
Gastrointestinal Manifestations in Systemic Mastocytosis: The Need of a Multidisciplinary Approach

 ,
,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mast Cells and Mast Cell-Related Disorders
2.1. Mast Cell Physiology
2.2. Mast Cell Activation Syndrome: Consensus Criteria and Classification
- typical clinical features of severe, acute systemic MCA (especially signs and symptoms of anaphylaxis);
- increase in serum total tryptase level by at least 20% above baseline plus 2 ng/mL during or within four hours after a symptomatic period;
- response of symptoms to MC blocking agents. MCA symptoms may range from mild to severe and sometimes life-threatening, correlating with the extent of mediator release from MCs [4].
2.3. 2017 WHO Classification of Mastocytosis
2.4. 2017 WHO Diagnostic Criteria of Mastocytosis
2.5. “B” and “C” Findings and 2017 WHO Classification of Mastocytosis
2.6. Symptoms and Signs in Systemic Mastocytosis
2.7. Gastrointestinal Symptomatology in Systemic Mastocytosis
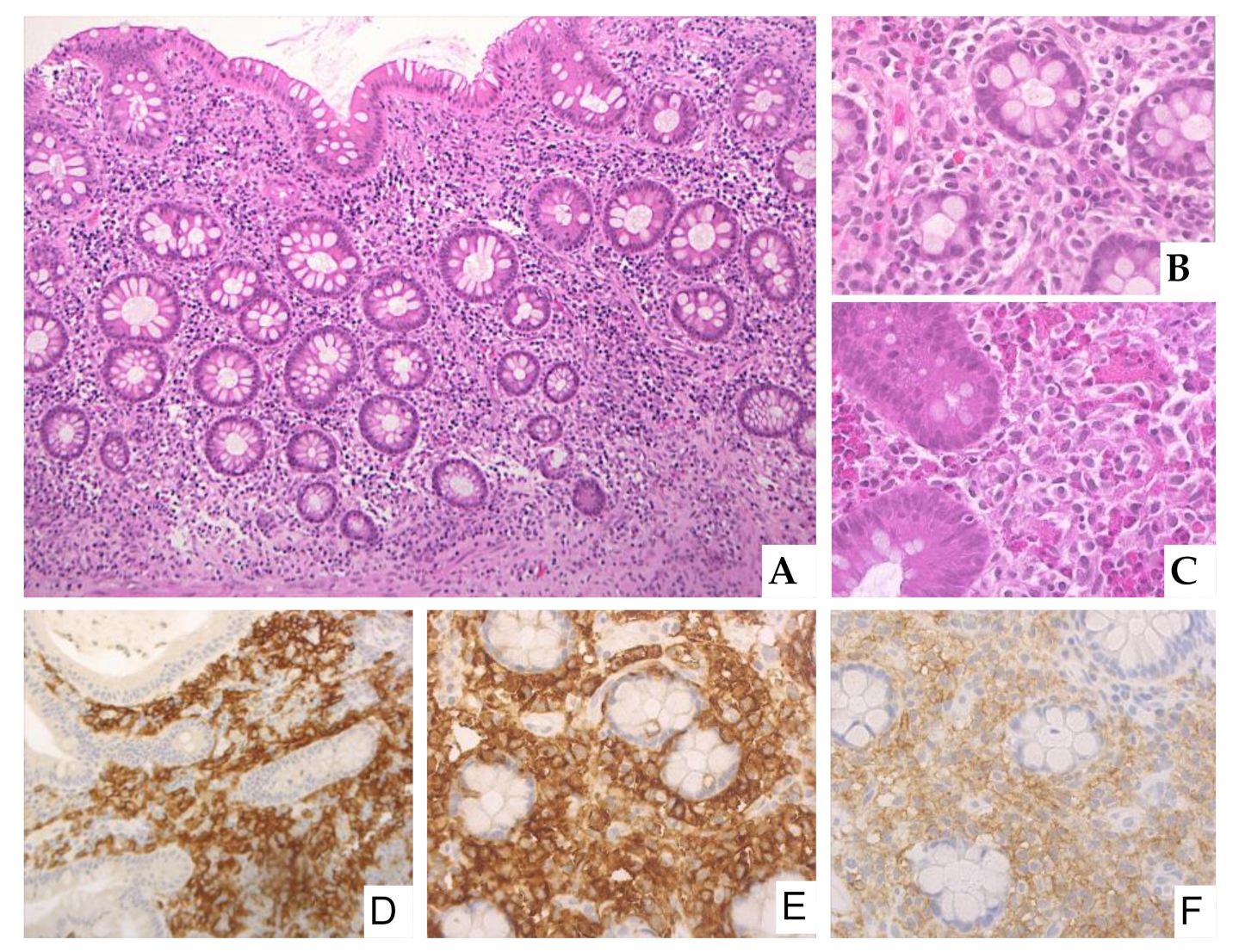
2.8. Gastrointestinal Involvement by Systemic Mastocytosis: Endoscopic and Histological Features
2.9. Diagnostic Issues
2.10. Molecular Features
2.11. Treatment
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P.; Akin, C.; Arock, M.; Brockow, K.; Butterfield, J.H.; Carter, M.C.; Castelss, M.; Escribano, L.; Hartmann, K.; Lieberman, P.; et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: A consensus proposal. Int. Arch. Allergy Immunol. 2012, 157, 215–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valent, P.; Akin, C.; Bonadonna, P.; Hartmann, K.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Siebenhaar, F.; Sperr, W.R.; Elberink, J.N.O.; et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J. Allergy Clin. Immunol. Pr. 2019, 7, 1125–1133.e1. [Google Scholar] [CrossRef]
- Horny, H.P.; Akin, C.; Arber, D.A.; Peterson, L.C.; Tefferi, A.; Metcalfe, D.D.; Bennet, J.M.; Bain, B.J.; Escribano, L.; Valent, P. WHO Classification of Tumours Haematopoietic and Lymphoid Tissues, Revised 4th ed.; WHO Classification of Tumours Editorial Board, Ed.; IARC: Lyon, France, 2017; pp. 62–69. [Google Scholar]
- Kirsch, R.; Geboes, K.; Shepherd, N.A.; de Hertogh, G.; Di Nicola, N.; Lebel, S.; Mickys, U.; Riddell, R.H. Systemic mastocytosis involving the gastrointestinal tract: Clinicopathologic and molecular study of five cases. Mod. Pathol. 2008, 21, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.T. Gastrointestinal abnormalities and involvement in systemic mastocytosis. Hematol. Oncol. Clin. 2000, 14, 579–623. [Google Scholar] [CrossRef]
- Ramsay, D.B.; Stephen, S.; Borum, M.; Voltaggio, L.; Doman, D.B. Mast Cells in Gastrointestinal Disease. Gastroenterol. Hepatol. 2010, 6, 772–777. [Google Scholar]
