Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
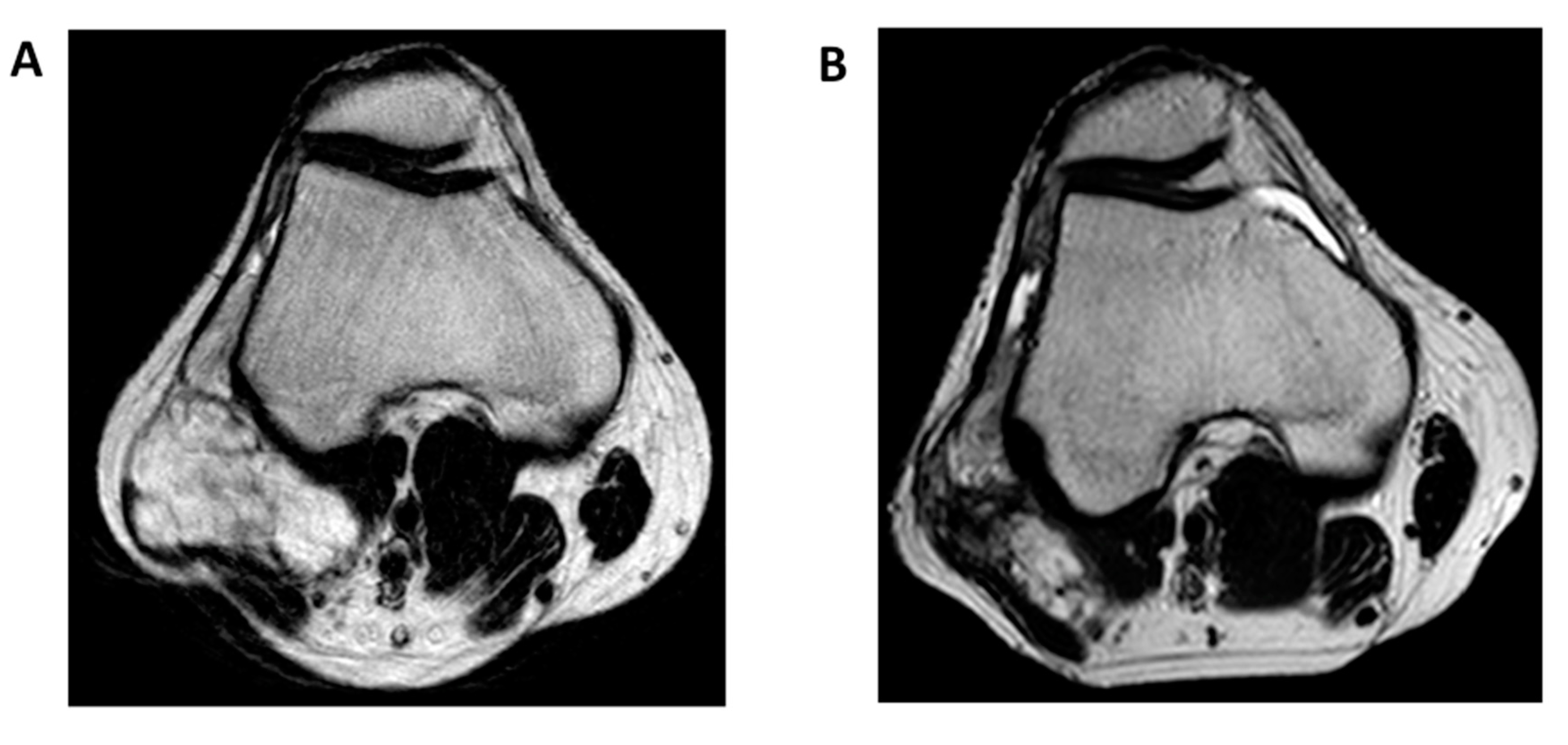
2. Diagnostic Criteria
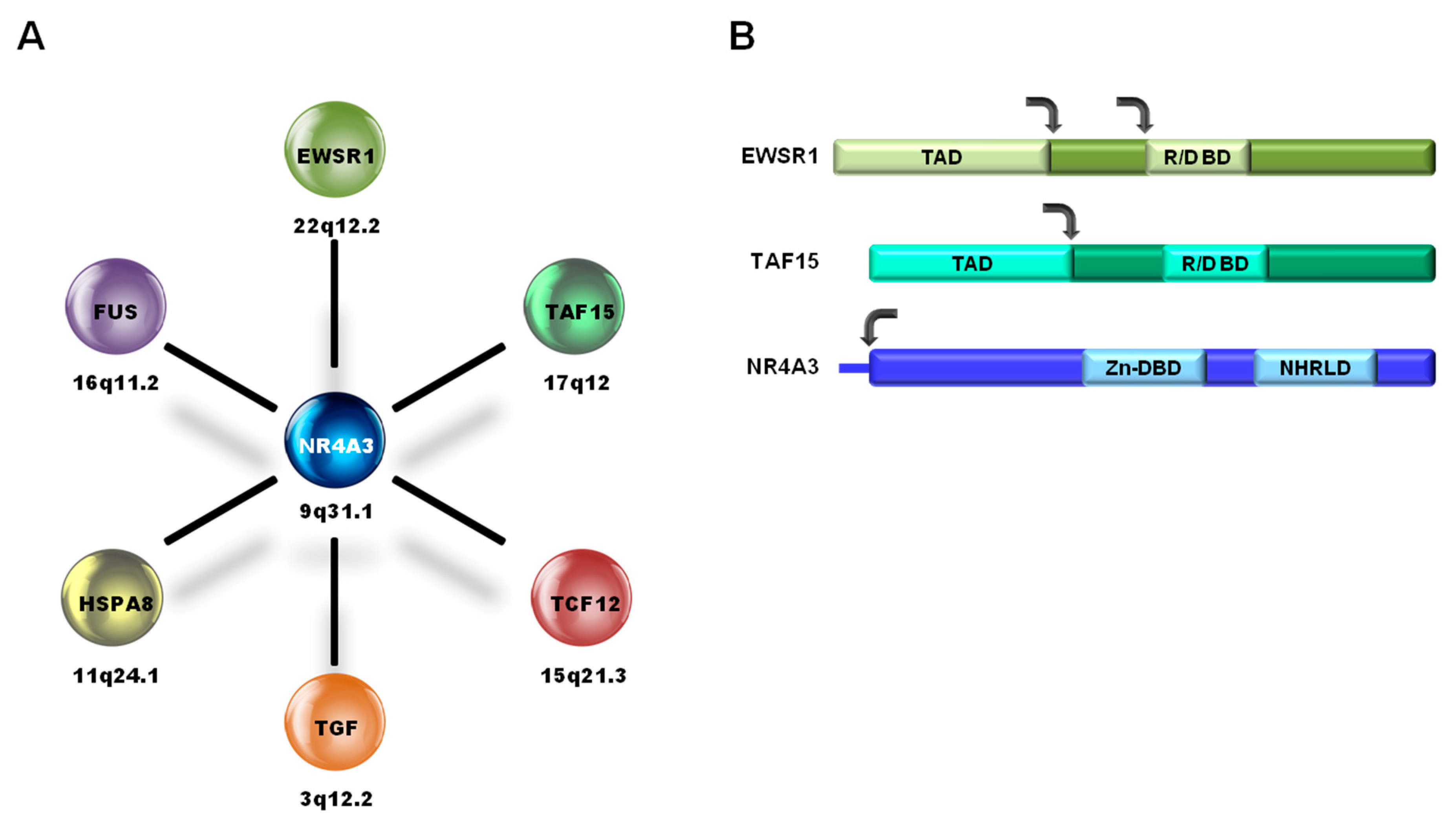
Pathological and Molecular Characteristics
3. State of the Art: Localized Disease
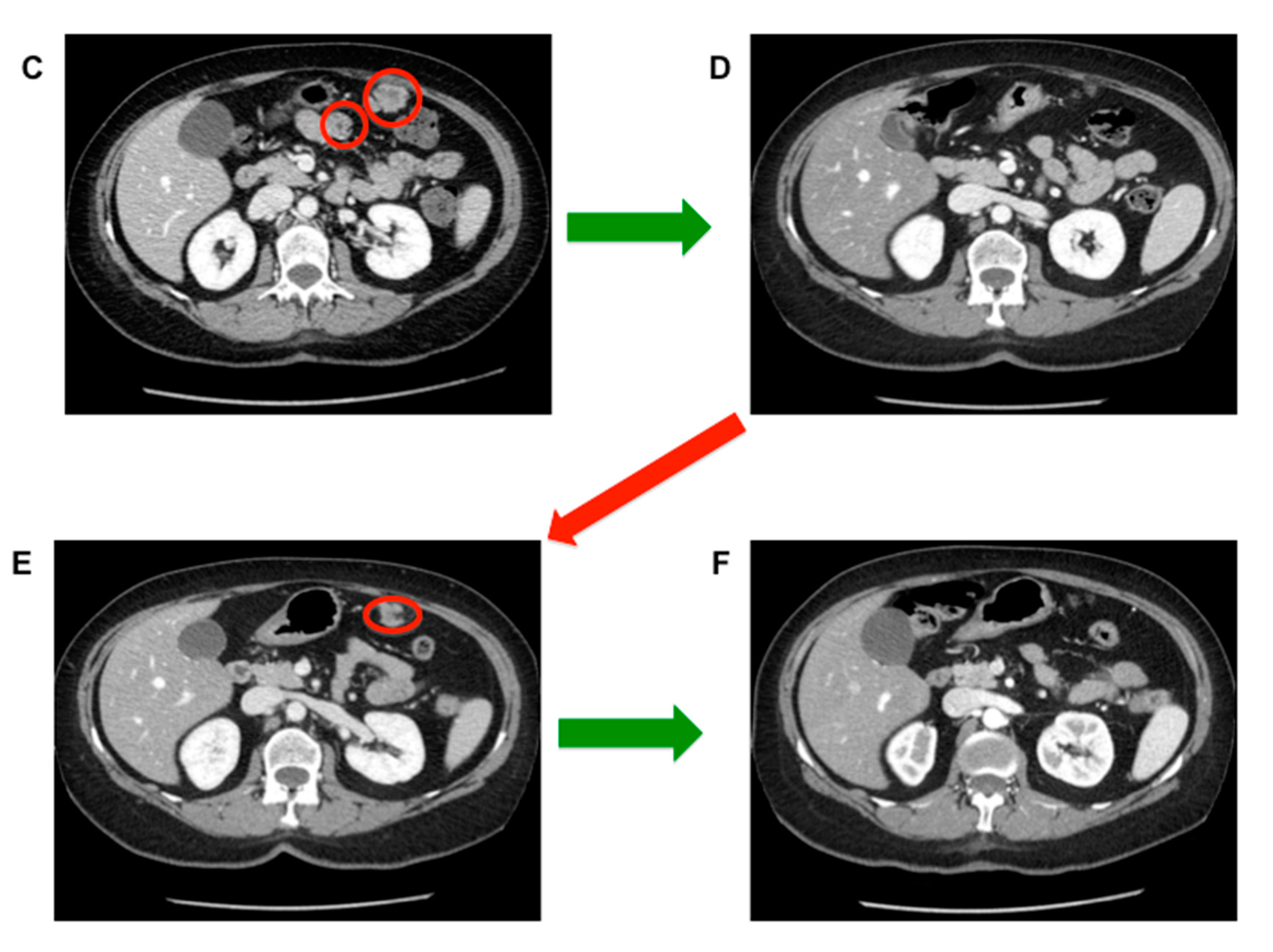
4. State of the Art: Advanced Disease
5. Current Research and Future Perspectives
6. Conclusions
Funding
Conflicts of Interest
References
- Horvai, A.E.; Agaram, N.P.; Lucas, D.R. Extraskeletal myxoid condrosarcoma. In World Health Organization (WHO) Classification of Soft Tissue and Bone Tumours, 5th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2020; pp. 303–305. [Google Scholar]
- Hachitanda, Y.; Tsuneyoshi, M.; Daimaru, Y.; Enjoji, M.; Nakagavara, A.; Ikeda, K.; Sueishi, K. Extraskeletal myxoid chondrosarcoma in children. Cancer 1988, 61, 2521–2526. [Google Scholar] [CrossRef]
- Stout, A.P.; Verner, E.W. Chondrosarcoma of the extraskeletal soft tissues. Cancer 1953, 6, 581–590. [Google Scholar] [CrossRef]
- Enzinger, F.M.; Shiraki, M. Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum. Pathol. 1972, 3, 421–435. [Google Scholar] [CrossRef]
- Meis-Kindblom, J.M.; Bergh, P.; Gunterberg, B.; Kindblom, L.G. Extraskeletal myxoid chondrosarcoma: A reappraisal of its morhologic spectrum and prognostic factors based on 117 cases. Am. J. Surg. Pathol. 1999, 23, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Brody, R.I.; Ueda, T.; Hamelin, A.; Jhanwar, S.C.; Bridge, J.A.; Healey, J.H.; Huvos, A.G.; Gerald, W.L.; Ladanyi, M. Molecular analysis of the fusion of EWS to an orphan nuclear receptor gene in extraskeletal myxoid condrosarcoma. Am. J. Pathol. 1997, 150, 1049–1058. [Google Scholar]
- Antonescu, C.R.; Argani, P.; Erlandson, R.A.; Healey, J.H.; Lasanyi, M.; Huvos, A.G. Skeletal and extraskeletal myxoid chondrosarcoma: A comparative clinico-pathologic, ultrastructural and molecular study. Cancer 1998, 83, 1504–1521. [Google Scholar] [CrossRef]
- Demicco, E.G.; Wang, W.L.; Madewell, J.E.; Huang, D.; Bui, M.M.; Bridge, J.A.; Meis, J.M. Osseous myxochondroid sarcoma: A detailed study of 5 cases of extraskeletal myxoid chondrosarcoma of the bone. Am. J. Surg. Pathol. 2013, 37, 752–762. [Google Scholar] [CrossRef]
