
Development of Multidrug Resistance in Acute Myeloid Leukemia Is Associated with Alterations of the LPHN1/GAL-9/TIM-3 Signaling Pathway

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples of Patients with MDS
2.2. Cell Lines and Cultivation Conditions
2.3. Determination of ADGRL1, LGALS9, HAVCR2, CD44, ABCB1, ABCC1 and ACTB Transcript Expression in Cell Lines and Patient Samples by RT-PCR and qRT-PCR
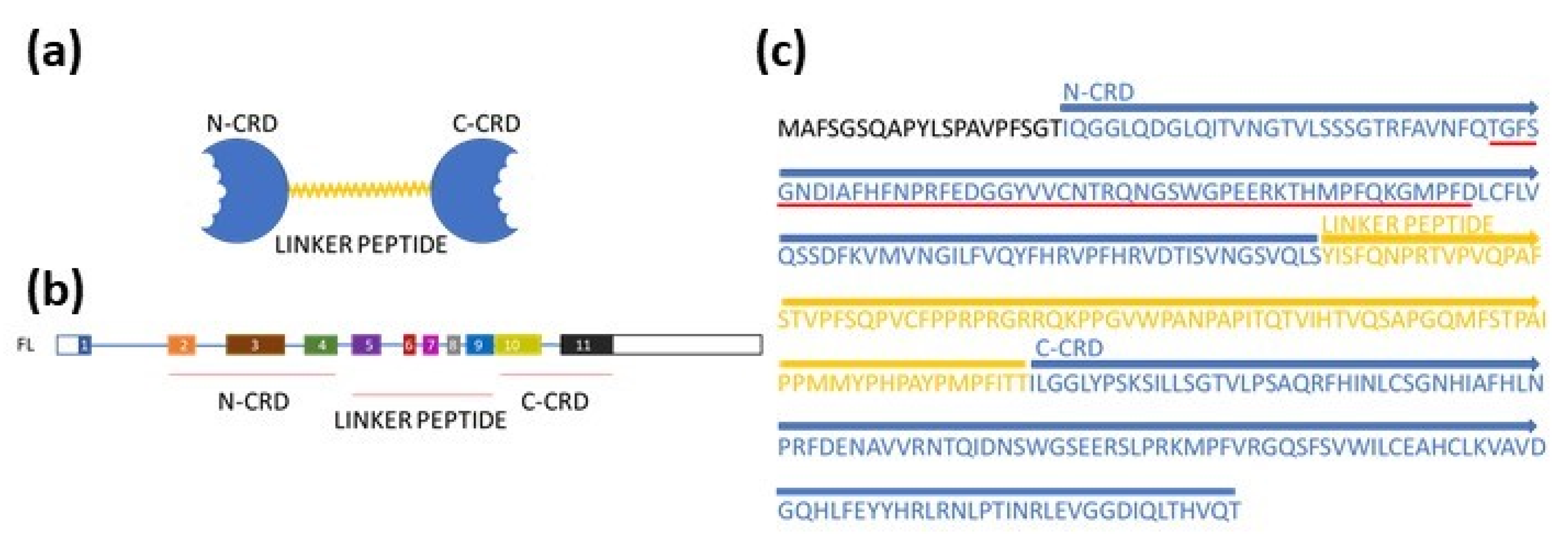
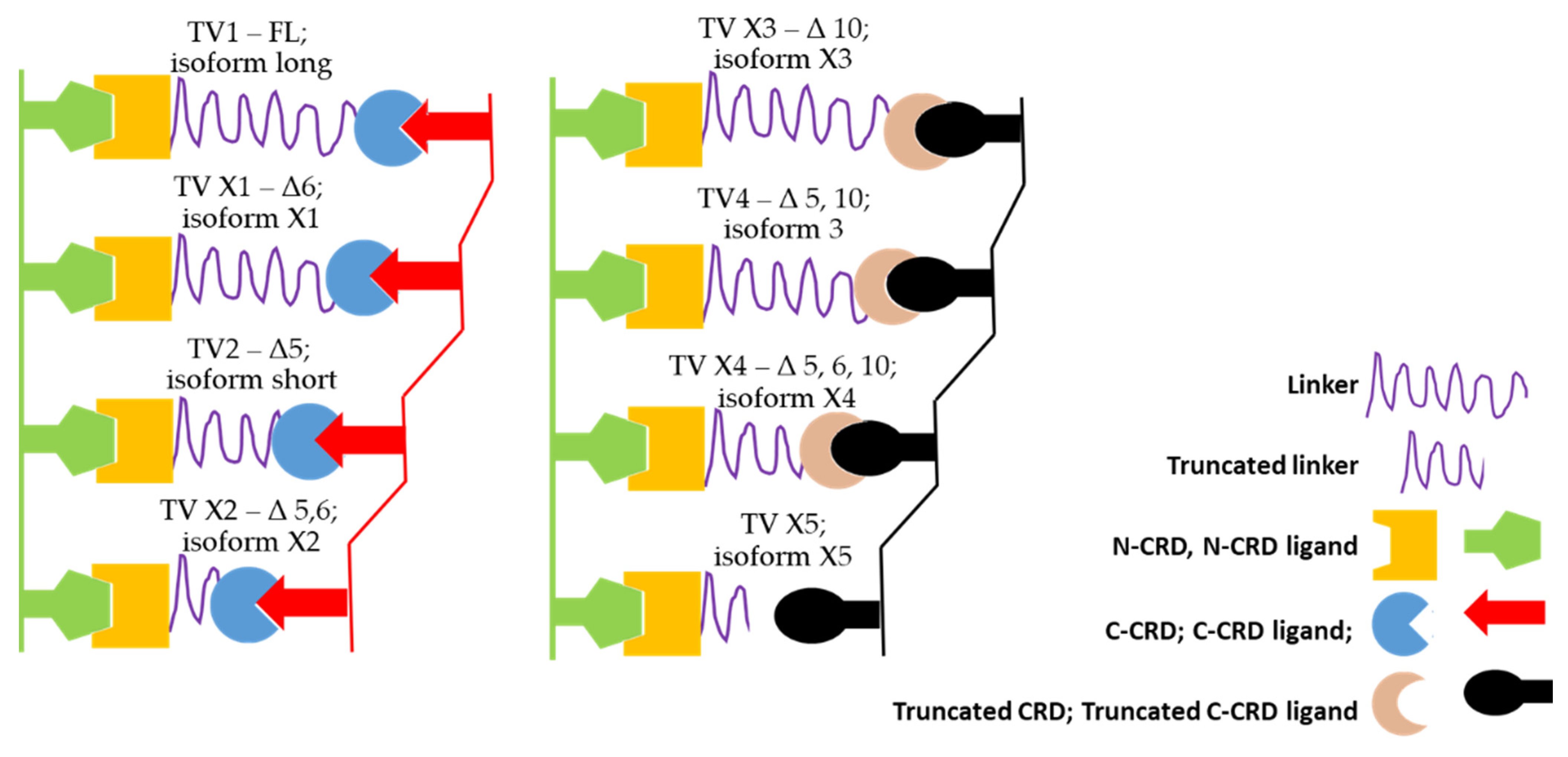
2.4. Alternative Splicing of LGALS9 and Dataset of Splice Variants
2.5. Detection of Proteins by Western Blotting
2.6. Statistical Analysis
3. Results
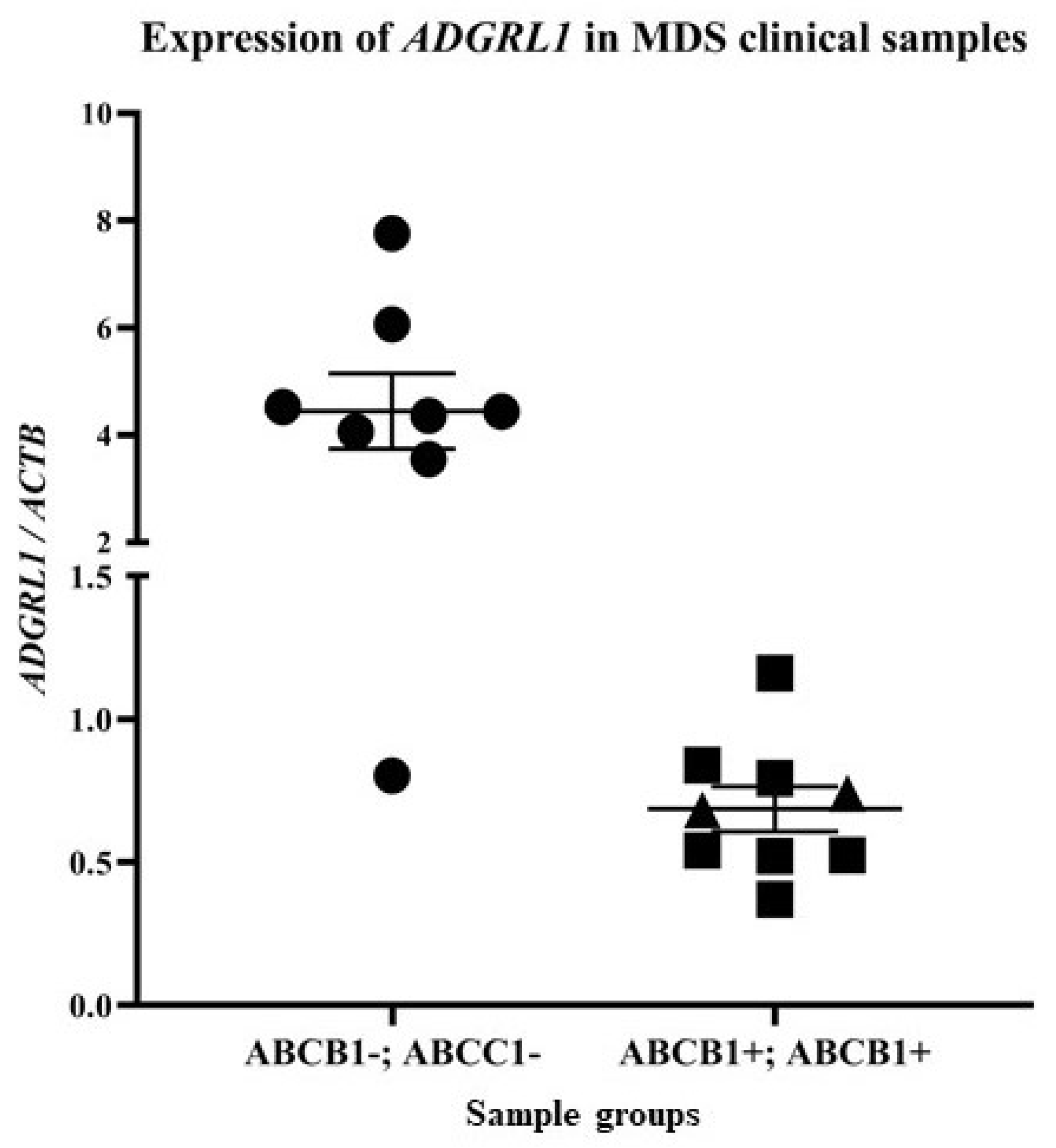
3.1. Latrophilin-1 Is Downregulated in Clinical Samples of Myelodysplastic Syndrome (MDS) with ABCB1 and ABCC1 Expression
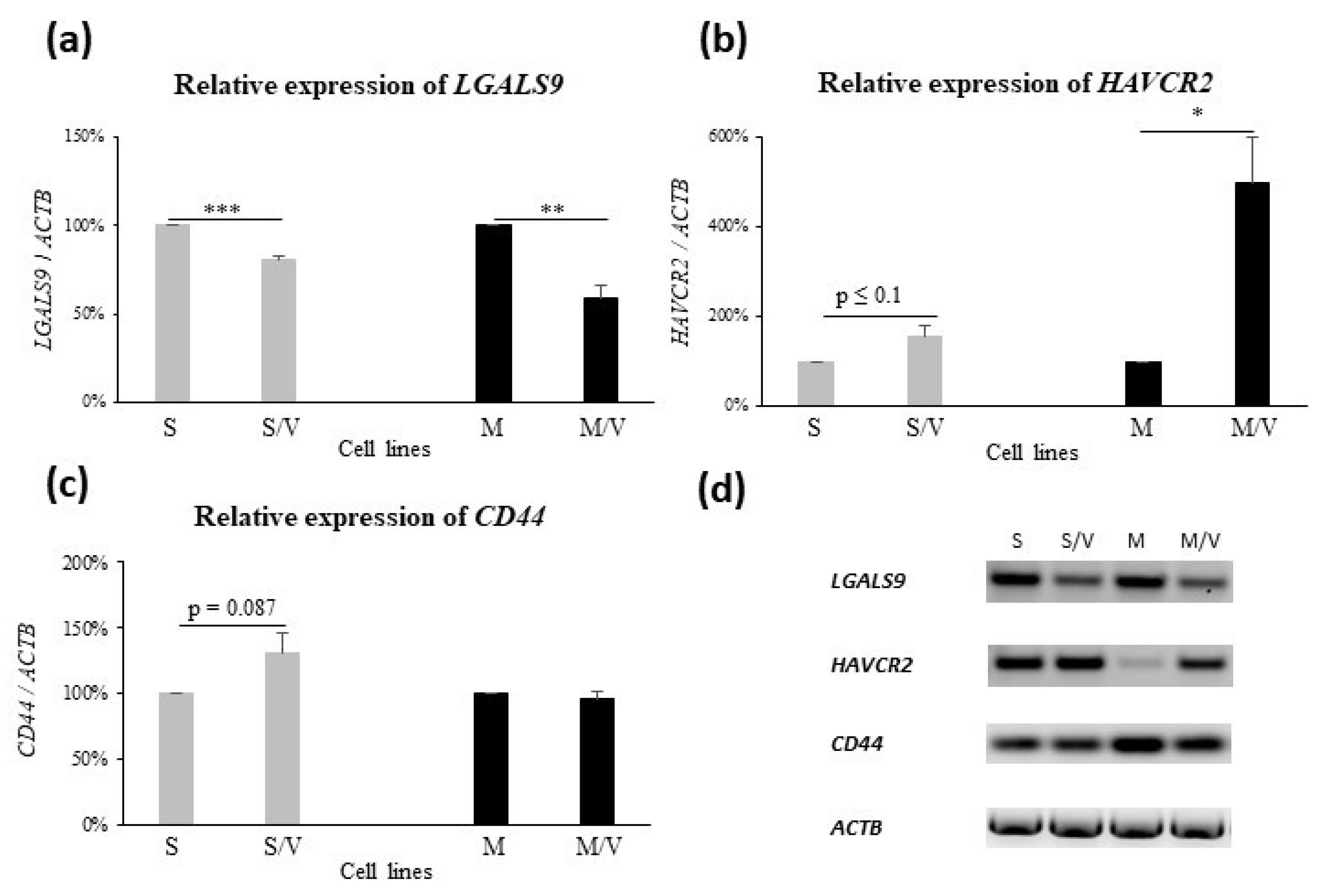
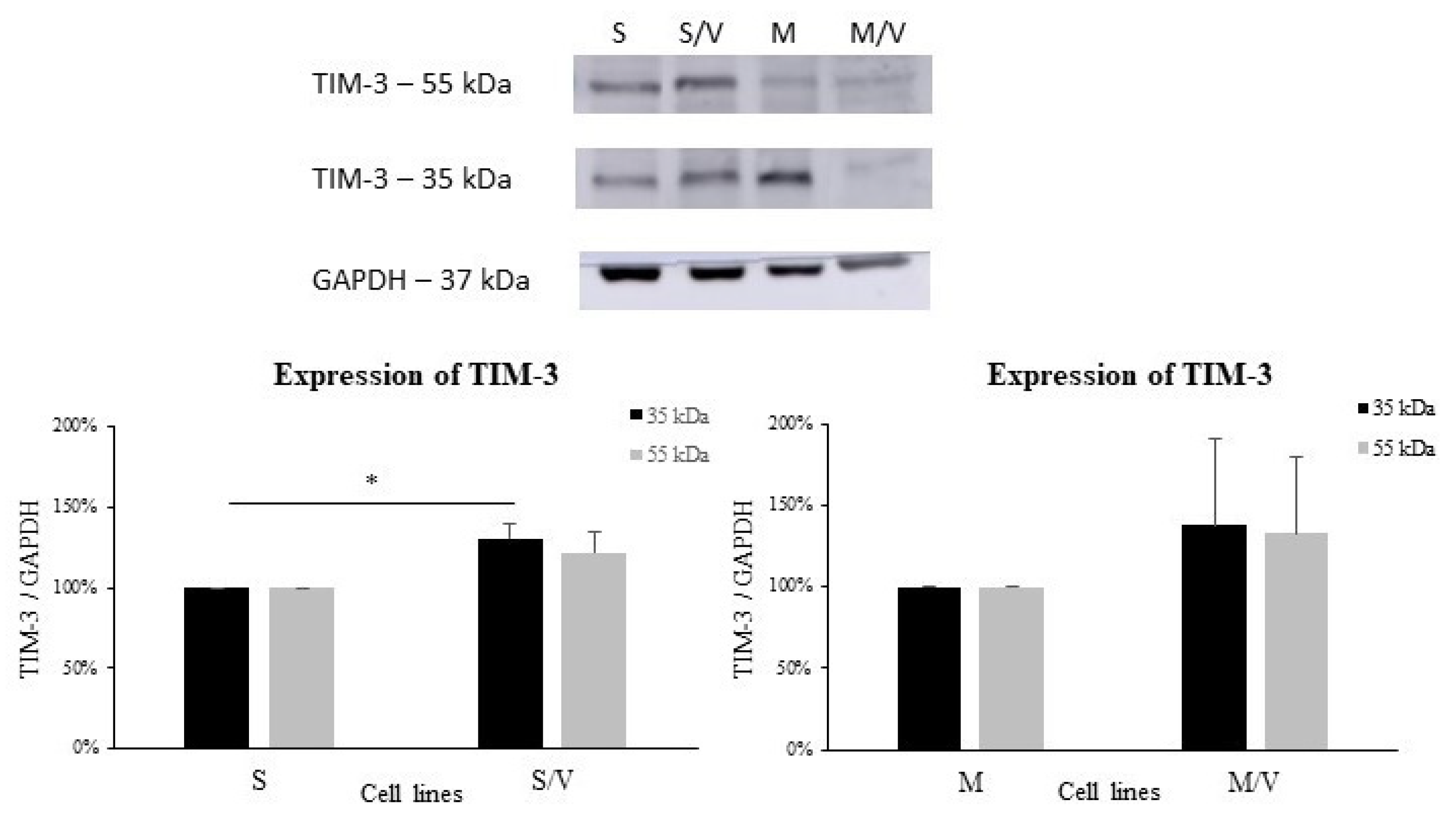
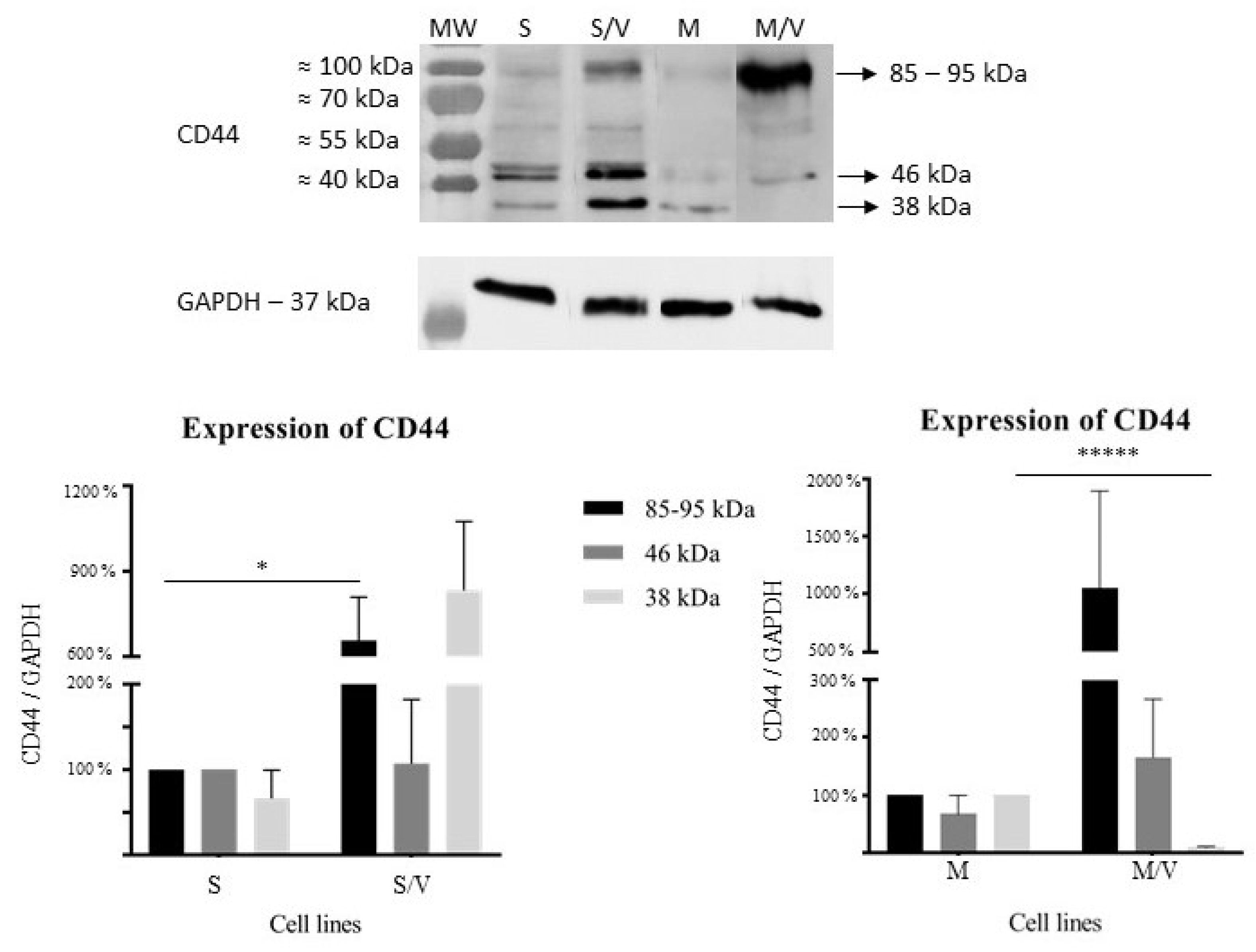
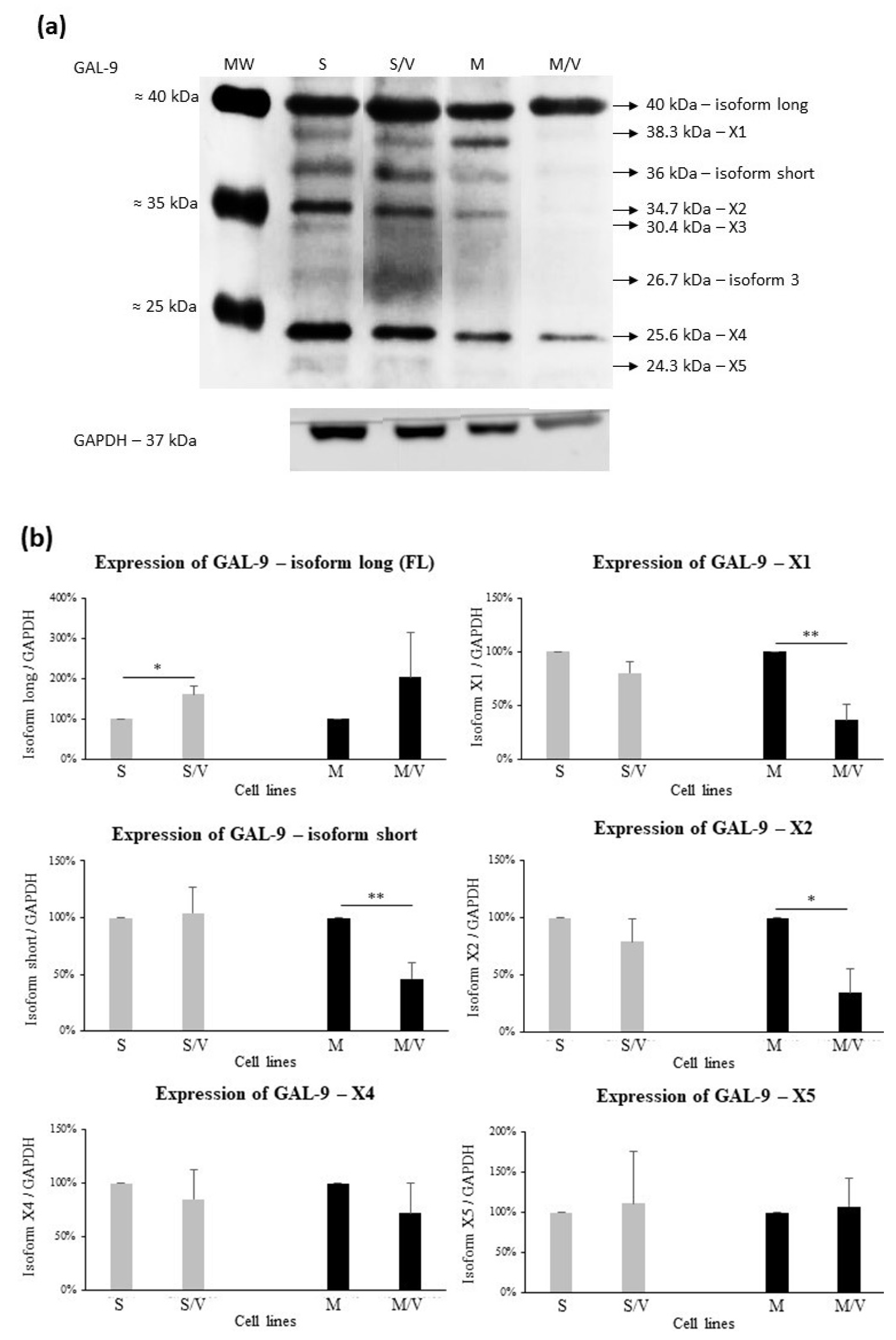
3.2. Expression of GAL-9, TIM-3, CD44, ABCB1 and ABCC1 in SKM-1 and MOLM-13 Cells and Their Resistant Counterparts
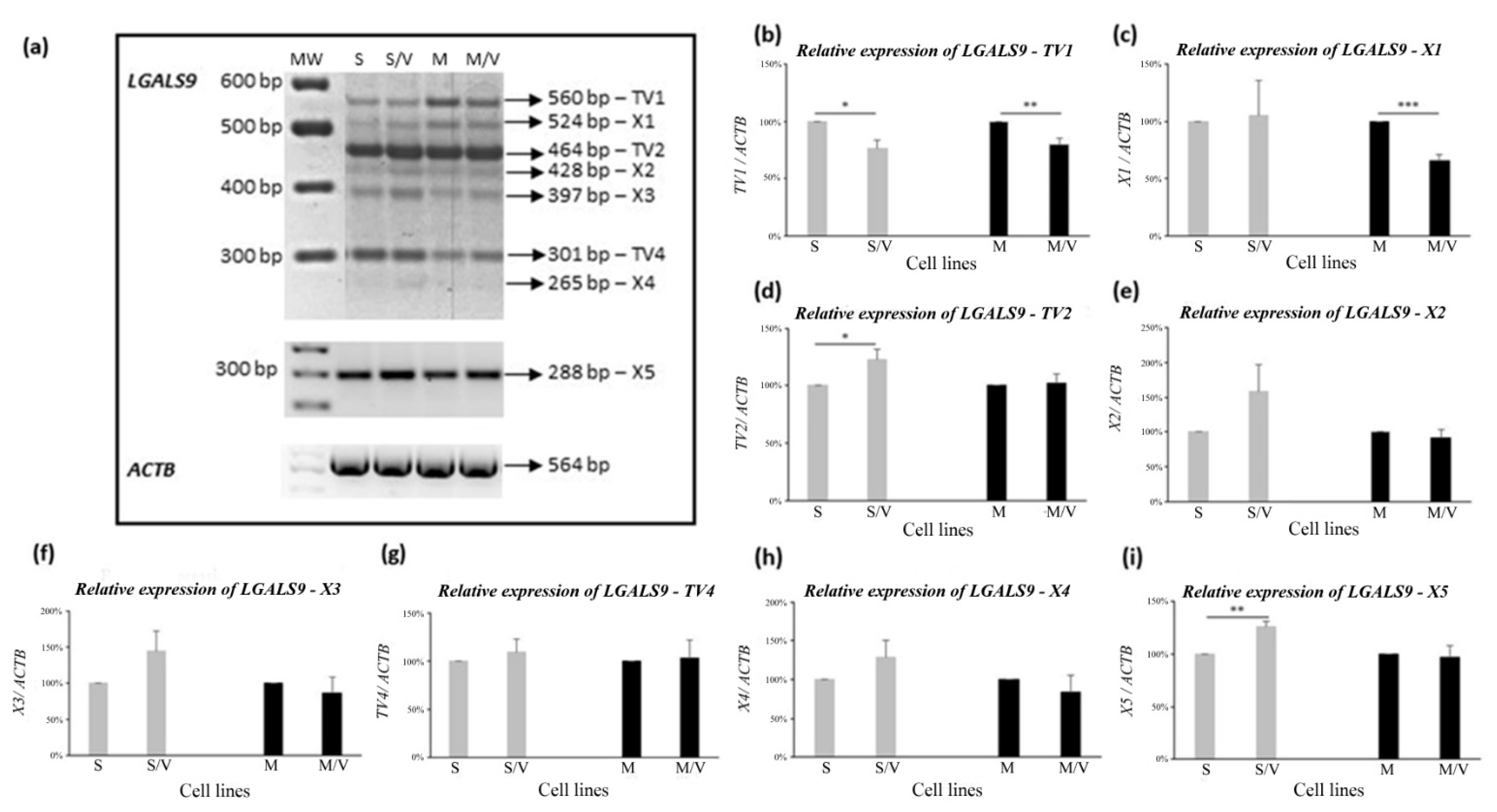
3.3. Alternative Splicing of LGALS9 Pre-mRNA in SKM-1 and MOLM-13 Cells
4. Discussion
- The induction of transcription may not be the sole regulatory mechanism for GAL-9 expression in AML cells; other mechanisms may be involved. The level of translation can be significantly limited by RNA interference, e.g., microRNA-22 decreases GAL-9 levels [65]. There is considerable ABCB1 expression in SKM-1/VCR and MOLM-13/VCR cells, and it has been shown that mRNA turnover and translation initiation may play a crucial role in its expression, not just the induction of transcription [66]. It is possible that the expression of other genes in ABCB1-positive cells may behave similarly. The protein level in cells can be controlled by ubiquitination and degradation in the proteasome. This pathway may be differentially active in ABCB1-positive and ABCB1-negative leukemia cells [67]. Different levels of proteasomal degradation of individual GAL-9 isoforms can also be expected.
- There are lines of evidence that GAL-9 is secreted into the ECM, where it might play a role in intercellular junctions [45,68]. GAL-9 affects AML cells through an autocrine loop with TIM-3, leading to activation of survival signaling pathways and directing the ability of AML blasts to self-renew [31] and/or affect immune cells, thus participating in the mechanisms by which AML blasts escape control of the immune system [24,69].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Bhatt, V.; Bixby, D.; Blum, W.; Coutre, S.E.; et al. Acute myeloid leukemia, version 3.2017, nccn clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 926–957. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Green, S.D.; Konig, H. Treatment of acute myeloid leukemia in the era of genomics-achievements and persisting challenges. Front. Genet. 2020, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.U.; Seipel, K.; Bacher, U.; Pabst, T. Autologous transplantation for older adults with aml. Cancers 2018, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Jones, R.J. Acute myeloid leukemia stem cell heterogeneity and its clinical relevance. In Stem Cells Heterogeneity in Cancer; Birbrair, A., Ed.; Springer Nature Switzerland AG: Cham, Switzerland, 2019; Volume 1139, pp. 153–169. [Google Scholar]