- Sokol, H.; Georgin-Lavialle, S.; Grandpeix-Guyodo, C.; Canioni, D.; Barete, S.; Dubreuil, P.; Lortholary, O.; Beaugerie, L.; Hermine, O. Gastrointestinal involvement and manifestations in systemic mastocytosis. Inflamm. Bowel Dis. 2010, 16, 1247–1253. [Google Scholar] [CrossRef]
- Siegert, S.I.; Diebold, J.; Ludolph-Hauser, D.; Lohrs, U. Are gastrointestinal mucosal mast cells increased in patients with systemic mastocytosis? Am. J. Clin. Pathol. 2004, 122, 560–565. [Google Scholar] [CrossRef]
- Nanagas, V.C.; Kovalszki, A. Gastrointestinal Manifestations of Hypereosinophilic Syndromes and Mast Cell Disorders: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2019, 57, 194–212. [Google Scholar] [CrossRef]
- Gülen, T.; Akin, C. Pharmacotherapy of mast cell disorders. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 295–303. [Google Scholar] [CrossRef]
- Doyle, L.A.; Sepehr, G.J.; Hamilton, M.J.; Akin, C.; Castells, M.C.; Hornick, J.L. A Clinicopathologic Study of 24 Cases of Systemic Mastocytosis Involving the Gastrointestinal Tract and Assessment of Mucosal Mast Cell Density in Irritable Bowel Syndrome and Asymptomatic Patients. Am. J. Surg. Pathol. 2014, 38, 832–843. [Google Scholar] [CrossRef] [Green Version]
- Zanelli, M.; Ricci, S.; Zizzo, M.; Sanguedolce, F.; De Giorgi, F.; Palicelli, A.; Martino, G.; Ascani, S. Systemic Mastocytosis Associated with “Smoldering” Multiple Myeloma. Diagnostics 2021, 11, 88. [Google Scholar] [CrossRef]
- Sokol, H.; Georgin-Lavialle, S.; Canioni, D.; Barete, S.; Damaj, G.; Soucie, E.; Bruneau, J.; Chandesris, M.-O.; Suarez, F.; Launay, J.-M.; et al. Gastrointestinal manifestations in mastocytosis: A study of 83 patients. J. Allergy Clin. Immunol. 2013, 132, 866–873. [Google Scholar] [CrossRef]
- Joris, M.; Georgin-Lavialle, S.; Chandesris, M.-O.; Lhermitte, L.; Claisse, J.-F.; Canioni, D.; Hanssens, K.; Damaj, G.; Hermine, O.; Hamidou, M. Mast Cell Leukaemia: C-KIT Mutations Are Not Always Positive. Case Rep. Hematol. 2012, 1–6. [Google Scholar] [CrossRef]
- Travis, W.D.; Li, C.Y.; Bergstralh, E.J.; Yam, L.T.; Swee, R.G. Systemic mast cell disease. Analysis of 58 cases and literature review. Medicine 1988, 67, 345–368. [Google Scholar] [CrossRef]
- Cherner, J.A.; Jensen, R.T.; Dubois, A.; O’Dorisio, T.M.; Gardner, J.D.; Metcalfe, D.D. Gastrointestinal dysfunction in systemic mastocytosis: A prospective study. Gastroenterology 1988, 95, 657–667. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Lhermitte, L.; Dubreuil, P.; Chandesris, M.-O.; Hermine, O.; Damaj, G. Mast cell leukemia. Blood 2013, 121, 1285–1295. [Google Scholar] [CrossRef] [Green Version]
- Hadjivasilis, A.; Joakim, K.J.; Neocleous, A.; Demetriou, K.; Paniivar, S.; Iacovu, F.; Michaelides, D.; Potamitis, G. Indolent systemic mastocytosis mimicking Crohn’s disease. Ann. Gastroenterol. 2019, 32, 208–210. [Google Scholar] [CrossRef]