- Finos, L.; Righi, A.; Frisoni, T.; Gambarotti, M.; Ghinelli, C.; Benini, S.; Vanel, D.; Picci, P. Primary extraskeletal myxoid chondrosarcoma of bone: Report of three cases and review of the literature. Pathol. Res. Pract. 2017, 2013, 461–466. [Google Scholar] [CrossRef]
- Filion, C.; Labelle, Y. Identification of genes regulated by the EWS/NR4A3 fusion protein in extraskeletal myxoid condrosarcoma. Tumour Biol. 2012, 33, 1599–1605. [Google Scholar] [CrossRef]
- Murphey, M.D.; Walker, E.A.; Wilson, A.J.; Kransdorf, M.J.; Temple, H.T.; Gannon, F.H. From the archives of the AFIP: Imaging of primary chondrosarcoma: Radiologic-pathologic correlation. Radiographics 2003, 23, 1245–1278. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, N.; Shinagare, A.B.; Jagannathan, J.P.; Shah, S.H.; Krajewski, K.M.; Hornick, J.L.; Ramaiya, N.H. Clinical and radiologic features of extraskeletal myxoid chondrosarcoma including initial presentation, local recurrence and metastases. Radiol. Oncol. 2014, 48, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateishi, U.; Hasegawa, T.; Nojima, T.; Takegami, T.; Arai, Y. MRI features of extraskeletal myxoid chondrosarcoma. Skelet. Radiol. 2006, 35, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Paioli, A.; Stacchiotti, S.; Campanacci, D.; Palmerini, E.; Frezza, A.M.; Longhi, A.; Radaelli, S.; Donati, D.M.; Beltrami, G.; Bianchi, G.; et al. Extrascheletal myxoid chondrosarcoma with molecularly confirmed diagnosis: A multicentre retrospective study within the Italian Sarcoma Group. Ann. Surg. Oncol. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Chiusole, B.; Le Cesne, A.; Rastrelli, M.; Maruzzo, M.; Cappellesso, R.; Del Fiore, B.; Imbevaro, S.; Sbaraglia, M.; Therrier, P.; Ruggieri, P.; et al. Extraskeletal Myxoid Chondrosarcoma: Clinical and Molecular Characteristics and Outcomes of Patients Treated at Two Institutions. Front. Oncol. 2020, 10, 828. [Google Scholar] [CrossRef]
- Saleh, G.; Evans, H.L.; Ro, Y.J.; Ayala, A.G. Extraskeletal myxoid chondrosarcoma. A clinicopathologic study of ten patients with long-term follow-up. Cancer 1992, 70, 2827–2830. [Google Scholar] [CrossRef]