- Ossenkoppele, G.J.; Breems, D.A.; Stuessi, G.; van Norden, Y.; Bargetzi, M.; Biemond, B.J.; Borne, P.A.V.D.; Chalandon, Y.; Cloos, J.; Deeren, D.; et al. Lenalidomide added to standard intensive treatment for older patients with aml and high-risk mds. Leukemia 2020, 34, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulos, P.; Zervakis, K.; Papadopoulou, V.; Iliakis, T.; Kalala, F.; Giannakopoulou, N.; Rougala, N.; Galanopoulos, A.; Bakarakos, P.; Variami, E.; et al. 5-azacytidine in the treatment of intermediate-2 and high-risk myelodysplastic syndromes and acute myeloid leukemia. A five-year experience with 44 consecutive patients. Anticancer Res. 2015, 35, 5141–5147. [Google Scholar]
- Janotka, L.; Messingerova, L.; Simonicova, K.; Kavcova, H.; Elefantova, K.; Sulova, Z.; Breier, A. Changes in apoptotic pathways in molm-13 cell lines after induction of resistance to hypomethylating agents. Int. J. Mol. Sci. 2021, 22, 2076. [Google Scholar] [CrossRef]
- Bertaina, A.; Vinti, L.; Strocchio, L.; Gaspari, S.; Caruso, R.; Algeri, M.; Coletti, V.; Gurnari, C.; Romano, M.; Cefalo, M.G.; et al. The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br. J. Haematol. 2017, 176, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazandjian, D.; Blumenthal, G.M.; Yuan, W.; He, K.; Keegan, P.; Pazdur, R. Fda approval of gefitinib for the treatment of patients with metastatic egfr mutation-positive non-small cell lung cancer. Clin. Cancer Res. 2016, 22, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Prebet, T.; Vey, N. Vorinostat in acute myeloid leukemia and myelodysplastic syndromes. Expert Opinion on Investigational Drugs 2011, 20, 287–295. [Google Scholar] [CrossRef]
- Blum, W.G.; Mims, A.S. Treating acute myeloid leukemia in the modern era: A primer. Cancer 2020, 126, 4668–4677. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.; Briz, O.; Rodriguez-Macias, G.; Diez-Martin, J.L.; Macias, R.I. Role of drug transport and metabolism in the chemoresistance of acute myeloid leukemia. Blood Rev. 2016, 30, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [Green Version]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med. Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Coculova, M.; Imrichova, D.; Seres, M.; Messingerova, L.; Bohacova, V.; Sulova, Z.; Breier, A. The expression of p-glycoprotein in leukemia cells is associated with the upregulated expression of nestin, a class 6 filament protein. Leuk. Res. 2016, 48, 32–39. [Google Scholar] [CrossRef]