- Zanelli, M.; Pai, R.K.; Zorzi, M.G.; Zizzo, M.; Martino, G.; Morelli, L.; Parisi, A.; De Marco, L.; Annessi, V.; Ascani, S. Gastrointestinal Mastocytosis: A Potential Diagnostic Pitfall to Be Aware. Int. J. Surg. Pathol. 2019, 27, 643–646. [Google Scholar] [CrossRef]
- Sperr, W.R.; Jordan, J.H.; Fiegl, M.; Escribano, L.; Bellas, C.; Dirhofer, S.; Semper, H.; Simontisch-Klupp, I.; Horny, H.P.; Valent, P. Serum tryptase levels in patients with mastocytosis: Correlation with mast cell burden and implication for defining the category of disease. Int. Arch. Allergy Immunol. 2002, 128, 136–141. [Google Scholar] [CrossRef]
- Zanelli, M.; Negro, A.; Santi, R.; Bisagni, A.; Ragazzi, M.; Ascani, S.; De Marco, L. Letter: Sprue-like enteropathy associated with angiotensin II receptor blockers other than olmesartan. Aliment. Pharmacol. Ther. 2017, 46, 471–473. [Google Scholar] [CrossRef] [Green Version]
- Negro, A.; De Marco, L.; Cesario, V.; Santi, R.; Boni, M.C.; Zanelli, M. A case of moderate sprue-like enteropathy associated with telmisartan. J. Clin. Med. Res. 2017, 9, 1022–1025. [Google Scholar] [CrossRef] [Green Version]
- Zanelli, M.; Ragazzi, R.; Fiorino, S.; Foroni, M.; Cecinato, P.; Sanchez, J.; Ascani, S.; De Marco, L. An Italian case of intestinal anisakiasis with a presurgical diagnosis: Could this parasite represent an emerging disease? Pathol. Res. Pract. 2017, 213, 558–564. [Google Scholar] [CrossRef]
- Johncilla, M.; Jessurun, J.; Brown, I.; Hornick, J.L.; Bellizzi, A.; Shia, J.; Yantiss, R.K. Are Enterocolic Mucosal Mast Cell Aggregates Clinically Relevant in Patients Without Suspected or Established Systemic Mastocytosis? Am. J. Surg. Pathol. 2018, 42, 1390–1395. [Google Scholar] [CrossRef]
- Lyons, J.J.; Sun, G.; Stone, K.D.; Nelson, C.; Wisch, L.; O’Brien, M. Mendelian inheritance of elevated serum tryptase associated with atopy and connective tissue abnormalities. J. Allergy Clin. Immunol. 2014, 133, 1471–1474. [Google Scholar] [CrossRef] [Green Version]
- Giannetti, A.; Filice, E.; Caffarelli, C.; Ricci, G.; Pession, A. Mast Cell Activation Disorders. Medicina 2021, 57, 124. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Zhao, M.; Giannetti, M.P.; Weller, E.; Hufdhi, R.; Novak, P.; Mendoza-Alvarez, L.B.; Hornick, J.; Lyons, J.J.; Glover, S.C.; et al. Distinct Small Intestine Mast Cell Histologic Changes in Patients With Hereditary Alpha-tryptasemia and Mast Cell Activation Syndrome. Am. J. Surg. Pathol. 2021, 45, 997–1004. [Google Scholar] [CrossRef]
- Wang, S.A.; Hutchinson, L.; Tang, G.; Chen, S.S.; Miron, P.M.; Huh, Y.O.; Jones, D.M.; Bueso-Ramos, C.; Verstovsek, S.; Medeiros, L.J.; et al. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: Clinical significance and comparison of chromosomal abnormalities in SM and AHNMD components. Am. J. Hematol. 2013, 88, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Ghosh, J.; Kapur, R. Mastocytosis: A mutated KIT receptor induced myeloproliferative disorder. Oncotarget 2015, 6, 18250–18264. [Google Scholar] [CrossRef]
