
Immune Microenvironment Features and Dynamics in Hodgkin Lymphoma


1
Haematopathology Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, 40138 Bologna, Italy
2
Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, 40138 Bologna, Italy
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(14), 3634; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143634
Submission received: 14 May 2021
/
Revised: 12 July 2021
/
Accepted: 14 July 2021
/
Published: 20 July 2021
(This article belongs to the Special Issue Recent Advances in Hodgkin’s Lymphoma)
Abstract
:Simple Summary
As happens in all neoplasms, the many reciprocal interactions taking place between neoplastic cells and the other reactive cells impact the course of the disease. Hodgkin Lymphoma is an haematologic malignancy where most of the pathological tissue is indeed composed by reactive cells and few neoplastic cells. Consequently, it represents an interesting subject for the description of the neoplastic and non-neoplastic cells interaction. In this review we report and discuss the more recent findings of microenvironmental studies about this disease.
Abstract
Classical Hodgkin’s lymphoma (cHL) accounts for 10% of all lymphoma diagnosis. The peculiar feature of the disease is the presence of large multinucleated Reed–Sternberg and mononuclear Hodgkin cells interspersed with a reactive microenvironment (ME). Due to the production of a large number of cytokines, Hodgkin cells (HCs) and Hodgkin Reed–Sternberg cells (HRSCs) attract and favour the expansion of different immune cell populations, modifying their functional status in order to receive prosurvival stimuli and to turn off the antitumour immune response. To this purpose HRSCs shape a biological niche by organizing the spatial distribution of cells in the ME. This review will highlight the contribution of the ME in the pathogenesis and prognosis of cHL and its role as a possible therapeutic target.
1. Introduction
Starting from its description in 1832 [1]. Hodgkin lymphoma (HL) captured the attention of scientists that clarified the nature of the disease throughout time. Classical Hodgkin’s lymphoma (cHL) accounts for 10% of all lymphoma diagnosis. The peculiar feature of the disease is the presence of large multinucleated Reed–Sternberg and mononuclear Hodgkin cells interspersed with a reactive microenvironment (ME) [1]. The latter can represent more than 95% of the entire tumoral mass and consists of T and B lymphocytes, neutrophils, eosinophils, macrophages, plasma cells, mast cells, fibroblasts and vessels [2]. The type and the number of these populations in the inflammatory background determine the distinction of cHL in histological subtypes: nodular sclerosis, mixed cellularity, lymphocyte depleted and lymphocyte rich [2]. The origin of the Hodgkin RS cells (HRSCs) has long been unknown, until molecular studies conducted by the microdissection of tissue sections and PCR analysis for rearranged immunoglobulin (IG) genes have shown that most HRSC and H cells (HCs) derived from peripheral B-lymphocytes, although a clonal rearrangement of the T-cell receptor genes was occasionally demonstrated (2% of cases) [2]. The immunophenotype and the genetic features of HC and HRSC are identical in these subtypes, but their clinical features are different. HRSC and HC express the CD30 and CD15 molecule in more than 98% and in about 75% of cases respectively [2]. Despite their mature B cells derivation, HRSC and HC are usually negative for the canonical B cells markers [1]. This feature was related to the downregulation, by epigenetic processes, of B-cell-specific transcription factors including PU.1, Oct.2 and its coactivator BOB.1 [3]. The B-cell origin of HRSC is demonstrated only by the expression of the transcription factor PAX5 [4].