- Gibalova, L.; Seres, M.; Rusnak, A.; Ditte, P.; Labudova, M.; Uhrik, B.; Pastorek, J.; Sedlak, J.; Breier, A.; Sulova, Z. P-glycoprotein depresses cisplatin sensitivity in l1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol. Vitr. 2012, 26, 435–444. [Google Scholar] [CrossRef]
- Imrichova, D.; Messingerova, L.; Seres, M.; Kavcova, H.; Pavlikova, L.; Coculova, M.; Breier, A.; Sulova, Z. Selection of resistant acute myeloid leukemia skm-1 and molm-13 cells by vincristine-, mitoxantrone- and lenalidomide-induced upregulation of p-glycoprotein activity and downregulation of cd33 cell surface exposure. Eur. J. Pharm. Sci. 2015, 77, 29–39. [Google Scholar] [CrossRef]
- Seres, M.; Pavlikova, L.; Bohacova, V.; Kyca, T.; Borovska, I.; Lakatos, B.; Breier, A.; Sulova, Z. Overexpression of grp78/bip in p-glycoprotein-positive l1210 cells is responsible for altered response of cells to tunicamycin as a stressor of the endoplasmic reticulum. Cells 2020, 9, 890. [Google Scholar] [CrossRef] [Green Version]
- Sulova, Z.; Ditte, P.; Kurucova, T.; Polakova, E.; Rogozanova, K.; Gibalova, L.; Seres, M.; Skvarkova, L.; Sedlak, J.; Pastorek, J.; et al. The presence of p-glycoprotein in l1210 cells directly induces down-regulation of cell surface saccharide targets of concanavalin a. Anticancer Res. 2010, 30, 3661–3668. [Google Scholar] [PubMed]
- Sulova, Z.; Mislovicova, D.; Gibalova, L.; Vajcnerova, Z.; Polakova, E.; Uhrik, B.; Tylkova, L.; Kovarova, A.; Sedlak, J.; Breier, A. Vincristine-induced overexpression of p-glycoprotein in l1210 cells is associated with remodeling of cell surface saccharides. J. Proteome Res. 2009, 8, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Goncalves Silva, I.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine 2017, 22, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Maiga, A.; Lemieux, S.; Pabst, C.; Lavallee, V.P.; Bouvier, M.; Sauvageau, G.; Hebert, J. Transcriptome analysis of g protein-coupled receptors in distinct genetic subgroups of acute myeloid leukemia: Identification of potential disease-specific targets. Blood Cancer J. 2016, 6, e431. [Google Scholar] [CrossRef]
- Sumbayev, V.V.; Goncalves Silva, I.; Blackburn, J.; Gibbs, B.F.; Yasinska, I.M.; Garrett, M.D.; Tonevitsky, A.G.; Ushkaryov, Y.A. Expression of functional neuronal receptor latrophilin 1 in human acute myeloid leukaemia cells. Oncotarget 2016, 7, 45575–45583. [Google Scholar] [CrossRef] [Green Version]
- Meza-Aguilar, D.G.; Boucard, A.A. Latrophilins updated. Biomol. Concepts 2014, 5, 457–478. [Google Scholar] [CrossRef]
- Silva, J.P.; Ushkaryov, Y.A. The latrophilins, “split-personality” receptors. Adv. Exp. Med. Biol. 2010, 706, 59–75. [Google Scholar]
- Aust, G. Adhesion-gpcrs in tumorigenesis. Adv. Exp. Med. Biol. 2010, 706, 109–120. [Google Scholar]