- Kristensen, T.; Vestergaard, H.; Møller, M.B. Improved Detection of the KIT D816V Mutation in Patients with Systemic Mastocytosis Using a Quantitative and Highly Sensitive Real-Time qPCR Assay. J. Mol. Diagn. 2011, 13, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Scherber, R.M.; Borate, U. How we diagnose and treat systemic mastocytosis in adults. Br. J. Haematol. 2017, 180, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Galura, G.M.; Cherukuri, S.V.; Hakim, N.; Gaur, S.; Orazi, A. Acute aleukemic mast cell leukemia: Report of a case and review of the literature. Leuk. Res. Rep. 2020, 14, 100230. [Google Scholar] [CrossRef] [PubMed]
- Reiter, A.; George, T.I.; Gotlib, J.R. New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood 2020, 135, 1365–1376. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Pardani, A. How I treat patients with indolent and smoldering mastocytosis (rare conditions difficult to manage). Blood 2013, 20, 2013–3094. [Google Scholar]
- Pardani, A. Systemic mastocytosis in adults: 2021 update on diagnosis, risk stratification and management. Am. J. Hematol. 2021, 96, 508–525. [Google Scholar] [CrossRef]
- Horan, R.F.; Sheffer, A.L.; Austen, K. Cromolyn sodium in the management of systemic mastocytosis. J. Allergy Clin. Immunol. 1990, 85, 852–855. [Google Scholar] [CrossRef]
- Akin, C. Mast cell activation syndromes. J. Allergy Clin. Immunol. 2017, 140, 349–355. [Google Scholar] [CrossRef] [Green Version]
- González-De-Olano, D.; Matito, A.; Orfao, A.; Escribano, L. Advances in the understanding and clinical management of mastocytosis and clonal mast cell activation syndromes. F1000Research 2016, 5, 2666. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, D.D. The treatment of mastocytosis: An overview. J. Investig. Dermatol. 1991, 96, 55S–56S. [Google Scholar] [CrossRef] [Green Version]
- Gilreath, J.; Tchertanov, L.; Deininger, M. Novel approaches to treating advanced systemic mastocytosis. Clin. Pharmacol. Adv. Appl. 2019, 11, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Ustun, C.; Reiter, A.; Scott, B.L.; Nakamura, R.; Damaj, G.; Kreil, S.; Shanley, R.; Hogan, W.J.; Perales, M.A.; Shore, T.; et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J. Clin. Oncol. 2014, 32, 3264–3274. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Advances in the classification and treatment of mastocytosis: Current status and outlook toward the future. Cancer. Res. 2017, 77, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Mital, A.; Piskorz, A.; Lewandowski, K.; Wasag, B.; Limon, J.; Hellmann, A. A case of mast cell leukemia with exon 9 KIT mutation and good response to imatinib. Eur. J. Hematol. 