Genetic instability is a characteristic of cHL that exhibits numerical chromosome aberrations usually in the hyperploid side. Several studies demonstrated that the constitutive activation of NF-kappaB sustains proliferation and prevents apoptosis in HRSC and HC. Different mechanisms underlie this phenomenon: gain/amplifications of REL (in 30% of the cases) and BCL3 oncogenes, aberrant activation of the natural inhibitors of NF-kappaB as the I-kappaB family or A20, the constitutive activation of AP-1 and LMP-1 overexpression (in EBV+ cases) [5,6,7,8,9,10]. In about 90% of cases cHL shows dysregulation, resulting in increased activation, of the JAK-STAT pathway by genetic alterations in multiple genes, such as STAT6 (32% of cases) STAT3, STAT5B, JAK1, JAK2 and the negative regulator PTPN1 [11]. Additional lower frequency somatic mutations of GNA13, XPO1 and ITPKB genes have also been reported [11]. After standard chemotherapy regimens with ABVD (doxorubicin, bleomycin, vinblastine and dacarbazine) and radiotherapy when indicated, almost all patients with cHL have a remission; however, 30% of the advanced stages and 15% of early stages have a relapse after treatment [12]. Early relapse defines a drug resistant subgroup associated to poor prognosis throughout all subsequent treatments. The introduction of the highly targeted antibody–drug conjugate brentuximab vedotin (BV), which delivers the cytotoxic drug monomethyl auristatin E (MMAE) to CD30-positive cells, represented an important step forward in the treatment of relapsed and refractory disease and the addition of BV to an AVD scheme is effective in advanced disease cHL, particularly in patients with a contraindication for receiving bleomycin [13]. Furthermore, in refractory cHL the immunotherapy with immune checkpoint inhibitors pembrolizumab or nivolumab, reverting the block of PD1 signalling pathway, has reached outstanding results with high response rates and quite durable disease control [14,15]. However, their precise mechanisms have not been clarified as they target not only the neoplastic cells but also the ME and, therefore, further studies are needed to explain the microenvironmental dynamics and the interaction between the tumour and immune system. This review will highlight the contribution of the ME in the pathogenesis and prognosis of cHL. The ME configuration depends on several complex and entangled mechanisms through which HRSC shape their own biological niche: they first recruit the immune population, then functionally reprogram them to receive survival stimuli and to escape the immune surveillance.
2. Recruitment and Constitution of the Microenvironment
As happens in all neoplasms, the many reciprocal interactions taking place between HRSC and the other reactive cells impact the course of the disease.
The HL ME is constituted by B and T lymphocytes, plasma cells, eosinophils, macrophages, mast cells myeloid suppressor derived cells and NK cells in a variable proportion.
HRSCs play a major role in the organization of the ME milieu: they are able to reshape not only the functional status of immune populations but also the spatial distribution of the different cell subsets, creating a specialized biological niche that provides prosurvival signals to HRSC and shelters them from the antitumour immune response (Figure 1).
They can directly recruit several immune cell types from the peripheral circulation and trigger the local expansion of these cellular subsets and this recruitment of immune cells is also stimulated by reactive macrophages and mast cells. Specifically, HRS cells synthesize and release many cytokines and chemokines such as IL-5, IL-7, IL-8, IL-9, CCL-5, CCL-17, CCL-20 and CCL-22 that are involved in the recruitment of granulocytes, lymphocyte, mast cells and macrophages [16,17]. HRSC also express a broad range of receptors including CD30, CD40, IL-7R, IL-9R, IL-13R, TACI and CCR5 that are able to detect growth and survival signals coming from the background with the final effect of the delivery of prosurvival feedback to HRSC.
As previously stated, the effects on the spatial distribution of microenvironmental components represents a consequence of specific recruitment by HRSC and at the same times allows the progress of interactions themselves.