- Kocibalova, Z.; Guzyova, M.; Imrichova, D.; Sulova, Z.; Breier, A. Overexpression of the abcb1 drug transporter in acute myeloid leukemia cells is associated with downregulation of latrophilin-1. Gen. Physiol. Biophys. 2018, 37, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A tim-3/gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem. Cell 2015, 17, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Goncalves Silva, I.; Ruegg, L.; Gibbs, B.F.; Bardelli, M.; Fruehwirth, A.; Varani, L.; Berger, S.M.; Fasler-Kan, E.; Sumbayev, V.V. The immune receptor tim-3 acts as a trafficker in a tim-3/galectin-9 autocrine loop in human myeloid leukemia cells. Oncoimmunology 2016, 5, e1195535. [Google Scholar] [CrossRef]
- Heusschen, R.; Freitag, N.; Tirado-Gonzalez, I.; Barrientos, G.; Moschansky, P.; Munoz-Fernandez, R.; Leno-Duran, E.; Klapp, B.F.; Thijssen, V.L.; Blois, S.M. Profiling lgals9 splice variant expression at the fetal-maternal interface: Implications in normal and pathological human pregnancy. Biol. Reprod. 2013, 88, 22. [Google Scholar] [CrossRef]
- Heusschen, R.; Schulkens, I.A.; van Beijnum, J.; Griffioen, A.W.; Thijssen, V.L. Endothelial lgals9 splice variant expression in endothelial cell biology and angiogenesis. Biochim. Biophys. Acta 2014, 1842, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, V.R.; de Bruyn, M.; Helfrich, W.; Bremer, E. Therapeutic potential of galectin-9 in human disease. Med. Res. Rev. 2013, 33 (Suppl. 1), E102–E126. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, tim-3, and tigit: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Messingerova, L.; Jonasova, A.; Barancik, M.; Polekova, L.; Seres, M.; Gibalova, L.; Breier, A.; Sulova, Z. Lenalidomide treatment induced the normalization of marker protein levels in blood plasma of patients with 5q-myelodysplastic syndrome. Gen. Physiol. Biophys. 2015, 34, 399–406. [Google Scholar]
- Palau, A.; Mallo, M.; Palomo, L.; Rodriguez-Hernandez, I.; Diesch, J.; Campos, D.; Granada, I.; Junca, J.; Drexler, H.G.; Sole, F.; et al. Immunophenotypic, cytogenetic, and mutational characterization of cell lines derived from myelodysplastic syndrome patients after progression to acute myeloid leukemia. Genes Chromosomes Cancer 2017, 56, 243–252. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis tool web services from the embl-ebi. Nucleic. Acids. Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic. Acids. Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; MacLeod, R.A.; Uphoff, C.C.; Drexler, H.G.; Nishizaki, C.; Katayama, Y.; Kimura, G.; Fujii, N.; Omoto, E.; Harada, M.; et al. Two acute monocytic leukemia (aml-m5a) cell lines (molm-13 and molm-14) with interclonal phenotypic heterogeneity showing mll-af9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia 1997, 11, 1469–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Matozaki, S.; Murayama, T.; Nishimura, R.; Tsutsumi, M.; Kawaguchi, R.; Yokoyama, Y.; Hikiji, K.; Isobe, T.; Chihara, K. Establishment of a leukaemic cell line from a patient with acquisition of chromosomal abnormalities during disease progression in myelodysplastic syndrome. Br. J. Haematol. 1993, 85, 469–476. [Google Scholar] [CrossRef]