- Lucas, D.R.; Fletcher, C.D.; Adsay, N.V.; Zalupski, M.M. High-grade extraskeletal myxoid chondrosarcoma: A high-grade epithelioid malignancy. Histopathology 1999, 35, 201–208. [Google Scholar] [CrossRef]
- Goh, Y.V.; Spagnolo, D.V.; Platten, M.; Caterina, P.; Fisher, C.; Oliveira, A.M.; Nascimento, A.G. Extraskeletal Myxoid Chondrosarcoma: A Light Microscopic, Immunohistochemical, Ultrastructural and Immuno-Ultrastructural Study Indicating Neuroendocrine Differentiation. Hostopathology 2001, 39, 514–524. [Google Scholar] [CrossRef]
- Harris, M.; Coyne, J.; Tariq, M.; Eyden, B.P.; Atkinson, M.; Freemont, A.J.; Varley, J.; Attwool, C.; Telford, N. Extraskeletal myxoid chondrosarcoma with neuroendocrine differentiation: A pathologic, cytogenetic, and molecular study of a case with a novel translocation t(9;17)(q22;q11.2). Am. J. Surg. Pathol. 2000, 24, 1020–1026. [Google Scholar] [CrossRef]
- Okamoto, S.; Hisaoka, M.; Ishida, T.; Imamura, T.; Kanda, H.; Shimajiri, S.; Hashimoto, H. Extraskeletal myxoid chondrosarcoma: A clinicopathologic, immunohistochemical, and molecular analysis of 18 cases. Hum. Pathol. 2001, 32, 1116–1124. [Google Scholar] [CrossRef]
- Oliveira, A.M.; Sebo, T.J.; McGrory, J.E.; Gaffey, T.A.; Rock, M.G.; Nascimento, A.G. Extraskeletal myxoid chondrosarcoma: A clinicopathologic, immunohistochemical, and ploidy analysis of 23 cases. Mod. Pathol. 2000, 13, 900–908. [Google Scholar] [CrossRef]
- Stenman, G.; Andersson, H.; Mandahl, N.; Meis-Kindblom, J.M.; Kindblom, L.G. Translocation t(9;22)(q22;q12) Is a Primary Cytogenetic Abnormality in Extraskeletal Myxoid Chondrosarcoma. Int. J. Cancer 1995, 62, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Flucke, U.; Tops, B.B.; Verdijk, M.A.; van Cleef, P.G.; van Zwam, P.H.; Slootweg, P.J.; Bovée, J.V.; Riedl, R.G.; Creytens, D.H.; Suurmeijer, A.J.; et al. NR4A3 rearrangement realiably distinguished between the clinicopathologically overlapping entities myoepithelial carcinoma of soft tissue and cellular extraskeletal myxoid chondrosarcoma. Virchows Arch. 2012, 460, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichs, S.H.; Jaramillo, M.A.; Gumerlock, P.H.; Gardner, M.B.; Lewis, J.P.; Freeman, A.E. Myxoid chondrosarcoma with a translocation involving chromosomes 9 and 22. Cancer Genti. Cytogenet. 1985, 14, 219–226. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, J.; Rius, J.; Castello, A.; Cases-Langhoff, C.; Badimon, L. Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ. Res. 2003, 92, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Domanski, H.A.; Brosjo, O.; Heim, S.; Bjerkehagen, B.; Sciot, R.; Dal Cin, P.; Fletcher, J.A.; et al. Molecular genetic characterization of the EWS/CHN and RBP56/CHN fusion genes in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 2002, 35, 340–352. [Google Scholar] [CrossRef]
- Broehm, C.J.; Wu, J.; Gullapalli, R.R.; Bocklage, T. Extraskeletal myxoid chondrosarcoma with a t(9;16)(q22;p11.2) resulting in a NR4A3-FUS fusion. Cancer Genet. 2014, 207, 276–280. [Google Scholar] [CrossRef]
- Urbini, M.; Astolfi, A.; Pantaleo, M.A.; Serravalle, S.; Dei Tos, A.P.; Picci, P.; Indio, V.; Sbaraglia, M.; Benini, S.; Righi, A.; et al. HSPA8 as a novel fusion partner of NR4A3 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 2017, 56, 582–586. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Cech, T.R.; Parker, R.R. Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem. 2015, 84, 355–379. [Google Scholar] [CrossRef]
- Kovar, H. Dr. Jekyll and Mr. Hyde: The Two Faces of the FUS/EWS/TAF15 Protein Family. Sarcoma 2011, 2011, 837474. [Google Scholar] [CrossRef]
- Mohan, H.M.; Aherne, C.M.; Rogers, A.C.; Baird, A.W.; Winter, D.C.; Murphy, E.P. Molecular pathways: The role of NR4A orphan nuclear receptors in cancer. Clin. Cancer Res. 2012, 18, 3223–3228. [Google Scholar] [CrossRef] [Green Version]
- De Vera, I.M.; Giri, P.K.; Munoz-Tello, P.; Brust, R.; Fuhrmann, J.; Matta-Camacho, E.; Shang, J.; Campbell, S.; Wilson, H.D.; Granados, J.; et al. Identification of a Binding Site for Unsaturated Fatty Acids in the Orphan Nuclear Receptor Nurr1. ACS Chem. Biol. 2016, 11, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Herring, J.A.; Elison, W.S.; Tessem, J.S. Function of Nr4a Orphan Nuclear Receptors in proliferation, apoptosis and fuel utilization across tissue. Cells 2019, 8, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamoto, T.; Czerniak, B.A.; Dinney, C.P.; Kamat, A.M. Cytoplasmic mislocalization of the orphan nuclear receptor Nurr1 is a prognostic factor