In fact, although cHL neoplastic cells diffusely express PD-L1 (due to the presence of gain/amplification of locus 9p24.1), most of the tissue PD-L1 is localized on the cell surface of CD68+/CD163+ tumour associated macrophages (TAMs). PD-L1+ TAMs however are not spatially distributed in a random way, but they are significantly more abundant in the proximity of HRSC [18]. The expression of PD-L1 is induced by cytokines as INFγ or GM-CSF secreted by both HRSC and ME cells; interestingly a recent study demonstrated that PD-L1/L2 protein levels rapidly increase on monocytes via trogocytosis. The latter is an intercellular transfer of membrane patches from HRSC to monocytes in cHL and was not observed in monocytes cocultured with PD-L1/L2-deficient HRSC [19]. Since trogocytosis implies a cellular contact, this process could explain the enrichment of PD-L1+/PD-L2+ TAM close to HRSC and may further contribute to antitumour immunity evasion. Additionally, the several T cell subsets (Th-1, Th-2, Th-17, T-regs and cytotoxic lymphocytes) are not randomly distributed in the cHL ME. The CD4+ and CD8+ T cells including the PD-1+ subset are more numerous around and in contact with PDL-1+ TAM than PD-L1- TAMs suggesting that the PD-L1+ TAMs may either promote antitumour immunity through antigen presentation or immunosuppression through the engagement of PD-1 [18]. Moreover, CD4+ T lymphocytes, including PD-1+ ones, are enriched in proximity to HRSCs where they form rosettes in a fraction of cHL samples [18]. The low number of CD8+ T lymphocytes in the vicinity of or in contact with HRSC could be related to the frequent decrease or loss of β2M/MHC class I complex expression related to the inactivating mutations in β2M gene; conversely the CD4 richness around/in contact with HRSC may also be partly linked to the more frequent expression of MHC class II by the tumour cells. Recent studies have identified a high concentration of CD4+/CTLA-4+ T cells in the ME. Although CTLA-4 selectively identifies FOXP3+ regulatory cells, in cHL the CTLA-4+/FOXP3- T cells exceed the CTLA-4+/FOXP3+ ones and are highly enriched in proximity or in contact to HRSCs and to TAM. The CTLA-4+/FOXP3- cells are mostly PD-1- and engage the CD86, a cognate ligand for CTLA-4, which is expressed by HRSC and TAM. The expression of MHC class II can determine the T-reg cell types in the ME: the tissue of MHC-II positive cHL shows abundant CD4+/FOXP3+ T cells resetting the HRSCs, whereas in MCH class II negative cases the FOXP3+ T-regs are rare and HRSC are closely surrounded by CD4+/LAG-3+ T cells. As mentioned above, the latter are also CTLA-4+/CD25+ but negative for PD-1 and for the canonical T-reg marker FOXP3 suggesting they may exhibit a type 1 regulatory (Tr1) T-cell phenotype [20].
This biological niche not only allows an immune escape but transmits antiapoptotic and proproliferative stimuli to HRSC. The immune infiltrate, either through the release of cytokines that activate JAK/STAT signalling or by contact with the numerous surface receptors of HRSC, fuels the activation of NF-kB, PI3K/AKT and MAPK/ERK pathways. HRSCs express several tumour necrosis factor receptors: the CD40 ligand on CD4 T lymphocytes engages the CD40 on HRSC triggering the NF-κB pathway, CD30 binds the CD30 ligand on mast cells and eosinophils activating the PI3/AKT pathway, TACI and BCMA link BAFF and APRIL respectively on myeloid cells and, by recruiting TNF receptor-associated factor (TRAF) adaptor molecules, sustain NF-κB activation and upregulate Bcl-2, Bcl-xL and c-Myc. Moreover, HRSCs aberrantly express several receptor tyrosine kinases, despite the absence of activating mutations, such as DDR1, DDR2, EPHB1, TRKB, TRKA, c-MET and the MET-homologous receptor RON; the latter can be activated in a paracrine and autocrine fashions favouring the survival of the neoplastic clone [21,22,23].
Noteworthy, however, is the fact that the composition of the Hodgkinian niche is not uniform in all cHL samples, but it can be influenced by additional factors such as the presence of EBV or the expression pattern of MHC complexes. EBV-related cHL has a cytokine immune milieu dissimilar to that in EBV-cases, as it was associated to a high cytotoxic environment. In fact, EBV-related cHL shows an increased infiltration of CD8+ T cells associated with the expression of the Th1 transcription factor Tbet and Interferon gamma, displaying the profile of typical effective antitumour immunity. On the other hand, EBV− tumours manifested a Th17 profile and engagement of the IL-23/IL-17 axis with autoimmune and pro-oncogenic effects and the overexpression of GITR (TNF family member), which acts as a stimulator for T-regs cells [24,25].
Even if this finding sounds promising in the adult EBV-related cHL, some differences were detected in paediatric EBV positive cases where this effective response is counterbalanced by a microenvironmental niche enriched in PD-L1+ cells; underlining the age-related features of the immune system.