- Cole, S.P.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef] [PubMed]
- Katoh, S.; Ishii, N.; Nobumoto, A.; Takeshita, K.; Dai, S.Y.; Shinonaga, R.; Niki, T.; Nishi, N.; Tominaga, A.; Yamauchi, A.; et al. Galectin-9 inhibits cd44-hyaluronan interaction and suppresses a murine model of allergic asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 27–35. [Google Scholar] [CrossRef]
- Nobumoto, A.; Nagahara, K.; Oomizu, S.; Katoh, S.; Nishi, N.; Takeshita, K.; Niki, T.; Tominaga, A.; Yamauchi, A.; Hirashima, M. Galectin-9 suppresses tumor metastasis by blocking adhesion to endothelium and extracellular matrices. Glycobiology 2008, 18, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, M.; Carrillo, J.; Yeste, A.; Gutzeit, C.; Segura-Garzon, D.; Walland, A.C.; Pybus, M.; Grasset, E.K.; Yeiser, J.R.; Matthews, D.B.; et al. Secreted igd amplifies humoral t helper 2 cell responses by binding basophils via galectin-9 and cd44. Immunity 2018, 49, 709–724.e8. [Google Scholar] [CrossRef] [Green Version]
- Hertweck, M.K.; Erdfelder, F.; Kreuzer, K.A. Cd44 in hematological neoplasias. Ann. Hematol. 2011, 90, 493–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aanhane, E.; Schulkens, I.A.; Heusschen, R.; Castricum, K.; Leffler, H.; Griffioen, A.W.; Thijssen, V.L. Different angioregulatory activity of monovalent galectin-9 isoforms. Angiogenesis 2018, 21, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liang, Y.; Luo, X.; Hu, Q. Oxidative resistance of leukemic stem cells and oxidative damage to hematopoietic stem cells under pro-oxidative therapy. Cell Death Dis. 2020, 11, 291. [Google Scholar] [CrossRef]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lubbert, M. Hypomethylating agents (hma) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel hma-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- Williams, M.S.; Amaral, F.M.; Simeoni, F.; Somervaille, T.C. A stress-responsive enhancer induces dynamic drug resistance in acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1217–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davletov, B.A.; Shamotienko, O.G.; Lelianova, V.G.; Grishin, E.V.; Ushkaryov, Y.A. Isolation and biochemical characterization of a ca2+-independent alpha-latrotoxin-binding protein. J. Biol. Chem. 1996, 271, 23239–23245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnoperov, V.; Bittner, M.A.; Holz, R.W.; Chepurny, O.; Petrenko, A.G. Structural requirements for alpha-latrotoxin binding and alpha-latrotoxin-stimulated secretion. A study with calcium-independent receptor of alpha-latrotoxin (cirl) deletion mutants. J. Biol. Chem. 1999, 274, 3590–3596. [Google Scholar] [CrossRef] [Green Version]
- Krasnoperov, V.G.; Beavis, R.; Chepurny, O.G.; Little, A.R.; Plotnikov, A.N.; Petrenko, A.G. The calcium-independent receptor of alpha-latrotoxin is not a neurexin. Biochem. Biophys. Res. Commun. 1996, 227, 868–875. [Google Scholar] [CrossRef]
