Chicanne, G.; Mas, V.M.-D.; Cariven, C.; Mesange, F.; Salles, J.-P.; Demur, C.; Allouche, M.; Payrastre, B.; et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene 2006, 25, 3113–3122. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Expression of Wnt Molecules in AML Cell Lines | HL-60 | THP1 | U937 |
|---|---|---|---|
| Relative median of fluorescence intensity (rMFI) ± SEM | |||
| Total β-catenin | 2.466 ± 0.238 | 6.765 ± 1.508 | 2.781 ± 0.288 |
| Non-phospho-β-catenin | 1.676 ± 0.058 | 2.360 ± 0.209 | 1.442 ± 0.0677 |
| Ser675-phospho-β-catenin | 3.471 ± 0.202 | 7.847 ± 1.443 | 3.398 ± 0.566 |
| Ser33/37/Thr41-phospho-β-catenin | 2.135 ± 0.119 | 3.013 ± 0.395 | 2.232 ± 0.21 |
| GSK-3α | 2.275 ± 0.161 | 2.654 ± 0.298 | 2.194 ± 0.18 |
| pGSK-3α (Ser21) | 9.355 ± 1.641 | 1.640 ± 2.678 | 7.901 ± 1.643 |
| GSK-3β | 2.217 ± 0.139 | 2.456 ± 0.293 | 1.713 ± 0.064 |
| GSK-3β (Ser 9) | 4.600 ± 0.416 | 9.401 ± 3.046 | 3.935 ± 0.643 |
| (A) White Blood Cells (WBC) | ||||||||
| WBC | GSK3α | GSK3β | Non-Phospho-β-catenin | Total β-Catenin | Phospho-GSK-3α (Ser 21) | Phospho-GSK3β (Ser 9) | Ser33/37/Thr41-Phospho- β-Catenin | Ser675-Phospho-β-Catenin |
| r | 0.1117 | 0.3236 | 0.3205 | 0.2317 | 0.1109 | 0.2414 | 0.4311 | 0.4106 |
| p value | 0.2583 | 0.0271 | 0.0284 | 0.0870 | 0.2597 | 0.0780 | 0.0043 | 0.0064 |
| (B) Hemoglobin (Hb) | ||||||||
| Hb | GSK3α | GSK3β | Non-Phospho-β-Catenin | Total β-Catenin | Phospho-GSK-3α (Ser 21) | Phospho-GSK3β (Ser 9) | Ser33/37/Thr41-Phospho- β-Catenin | Ser675-Phospho-β-Catenin |
| r | −0.02664 | −0.02471 | 0.09962 | 0.002832 | 0.1640 | 0.2041 | 0.1749 | 0.2092 |
| p value | 0.4387 | 0.4431 | 0.2816 | 0.4935 | 0.1696 | 0.1162 | 0.1538 | 0.1104 |
| (C) Platelets (PLTS) | ||||||||
| PLTS | GSK3α | GSK3β | Non-Phospho-β-Catenin | Total β-Catenin | Phospho-GSK-3α (Ser 21) | Phospho-GSK3β (Ser 9) | Ser33/37/Thr41-Phospho- β-Catenin | Ser675-Phospho-β-Catenin |
| r | 0.2725 | 0.2478 | 0.4430 | 0.1463 | 0.2366 | 0.3473 | 0.3730 | 0.4452 |
| p value | 0.0539 | 0.0726 | 0.0034 | 0.1972 | 0.0824 | 0.0190 | 0.0125 | 0.0033 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takam Kamga, P.; Dal Collo, G.; Cassaro, A.; Bazzoni, R.; Delfino, P.; Adamo, A.; Bonato, A.; Carbone, C.; Tanasi, I.; Bonifacio, M.; et al. Small Molecule Inhibitors of Microenvironmental Wnt/β-Catenin Signaling Enhance the Chemosensitivity of Acute Myeloid Leukemia. Cancers 2020, 12, 2696. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092696
Takam Kamga P, Dal Collo G, Cassaro A, Bazzoni R, Delfino P, Adamo A, Bonato A, Carbone C, Tanasi I, Bonifacio M, et al. Small Molecule Inhibitors of Microenvironmental Wnt/β-Catenin Signaling Enhance the Chemosensitivity of Acute Myeloid Leukemia. Cancers. 2020; 12(9):2696. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092696
Chicago/Turabian StyleTakam Kamga, Paul, Giada Dal Collo, Adriana Cassaro, Riccardo Bazzoni, Pietro Delfino, Annalisa Adamo, Alice Bonato, Carmine Carbone, Ilaria Tanasi, Massimiliano Bonifacio, and et al. 2020. "Small Molecule Inhibitors of Microenvironmental Wnt/β-Catenin Signaling Enhance the Chemosensitivity of Acute Myeloid Leukemia" Cancers 12, no. 9: 2696. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092696