3. T Lymphocytes
T lymphocytes are the most represented component of cHL ME. The infiltrate is made up of both CD8+ and CD4+ T cells, the latter constituting often the largest population. At the site of disease, the recruited T lymphocytes surround HRSC and may form structures called “rosettes”. In the late 70s such structures were interpreted as an ineffective attempt to eliminate HRSC [30,31]. The inflammatory background contains abundant T helper (Th)-2 and IL-10 secreting T regulatory (T-reg) cells that are involved in the development and maintenance of an immunosuppressive ME [32,33]. In fact, HRSCs produce Th-2 and T-reg chemoattractants, including CCL17/TARC, CCL22, CCL5, IL-4, IL-5, IL-10 and IL-13 and induce a functional reprogramming of tumour-infiltrating T cells skewing CD4+ differentiation toward Th-2 and T-reg phenotypes [34,35]. In vitro experiments showed HRSC and HC selectively overexpress the immunoregulatory glycan-binding protein, galectin-1 (Gal1), through an AP1-dependent enhancer, and TGF-β: both proteins favour the secretion of Th-2 cytokines and the expansion of CD4+/CD25high/FOXP3+ T-reg cells [36]. However, several studies partially modified this picture of Hodgkin lymphoma ME demonstrating that Th-1/activation markers rather than Th-2/immunosuppression markers in T lymphocytes may play a major role. Graves et al. showed that T-lymphocytes express Th-1 associated CXCR3/CCR5 molecules and retain the capacity of TNFα/IFNγ/IL-2 cytokines secretion [37]. By applying mass cytometry to cell suspension from cHL biopsies, Cader and collaborators also identified selective expansion of more differentiated CD4+ Th-1 polarized T effector memory cells and Th-1 polarized T-regs in cHL cell suspensions [38]. New lymphocyte subpopulations have recently been identified in Hodgkin ME. Aoki et al. performed high-dimensional and spatial profiling of immune cells in cHL using scRNA-seq, multicolour immunohistochemistry and imaging mass cytometry identifying a unique regulatory CD4+/FOXP3+ T cell-like subset that expressed lymphocyte activation gene 3 (LAG3). These LAG3+ T cells demonstrated an immunosuppressive function mediated by the expression of IL-10 and TGF-β [20]. Moreover Ferrarini et al. found that HRS and surrounding T lymphocytes stained positive for IL-17 in 40% of cases and that the coculture of cHL cell lines with peripheral blood mononuclear cells promoted the enrichment of Th17 lymphocytes and FOXP3+/IL-17+ cells: these observations suggest the existence of an IL-17 tumour-shaped inflammatory milieu, at least in the subset of cHL cases [39]. At last, Le et al. described a previously unrecognized subset of CD8+ T cells exhibiting phenotypic and functional similarities with CD4+ follicular Th (Tfh) cells in a subgroup of patients. Similar to Tfh cells these CD8+ lymphocytes coexpress CXCR5, the inducible T-cell costimulator (ICOS) molecule, BCL6, PD-1, BTLA, CD200 and OX40 and downregulate CCR-7. They have deficient cytotoxicity, low INFγ secretion ability and functional properties in common with intratumoral CD4+ Tfh cells, such as the production of IL-4, IL-21, CXCL13 and capability to sustain B cells [40].