- Lelianova, V.G.; Davletov, B.A.; Sterling, A.; Rahman, M.A.; Grishin, E.V.; Totty, N.F.; Ushkaryov, Y.A. Alpha-latrotoxin receptor, latrophilin, is a novel member of the secretin family of g protein-coupled receptors. J. Biol. Chem. 1997, 272, 21504–21508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asayama, T.; Tamura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Heo, Y.M.; Hong, S.H.; Kim, K.; Park, S. The binding properties of glycosylated and non-glycosylated tim-3 molecules on cd4cd25 t cells. Immune Netw. 2009, 9, 58–63. [Google Scholar] [CrossRef]
- van de Weyer, P.S.; Muehlfeit, M.; Klose, C.; Bonventre, J.V.; Walz, G.; Kuehn, E.W. A highly conserved tyrosine of tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem. Biophys. Res. Commun. 2006, 351, 571–576. [Google Scholar] [CrossRef]
- Vega-Carrascal, I.; Reeves, E.P.; Niki, T.; Arikawa, T.; McNally, P.; O’Neill, S.J.; Hirashima, M.; McElvaney, N.G. Dysregulation of tim-3-galectin-9 pathway in the cystic fibrosis airways. J. Immunol. 2011, 186, 2897–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horlad, H.; Ohnishi, K.; Ma, C.; Fujiwara, Y.; Niino, D.; Ohshima, K.; Jinushi, M.; Matsuoka, M.; Takeya, M.; Komohara, Y. Tim-3 expression in lymphoma cells predicts chemoresistance in patients with adult t-cell leukemia/lymphoma. Oncol. Lett. 2016, 12, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Dama, P.; Tang, M.; Fulton, N.; Kline, J.; Liu, H. Gal9/tim-3 expression level is higher in aml patients who fail chemotherapy. J. Immunother. Cancer 2019, 7, 175. [Google Scholar] [CrossRef]
- Chabot, S.; Kashio, Y.; Seki, M.; Shirato, Y.; Nakamura, K.; Nishi, N.; Nakamura, T.; Matsumoto, R.; Hirashima, M. Regulation of galectin-9 expression and release in jurkat t cell line cells. Glycobiology 2002, 12, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Heusschen, R.; Griffioen, A.W.; Thijssen, V.L. Galectin-9 in tumor biology: A jack of multiple trades. Biochim. Biophys. Acta 2013, 1836, 177–185. [Google Scholar] [CrossRef]
- Spitzenberger, F.; Graessler, J.; Schroeder, H.E. Molecular and functional characterization of galectin 9 mrna isoforms in porcine and human cells and tissues. Biochimie 2001, 83, 851–862. [Google Scholar] [CrossRef]
- Yang, Q.; Jiang, W.; Zhuang, C.; Geng, Z.; Hou, C.; Huang, D.; Hu, L.; Wang, X. Microrna-22 downregulation of galectin-9 influences lymphocyte apoptosis and tumor cell proliferation in liver cancer. Oncol. Rep. 2015, 34, 1771–1778. [Google Scholar] [CrossRef] [Green Version]