in bladder cancer. Cancer 2010, 116, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Brenca, M.; Stacchiotti, S.; Fassetta, K.; Sbaraglia, M.; Janjusevic, M.; Racanelli, D.; Polano, M.; Rossi, S.; Brich, S.; Dagrada, G.P.; et al. NR3A4 fusion proteins trigger an axon guidance switch that marks the difference between EWSR1 and TAF15 translocated extraskeletal myxoid chondrosarcoma. J. Pathol. 2019, 249, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agaram, N.P.; Zhang, L.; Sung, Y.S.; Singer, S.; Antonescu, C.R. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum. Pathol. 2014, 45, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanomi, D.; Chivatakarn, O.; Bai, G.; Abdesselem, H.; Lettieri, K.; Marquardt, T.; Pierchala, B.A.; Pfaff, S.L. Ret is a multifunctional co receptor that integrates diffusible- and contact-axon guidance signals. Cell 2012, 148, 568–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, M.; Tamagnone, L. Tyrosine phosphorylation in semaphoring signalling: Shifting into overdrive. EMBO Rep. 2008, 9, 865–871. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Pantaleo, M.A.; Astolfi, A.; Dagrada, P.; Negri, T.; Dei Tos, A.P.; Indio, V.; Morosi, C.; Gronchi, A.; Colombo, C.; et al. Activity of sunitinib in extraskeletal myxoid condrosarcoma. Eur. J. Cancer 2014, 50, 1657–1664. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Ferrari, S.; Redondo, A.; Hindi, N.; Palmerini, E.; Vaz Salgado, M.A.; Frezza, A.M.; Casali, P.G.; Gutierrez, A.; Lopez-Pousa, A.; et al. Pazopanib for the treatment of advanced extraskeletal myxoid chondrosarcoma: A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 1252–1262. [Google Scholar] [CrossRef]
- Davis, E.J.; Wu, Y.M.; Robinson, D.; Schuetze, S.M.; Baker, L.H.; Athanikar, J.; Cao, X.; Kunju, L.P.; Chinnaiyan, A.M.; Chugh, R. Next generation sequencing in extraskeletal myxoid chondrosarcoma. Oncotarget 2017, 8, 21770–21777. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, S.; Dagrada, G.P.; Morosi, C.; Negri, T.; Romanini, A.; Pilotti, S.; Gronchi, A.; Casali, P.G. Extraskeletal myxoid condrosarcoma: Tumor response to sunitinib. Clin. Sarcoma Res. 2012, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morioka, H.; Takahashi, S.; Araki, N.; Sugiura, H.; Ueda, T.; Takahashi, M.; Yonemoto, T.