4. Macrophages
The finding that tumour associated macrophages (TAMs) and their interaction with neoplastic cells impacted on Hodgkin lymphoma prognosis dates back to 1985, when Ree and Kadin, described the immunohistochemical expression pattern of the peanut agglutinin (PNA). They concluded that a high number of macrophages was an important determinant in the clinical presentation and course of disease: the higher the number the worse the prognosis [41]. However, to date, despite several studies, the role of TAM in cHL has not yet been fully clarified. Macrophages are versatile cells characterized by an extraordinary functional plasticity: they can be immune-stimulatory or immune-suppressive, pro- or anti inflammation and in a tumoral setting they can favour or restrain disease development [42]. This heterogeneity has been oversimplified by the concept of functional polarization, which identifies two types of macrophages, M1 and M2, with distinct and opposite functions, induced by different types of cytokines present in the ME [43]. M1, or classically activated macrophages, are activated by Th-1 cytokines like INFγ and bacterial products; they secrete immunostimulatory cytokines that fuel the adaptive immune response and may acquire cytotoxic activity against transformed cells. M2, or alternatively activated macrophages, develop in a Th-2 cytokine-rich ME such as IL-4 and IL-13; they have high scavenging activity and produce several growth factors that activate the process of tissue repair and suppress adaptive immune responses [44,45]. The M2-like phenotype is often associated to protumour functions: in fact, these cells produce and release cytokines (IL-6) and growth factors (EGF, FGF, VEGF, PDGF and TGF-β), which stimulate proliferation and increase resistance to the apoptosis of tumour cells, activate fibroblasts and trigger angiogenesis. Furthermore, TAMs are the source of proteolytic enzymes that remodel the extracellular matrix favouring tissue invasion and metastasis [44]. In cHL, HRSC and HC recruit monocytes from the peripheral blood and can induce a M2 phenotype by the secretion of TNF, IL-10, TGF-β, GM-CSF, IL-13 and CCR5 [46,47]. The M2 macrophages may suppress cytotoxic T cell activity and attract T-reg lymphocytes, thus facilitating tumour growth and immune escape. In 2010 Steidl C et al. recognized a gene expression profiling signature of TAMs being associated with poor prognosis and correlated an increased number of CD68+ macrophages (as defined by immunohistochemistry) with a shorter PFS and a higher risk of relapse after autologous hematopoietic stem-cell transplantation [46]. Indeed, the prognostic value of the immunohistochemical quantification of TAMs exceeded IPS and was validated in the setting of multicentre phase III randomized controlled clinical trial E2496 [47]. However, although several studies confirmed that a higher density of TAMs is associated with inferior outcomes after first line treatment [48,49,50,51,52,53], others denied their negative impact in ABVD treated cHL. A possible explanation of these conflicting results can be ascribed to standardization of scoring methodologies. Jachimowicz et al. applied a whole-slide image analysis (WSI) that allows a quantitative, large-scale assessment of tumour composition utilizing conventional histopathology slides. They applied this approach to advanced-stage cHL samples from patients treated with BEACOPP-based regimens in the HD12, HD15 and HD18 trials of the German Hodgkin Study Group [54] and did not observe a correlation of macrophage content with outcome despite the cohort being enriched in vents [55]. A further issue might be the limits in the characterization of polarized macrophages in situ. CD163 was proposed as the marker of the M2-like phenotype and CD163+ TAMs and it indeed resulted in being a strong predictor of adverse outcomes in numerous series of ABVD treated cHL [48]; however, several authors challenge that CD163 can be considered a reliable M2 marker when used on its own. Barros et al. proposed a double-immunohistochemical labelling approach based on the detection of macrophage markers together with transcription factors regulating macrophage polarization, such as pSTAT1 and RBP-J for M1 and CMAF for M2 [21]. They demonstrated that predominant M1-like polarization, disclosed by the CD163+pSTAT1+/CD163+CMAF+ ratio > 1.5, was associated with better OS [21]. Since c-Myc was demonstrated to induce M2-like polarization by regulating genes associated with the alternative activation of macrophages, including SCARB1, ALOX15, MRC1 and CD209 [56], Werner et al. combined a macrophage-specific antibody (CD68 or CD163) to a reagent detecting c-MYC protein: they showed that MYC− macrophages (M1) are significantly higher in EBV+ cases while no differences exist in MYC+ macrophages (M2) between the EBV+ and EBV− cases [57]. Interestingly, they observed that a relative lack of TAM may allow cHL growth, intermediate numbers of TAM display an inhibitory effect and above a certain higher threshold, TAM may again support tumour growth [57]. These latter findings suggest that TAMs may have an hormetic rather than a linear relationship to outcome parameters and that their effects on survival may reflect underlying changes in macrophage polarization [57]. The mechanisms by which HRSC and HC modulate the TAM differentiation into alternatively activated tumour-promoting M2-like phenotypes are still not well defined. Functional studies showed that the global mRNA expression and protein quantity of macrophages in the cHL-conditioned medium differ from those of macrophages exposed to conditioned media from different diseases such as diffuse