- Yague, E.; Armesilla, A.L.; Harrison, G.; Elliott, J.; Sardini, A.; Higgins, C.F.; Raguz, S. P-glycoprotein (mdr1) expression in leukemic cells is regulated at two distinct steps, mrna stabilization and translational initiation. J. Biol. Chem. 2003, 278, 10344–10352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyca, T.; Pavlíková, L.; Boháčová, V.; Mišák, A.; Poturnayová, A.; Breier, A.; Sulová, Z.; Šereš, M. Insight into bortezomib focusing on its efficacy against p-gp-positive mdr leukemia cells. Int. J. Mol. Sci. 2021, 22, 5504. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Fujita, K.; Rosen, H.; Hirashima, M.; Masaki, T.; Hattori, T.; Hoshino, K. Plasma galectin-9 concentrations in normal and diseased condition. Cell Physiol. Biochem. 2018, 50, 1856–1868. [Google Scholar] [CrossRef] [PubMed]
- Yasinska, I.M.; Goncalves Silva, I.; Sakhnevych, S.; Gibbs, B.F.; Raap, U.; Fasler-Kan, E.; Sumbayev, V.V. Biochemical mechanisms implemented by human acute myeloid leukemia cells to suppress host immune surveillance. Cell. Mol. Immunol. 2018, 15, 989–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earl, L.A.; Bi, S.; Baum, L.G. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology 2011, 21, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zheng, M.; Qu, Y.; Li, J.; Ji, J.; Feng, B.; Lu, A.; Li, J.; Wang, M.; Liu, B. Different roles of galectin-9 isoforms in modulating e-selectin expression and adhesion function in lovo colon carcinoma cells. Mol. Biol. Rep. 2009, 36, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Kashio, Y.; Nakamura, K.; Abedin, M.J.; Seki, M.; Nishi, N.; Yoshida, N.; Nakamura, T.; Hirashima, M. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunol. 2003, 170, 3631–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Yamamoto, M.; Nagoshi, H.; Kobayashi, T.; Sasaki, N.; Shimura, Y.; Horiike, S.; Kimura, S.; Yamauchi, A.; Hirashima, M.; et al. Targeting activating transcription factor 3 by galectin-9 induces apoptosis and overcomes various types of treatment resistance in chronic myelogenous leukemia. Mol. Cancer Res. 2010, 8, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Legras, S.; Gunthert, U.; Stauder, R.; Curt, F.; Oliferenko, S.; Kluin-Nelemans, H.C.; Marie, J.P.; Proctor, S.; Jasmin, C.; Smadja-Joffe, F. A strong expression of cd44-6v correlates with shorter survival of patients with acute myeloid leukemia. Blood 1998, 91, 3401–3413. [Google Scholar] [CrossRef]
- Quere, R.; Andradottir, S.; Brun, A.C.; Zubarev, R.A.; Karlsson, G.; Olsson, K.; Magnusson, M.; Cammenga, J.; Karlsson, S. High levels of the adhesion molecule cd44 on leukemic cells generate acute myeloid leukemia relapse after withdrawal of the initial transforming event. Leukemia 2011, 25, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Bendall, L.J.; Bradstock, K.F.; Gottlieb, D.J. Expression of cd44 variant exons in acute myeloid leukemia is more common and more complex than that observed in normal blood, bone marrow or cd34+ cells. Leukemia 2000, 14, 1239–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jiang, G. Cd44 and hematologic malignancies. Cell Mol. Immunol. 2006, 3, 359–365. [Google Scholar] [PubMed]
- Prochazka, L.; Tesarik, R.; Turanek, J. Regulation of alternative splicing of cd44 in cancer. Cell Signal. 2014, 26, 2234–2239. [Google Scholar] [CrossRef]