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Results of a sub-analysis of a phase 2 study on trabectedin treatment for extraskeletal myxoid chondrosarcoma and mesenchymal chondrosarcoma. BMC Cancer 2016, 16, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurzrock, R.; Patnaik, A.; Aisner, J.; Warren, T.; Leong, S.; Benjamin, R.; Eckhardt, S.G.; Eid, J.E.; Greig, G.; Habben, K.; et al. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 2458–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; West, R.B.; Marinelli, R.J.; Nielsen, T.O.; Rubin, B.P.; Goldblum, J.R.; Patel, R.M.; Zhu, S.; Montgomery, K.; Ng, T.L.; et al. The gene expression profile of extraskeletal myxoid chondrosarcoma. J. Pathol. 2005, 206, 433–444. [Google Scholar] [CrossRef]
- Poulin, H.; Filion, C.; Ladanyi, M.; Labelle, Y. Serum- and glucocorticoid-regulated kinase 1 (SGK1) induction by the EWS/NOR1(NR4A3) protein. Biochem. Biophys. Res. Commun. 2006, 346, 306–313. [Google Scholar] [CrossRef]
- Sjogren, H.; Meis-Kindblom, J.M.; Orndal, C.; Bergh, P.; Ptaszynski, K.; Aman, P.; Kindblom, L.G.; Stenman, G. Studies on the molecular pathogenesis of extraskeletal myxoid chondrosarcoma-cytogenetic, molecular genetic, and cDNA microarray analyses. Am. J. Pathol. 2003, 162, 781–792. [Google Scholar] [CrossRef]
- Enneking, W.F.; Spainer, S.S.; Goodman, M.A. A system for the surgical staging of muscoloskeletal sarcoma. Clin. Orthop. Relat. Res. 2003, 415, 4–18. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; et al. ESMO Guidelines Committee and EURACAN. Soft tissue and visceral sarcomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29 (Suppl. 4), iv51–iv67. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Wada, T.; Nagoya, S.; Ikeda, T.; Isu, K.; Yamashiro, K.; Kawai, A.; Ishii, T.; Araki, N.; Myoui, A.; et al. Extraskeletal myxoid chondrosarcoma: A multi-institutional study of 42 cases in Japan. Cancer 2003, 97, 1285–1292. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology: Soft Tissue Sarcoma. Version 2.2020, 28 May 2020. Available online: https://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf (accessed on 19 September 2020).