large B-cell lymphoma (DLBCL) cells or by a macrophage colony-stimulating factor (M-CSF). Conditioned medium from cHL cells triggers TAM differentiation into a M2-like phenotype via JAK/STAT signalling activation by the upregulation of surface antigens such as CD40, CD163, CD206 and PDL1 [58]. In particular, high mannose receptor C type 1 (MRC1 or CD206) enhances mannose-dependent endocytosis and type I collagen uptake contributing to matrix-remodelling and preclinical models revealed that cocultures of cHL cells with monocytes/macrophages support the dissemination of lymphoma cells via lymphatic vessels [58]. Finally, CD206 high IHC expression displays a positive correlation with an advanced stage of HL patients [58]. Given that M2-like TAM likely favour cHL progression, the possibility of reconverting the macrophage phenotype towards a proinflammatory M1 status could be an intriguing therapeutic perspective. An anticipation of this possible scenario comes from trabectedin (ET-743, Yondelis). Trabectedin is an alkylating agent that binds to DNA at the N2 position of guanine promoting degradation of RNA polymerase and generating DNA double-strand breaks, blocking the cell cycle and decreasing cytokines production. This anticancer drug is already approved as second-line therapy for ovarian cancer and soft tissue sarcomas. Its immunomodulating activities have been also demonstrated in preclinical models of hematologic malignancies. Casagrande et al. recently demonstrated that it overcomes doxorubicin-resistance and decreases xenograft growth in cHL [59]. Moreover, conditioned medium from trabectedin-treated HRSC showed less chemoattractivity toward monocytes, which also expressed lower levels of indoleamine 2,3-dioxygenase-1, CD206 and PD-L1, and showed less production of IL-10, TARC and TGF-β. This lead to less inhibition of activated lymphocyte growth [59]. Promising results are also coming from the usage of the PI3Kδ/γ inhibitor RP6530 both in preclinical and clinical models [60]. Since recent data in solid tumours show that the selective targeting of the γ isoform of PI3K in TAMs modulates the immunosuppressive tissue ME resulting in tumour regression, Locatelli et al. investigated the activity of RP6530 in vitro and in vivo in cHL cell line xenografts [60]. They demonstrated that in vitro RP6530 not only had a direct cytotoxic effect on Hodgkin lymphoma cells, switching the activation of macrophages from an immunosuppressive M2-like phenotype to a more inflammatory M1-like state, through the downregulation of lactic acid metabolism. In addition, in human tumour xenografts, RP6530 repolarized TAMs into proinflammatory macrophages inhibiting neoangiogenesis, leading to tumour regression [60]. Furthermore, patients enrolled in a phase I trial using RP6530 that achieved a complete and partial response and showed a significant inhibition of circulating myeloid-derived suppressor cells (MDSCs) and an average mean reduction in the serum TARC level [60]. These data represent the first evidence of therapeutic-induced suppression of the malignant cell growth and ME reshape in CHL.
5. B-Lymphocytes and Plasma Cells
The role of B cells in cHL is significantly less clear and defined and the insights we have derive more from the clinical scenario than from specific biological assays. Non-malignant B cells are prevalent in nodular lymphocyte predominant HL, which can be successfully treated with anti-CD20 monoclonal antibodies [61], however, they are also present in the ME of cHL and targeting B cells with rituximab both as a single agent and in combination with a standard scheme as ABVD could bring benefits, particularly on B symptoms and in the advanced stage of the neoplasm [62,63,64]. Several studies utilizing gene expression profiling and/or immunohistochemistry suggested a favourable effect for reactive B cells in cHL. Greaves et al. demonstrated that a high non-follicular CD20+ cell density confers a superior 5-year OS in cHL patients but not a longer FFTF (freedom from first-line treatment failure) and disease specific survival [37]. In line with this latter finding Panico et al. reported that high CD20+ background cells predicted a favourable outcome in cHL and antagonized CD68+ macrophages, while depletion of CD20+ cells together with an increase of TAM identifies a group of patients with high-risk disease [65]. Chetaille et al. profiled cHL samples, using DNA microarrays, and observed that cases with a favourable outcome overexpressed genes specific for B cells [66] and recently Jachimowicz et al. found that low B cell count was associated with a significantly reduced PFS and OS in cHL patients treated with BEACOPP-based regimens [54]. These studies suggest an antitumoral role for B cell in cHL and indicate that they could be central actors rather than bystanders in the local immune reaction. However, existing data on the role of tumour-associated B cells (TAB) indicate a tumour promoting function in solid tumour where their immunoinhibitory function resembles that of regulatory B cells (B-regs), which increase in tumour progression and are a proven source of inhibitory cytokines such as IL-10 and TGF-β [67]. Additionally, in mouse cancer models, TABs are required for establishing a chronic inflammatory state that promote de novo carcinogenesis [68] and are able to orchestrate and sustain melanoma inflammation. In this setting, TABs represent a predictor for survival and response to immune checkpoint blockade therapy [69]. Increased IL-10+ B cell numbers in ME can also be associated by increased numbers of Foxp3+ Tregs, which are independently associated with tumour progression or reduced patient survival [70].