- Azevedo, R.; Gaiteiro, C.; Peixoto, A.; Relvas-Santos, M.; Lima, L.; Santos, L.L.; Ferreira, J.A. Cd44 glycoprotein in cancer: A molecular conundrum hampering clinical applications. Clin. Proteom. 2018, 15, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.S.; Wei, M.; Ma, W.L.; Meng, W.; Zheng, W.L. Knockdown of cd44 enhances chemosensitivity of acute myeloid leukemia cells to adm and ara-c. Tumor Biol. 2014, 35, 3933–3940. [Google Scholar] [CrossRef]
- Hoofd, C.; Wang, X.; Lam, S.; Jenkins, C.; Wood, B.; Giambra, V.; Weng, A.P. Cd44 promotes chemoresistance in t-all by increased drug efflux. Exp. Hematol. 2016, 44, 166–171.e17. [Google Scholar] [CrossRef]
- Ohashi, R.; Takahashi, F.; Cui, R.; Yoshioka, M.; Gu, T.; Sasaki, S.; Tominaga, S.; Nishio, K.; Tanabe, K.K.; Takahashi, K. Interaction between cd44 and hyaluronate induces chemoresistance in non-small cell lung cancer cell. Cancer Lett. 2007, 252, 225–234. [Google Scholar] [CrossRef]
- Roy, S.; Kar, M.; Roy, S.; Padhi, S.; Kumar, A.; Thakur, S.; Akhter, Y.; Gatto, G.; Banerjee, B. Inhibition of cd44 sensitizes cisplatin-resistance and affects wnt/beta-catenin signaling in hnscc cells. Int. J. Biol. Macromol. 2020, 149, 501–512. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alternative Splicing | Transcript Variant Name and ID | Protein Isoform Name and ID |
|---|---|---|
| Full-length (FL) | TV1; NM_009587.3 | Isoform long; NP_033665.1 |
| Δ6 | X1; XM_017024623.2 | Isoform X1; XP_016880112.1 |
| Δ5 | TV2; NM_002308.4 | Isoform short; NP_002299.2 |
| Δ5,6 | X2; XM_006721892.3 | Isoform X2; XP_006721955.1 |
| Δ10 | X3; XM_006721893.3 | Isoform X3; XP_006721956.1 |
| Δ5,10 | TV4; NM_001330163.2 | Isoform 3; NP_001317092.1 |
| Δ5,6,10 | X4; XM_006721895.4 | Isoform X4; XP_006721958.1 |
| Exon 6′; premature stop | X5; 011524796.2 | Isoform X5; XP_011523098.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kocibalova, Z.; Guzyova, M.; Borovska, I.; Messingerova, L.; Copakova, L.; Sulova, Z.; Breier, A. Development of Multidrug Resistance in Acute Myeloid Leukemia Is Associated with Alterations of the LPHN1/GAL-9/TIM-3 Signaling Pathway. Cancers 2021, 13, 3629. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143629
Kocibalova Z, Guzyova M, Borovska I, Messingerova L, Copakova L, Sulova Z, Breier A. Development of Multidrug Resistance in Acute Myeloid Leukemia Is Associated with Alterations of the LPHN1/GAL-9/TIM-3 Signaling Pathway. Cancers. 2021; 13(14):3629. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143629
Chicago/Turabian StyleKocibalova, Zuzana, Martina Guzyova, Ivana Borovska, Lucia Messingerova, Lucia Copakova, Zdena Sulova, and Albert Breier. 2021. "Development of Multidrug Resistance in Acute Myeloid Leukemia Is Associated with Alterations of the LPHN1/GAL-9/TIM-3 Signaling Pathway" Cancers 13, no. 14: 3629. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143629