- Drilon, A.D.; Popat, S.; Bhuchar, G.; D’Adamo, D.R.; Keohan, M.L.; Fisher, C.; Antonescu, C.R.; Singer, S.; Brennan, M.F.; Judson, J.; et al. Extraskeletal myxoid chondrosarcoma: A retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 2008, 113, 3364–3371. [Google Scholar] [CrossRef] [Green Version]
- Bishop, A.J.; Bird, J.E.; Conley, A.P.; Roland, C.L.; Moon, B.S.; Satcher, R.L.; Livingston, J.A.; Patel, S.; Wang, W.L.; Lazar, A.J.; et al. Extraskeletal myxoid chondrosarcoma: Combined modality therapy with both radiation and surgery improves local control. Am. J. Clin. Oncol. 2019, 42, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Kemmerer, E.J.; Gleeson, E.; Poli, J.; Ownbey, R.T.; Brady, L.W.; Bowne, W.B. Benefit of radiotherapy in extraskeletal myxoid chondrosarcoma: A propensity score weighted population-based analysis of the SEER database. Am. J. Clin. Oncol. 2018, 41, 674–680. [Google Scholar] [CrossRef] [PubMed]
- McGrory, J.E.; Rock, M.G.; Nascimento, A.G.; Oliveira, A.M. Extraskeletal myxoid chondrosarcoma. Clin. Orthop. Relat. Res. 2001, 382, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Gronchi, A.; Palmerini, E.; Quagliolo, V.; Martin-Broto, J.; Lopez-Pousa, A.; Grignani, G.; Brunello, A.; Blay, J.Y.; Tendero, O.; Diaz Beveridge, R.; et al. Neoadjuvant Chemotherapy in High-Risk Soft Tissue Sarcomas: Final Results of a Randomized Trial from Italian (ISG), Spanish (GEIS), French (FSG), and Polish (PSG) Sarcoma Groups. J. Clin. Oncol. 2020, 8, 2178–2186. [Google Scholar] [CrossRef]
- Patel, S.R.; Burgess, M.A.; Papadopoulos, N.E.; Linke, K.A.; Benjamin, R.S. Extraskeletal myxoid chondrosarcoma. Long term experience with chemotherapy. Am. J. Clin. Oncol. 1995, 18, 161–163. [Google Scholar]
- Ogura, K.; Fujiwara, T.; Beppu, Y.; Chuman, H.; Yoshida, A.; Kawano, H.; Kawai, A. Extraskeletal myxoid chondrosarcoma: A review of 23 patients trated at a single referral center with long-term follow-up. Arch. Orthop. Trauma Surg. 2012, 9, 4132–4138. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Dagrada, G.P.; Sanfilippo, R.; Negri, T.; Vittimberga, I.; Ferrari, S.; Grosso, F.; Apice, G.; Tricomi, M.; Colombo, C.; et al. Anthracycline-based chemotherapy in extraskeletal myxoid chondrosarcoma: A retrospective study. Clin. Sarcoma Res. 2013, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Chow, W.; Frankel, P.; Ruel, C.; Araujo, D.M.; Milhem, M.; Okuno, S.; Hartner, L.; Undevia, S.; Staddon, A. Results of a prospective phase 2 study of pazopanib in patients with surgically unresectable or metastatic chondrosarcoma. Cancer 2020, 126, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Xu, J.; Sun, X.; Liu, K.; Li, X.; He, F.; Liu, X.; Gu, J.; Lv, Z.; Yang, R.; et al. Apatinib for the treatment of inoperable metastatic or locally advanced chondrosarcoma: What we can learn about the biological behavior of chondrosarcoma from two-center study. Cancer Manag. Res. 2020, 12, 3513–3525. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Grignani, G.; Martinez Trufero, J.; Redondo, A.; Valverde, C.; Lopez Pousa, A.; Stacchiotti, S.; Palmerini, E.; de Alava, E.; et al. IMMUNOSARC: A collaborative Spanish (GEIS) and Italian (ISG) Sarcoma Groups phase I/II trial of sunitinib plus nivolumab in selected bone and soft tissue sarcoma subtypes-Results of the phase II-soft tissue sarcoma cohort. Ann. Oncol. 2019, 30 (Suppl. 5), 683–709. [Google Scholar] [CrossRef]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the Insulin-Like growth Factor-1 Receptor (IGF-1R) in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma and other soft tissue sarcoma. Results of a Sarcoma Alliance for Research through Collaboration (SARC) study. Cancer 2014, 120, 2448–2456. [Google Scholar] [PubMed] [Green Version]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Sarantopoulos, J.; Patnaik, A.; Papadopoulos, K.; Lin, C.C.; Rodon, J.; Murphy, B.; Roth, B.; McCaffery, I.; Gorski, K.S.; et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J. Clin. Oncol. 2009, 27, 5800–5807. [Google Scholar] [CrossRef] [PubMed]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the antiinsulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, K.P.; Romero, R.S.; Gonzalez, G.; Dix, J.E.; Lowy, I.; Furi, M. Anti-Hu-associated autoimmune limbic encephalitis in a patient with PD-1 inhibitor-responsive myxoid chondrosarcoma. Oncologist 2018, 23, 118–120. [Google Scholar] [CrossRef] [Green Version]