2011, 86, 531–535. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Lubke, J.; Naumann, N.; Kluger, S.; Schwaab, J.; Metzgeroth, G.; Evans, E.; Gardino, A.K.; Lengauer, C.; Hofmann, W.K.; Farius, A.; et al. Inhibitory effects of midostaurin and avapritinib on myeloid progenitor cells derived from patients with KIT D816V positive advanced systemic mastocytosis. Leukemia 2019, 33, 1195–1205. [Google Scholar] [CrossRef]
- Winger, B.A.; Cortopassi, W.A.; Ruiz, D.G.; Ding, L.; Jang, K.; Leyte-Vidal, A.; Zhang, N.; Esteve-Puig, R.; Jacobson, M.P.; Shah, N.P. ATP-Competitive Inhibitors Midostaurin and Avapritinib Have Distinct Resistance Profiles in Exon 17-Mutant KIT. Cancer Res. 2019, 79, 4283–4292. [Google Scholar] [CrossRef]
- Casassus, P.; Caillat-Vigneron, N.; Martin, A.; Simon, J.; Gallais, V.; Beaudry, P.; Eclache, V.; Laroche, L.; Lortholary, P.; Raphael, M.; et al. Treatment of adult systemic mastocytosis with interfero-alpha: Results of a multicenter phase II trial on 20 patients. Br. J. Haematol. 2002, 119, 1090–1097. [Google Scholar] [CrossRef]
- Barete, S.; Lortholary, O.; Damaj, G.; Hirsch, I.; Chandesris, M.-O.; Elie, C.; Hamidou, M.; Durieu, I.; Suarez, F.; Grosbois, B.; et al. Long-term efficacy and safety of cladribine (2-CdA) in adult patients with mastocytosis. Blood 2015, 126, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Ustun, C.; Gotlib, J.; Popat, U.; Artz, A.; Litzow, M.; Reiter, A.; Nakamura, R.; Kluin-Nelemans, H.C.; Verstovsek, S.; Gajewski, J.; et al. Consensus opinion on allogenenic hematopoietic cell transplantation in advanced system mastocytosis. Biol. Blood Marrow Transplant. 2016, 22, 1348–1356. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T.; Huntly, B. Targeting Epigenetic Readers in Cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Zanelli, M.; Ciarrocchi, A.; De Petris, G.; Zizzo, M.; Costantini, M.; Bisagni, A.; Torricelli, F.; Nicoli, D.; Ramundo, D.; Ricci, S.; et al. Acute Radiation Colitis after Preoperative Short-Course Radiotherapy for Rectal Cancer: A Morphological, Immunohistochemical and Genetic Study. Cancers 2020, 12, 2571. [Google Scholar] [CrossRef]
- Hodges, K.; Kennedy, L.; Meng, F.; Alpini, G.; Francis, H. Mast cells, disease and gastrointestinal cancer: A comprehensive review of recent findings. Transl. Gastrointest. Cancer 2012, 1, 138–150. [Google Scholar]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Metcalfe, D.D.; Mekori, Y.A. Pathogenesis and Pathology of Mastocytosis. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 487–514. [Google Scholar] [CrossRef]