- Hollmann, T.J.; Hornick, J.L. INI1-deficient tumors: Diagnostic features and molecular genetics. Am. J. Surg. Pathol. 2011, 35, e47–e63. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Title | Phase | Status | Conditions | Age at Study Entry | ClinicalTrials.Gov Identifiers |
|---|---|---|---|---|---|
| Phase I–II trial of sunitinib plus nivolumab after standard treatment in advanced soft tissue and bone sarcomas | I–II | Recruiting | Advanced STS Advanced BS | 12–80 yrs | NCT03277924 |
| A phase 1 study of the EZH2 inhibitor tazemetostat in paediatric subjects with relapsed or refractory INI1-negative tumors or synovial sarcoma | I | Recruiting | Advanced SS Advanced INI1-negative tumors | 6 mos–18 yrs | NCT02601937 |
| A phase II, multicenter study of the EZH2 inhibitor tazemetostat in adult subjects with INI1-negative tumor or relapsed/refractory synovial sarcoma | II | Recruiting | Advanced SS Advanced INI1-negative tumors | ≥18 yrs | NCT02601950 |
| A phase II open-label trial of pazopanib administered as a single agent in patients with unresectable or metastatic solitary fibrous tumour (SFT) or extraskeletal myxoid chondrosarcoma (EMC) | II | Completed | Advanced SFT Advanced EMC | ≥18 yrs | NCT02066285 |
| A phase II trial of perifosine in patients with chemo-insensitive sarcomas: Sarcoma Alliance for Research through Collaboration (SARC) multi-center trial | II | Completed | Advanced CS Advanced ASPS Advanced EMC | ≥13 yrs | NCT00401388 |
| A Phase II trial of R1507, a recombinant human monoclonal antibody to the insulin-like growth factor-1 receptor for the treatment of participants with recurrent or refractory Ewing’s sarcoma, osteosarcoma, synovial sarcoma, rhabdomyosarcoma and other sarcomas | II | Completed | Advanced STS Advanced BS | ≥2 yrs (Ewing’s sarcoma cohort 2–21 yrs) | NCT00642941 |
| Pazopanib neoadjuvant trial in non-rhabdomyosarcoma soft tissue sarcomas (PAZNTIS): A phase II/III randomized trial of preoperative chemoradiation or preoperative radiation plus or minus pazopanib | II–III | Active, not recruiting | Localized STS | ≥2 yrs | NCT02180867 |
| A phase 1 study of doxorubicin and A12 (cixutumumab) in advanced soft tissue sarcoma | I | Completed | Advanced STS | ≥16 yrs | NCT00720174 |
| A phase 1B/II study of GDC-0449 (NSC 747691-vismodegib) in combination with RO4929097, a gamma-secretase Inhibitor (GSI) in advanced/metastatic sarcomas | I–II | Completed | Advanced STS | ≥18 yrs | NCT01154452 |
| A randomized, double-blind phase II, study of Gemcitabine alone or in combination with pazopanib for refractory soft tissue sarcoma | II | Active, not recruiting | Advanced STS | ≥18 yrs | NCT01532687 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stacchiotti, S.; Baldi, G.G.; Morosi, C.; Gronchi, A.; Maestro, R. Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management. Cancers 2020, 12, 2703. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092703
Stacchiotti S, Baldi GG, Morosi C, Gronchi A, Maestro R. Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management. Cancers. 2020; 12(9):2703. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092703
Chicago/Turabian StyleStacchiotti, Silvia, Giacomo Giulio Baldi, Carlo Morosi, Alessandro Gronchi, and Roberta Maestro. 2020. "Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management" Cancers 12, no. 9: 2703. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092703