- Broesby-Olsen, S.; Farkas, D.K.; Vestergaard, H.; Hermann, A.P.; Møller, M.B.; Mortz, C.G.; Kristensen, T.K.; Bindslev-Jensen, C.; Sørensen, H.T.; Frederiksen, H. Risk of solid cancer, cardiovascular disease, anaphylaxis, osteoporosis and fractures in patients with systemic mastocytosis: A nationwide population-based study. Am. J. Hematol. 2016, 91, 1069–1075. [Google Scholar] [CrossRef]
- Shivarov, V.; Gueorguieva, R.; Ivanova, M.; Stoimenov, A. Incidence of second solid cancers in mastocytosis patients: A SEER database analysis. Leuk. Lymphoma 2017, 59, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Guilarte, M.; Santos, J.; de Torres, I.; Alonso, C.; Vicario, M.; Ramos, L.; Martinez, C.; Casellas, F.; Saperas, E.; Malegelada, J.R. Diarrhoea-predominant IBS patients show mast cell activation and hyperplasia in the jejunum. Gut 2007, 56, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, A.; Perez Ingles, D.; Myneedu, K.; Deoker, A.; Sarosiek, I.; Zuckerman, M.J.; Schmulson, M.J.; Bashashati, M. Mast cells are increased in the small intestinal mucosa of patients with irritable bowel syndrome: A systematic review and meta-analysis. Neurogastroenterol. Motil. 2019, 31, e13718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, A.; Jain, D.; Roland, B.C.; Kinzel, J.; Gibson, J.; Schrader, R.; Hanso, J.A. Performing colonic mast cell counts in patients with chronic diarrhea of unknown etiology has limited diagnostic use. Arch. Pathol. Lab. Med. 2015, 139, 225–232. [Google Scholar] [CrossRef]


{kind=link}
| Cutaneous mastocytosis |
|
| Systemic mastocytosis (SM) |
|
| Mast cell sarcoma Unifocal mast cell tumor. No evidence of SM. Destructive growth pattern. High grade cytology |
| The diagnosis of SM can be made when the major criterion and one minor criterion or at least 3 minor criteria are fulfilled |
| Major criterion |
|
| Minor criteria |
|
| “B” findings |
|
| “C” findings |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanelli, M.; Pizzi, M.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Soriano, A.; Bisagni, A.; Martino, G.; Caprera, C.; Moretti, M.; et al. Gastrointestinal Manifestations in Systemic Mastocytosis: The Need of a Multidisciplinary Approach. Cancers 2021, 13, 3316. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133316
Zanelli M, Pizzi M, Sanguedolce F, Zizzo M, Palicelli A, Soriano A, Bisagni A, Martino G, Caprera C, Moretti M, et al. Gastrointestinal Manifestations in Systemic Mastocytosis: The Need of a Multidisciplinary Approach. Cancers. 2021; 13(13):3316. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133316
Chicago/Turabian StyleZanelli, Magda, Marco Pizzi, Francesca Sanguedolce, Maurizio Zizzo, Andrea Palicelli, Alessandra Soriano, Alessandra Bisagni, Giovanni Martino, Cecilia Caprera, Marina Moretti, and et al. 2021. "Gastrointestinal Manifestations in Systemic Mastocytosis: The Need of a Multidisciplinary Approach" Cancers 13, no. 13: 3316. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133316