Whether or not plasma cells (PCs) play a role in the dynamics of the ME in cHL has not yet been the subject of in-depth research. Their clinical relevance diverges in the tumour ME of different malignancies: infiltration of PC is associated with poor prognosis in colorectal cancer, breast cancer and ovarian cancer but it is associated with a favourable outcome in the ME of non-small lung cancer [71,72,73,74]. Gholiha et al. investigated diagnostic tumour biopsies from patients with cHL and found that CD138+ PC were associated with the presence of B-symptoms, advanced stage and eosinophil infiltration [75]. Moreover, patients with high proportion of PCs (defined as ≥10%) have an inferior event free survival (EFS) and OS although significance was not maintained in the multivariate analysis [76]. PC and eosinophils are attracted by CCL28, produced by HRSC and HC and these cells also secrete interleukin-6 and IL-21, involved in plasma cell differentiation. Interestingly, Thompson et al. correlated elevated quantitates of serum free immunoglobulin kappa and lambda light chains (free light chain: FLC), present in 30% of CHL patients, with inferior EFS and OS [76]. Being the HRSC and HC incapable of secreting functional Ig molecules the authors suggested that polyclonal FLC were secreted by the polyclonal PC of the ME. It was hypothesized that patients with elevated FLC have elevations in inflammatory cytokines and chemokines. Although these studies indicate that PC can adversely affect prognosis, the way they facilitate the survival of the neoplastic clone is still unknown. It was demonstrated that that IgA+ immunosuppressive PCs infiltrate therapy-resistant prostate cancer, where it induces CD8+ cell exhaustion and suppresses antitumour cytotoxic T lymphocyte responses through PD-L1 and IL-10 [77]. In squamous carcinoma during tumour development, autoantibody production by PCs leads to the deposition of immune complexes within neoplastic tissue. These later, through activating FcγR, trigger several protumour pathways, including angiogenic, tissue remodelling and prosurvival pathways and regulate the response to chemotherapy [78].
6. Mast Cells
cHL is frequently associated with mast cell (MC) infiltration and quantitative studies showed that the population of MC is significantly higher in nodular sclerosis (NS) than in other subtypes [79]. Several experimental data indicate that in NS-cHL, MCs promote fibrosis releasing TGF-β, which induces fibroblast proliferation, migration, contraction and collagen production. It has been hypothesized that IL-13 production by HRSC may lead to fibrosis and furthermore, promote MC proliferation and infiltration. the MC in turn might further produce the fibrotic cytokines IL-13 and TGF-β, resulting in fibrosis typical of NS-cHL [80]. Moreover, in immunodeficient NOD/SCID mice inoculated with both MC and human HRSC, a more marked fibrotic component, was detected compared to tumours in mice that have been inoculated with HRSC alone [81]. Interestingly, the fibrosis inducing tension and compression among cells was demonstrated to help cancer progression and tumour cell invasion through the increase the local concentration of growth factors and cytokines, one can assume that fibrosis induced by MC could also contribute to cHL progression [82]. Furthermore, MC can promote the differentiation of T cells into FOXP3+/CD25+ Treg cells via the secretion of TGF-β and may be the major source of pro-tumorigenic angiogenic and lymphangiogenic factors [83]. However, depending on the cytokine milieu of tumour ME, MC can conversely release antitumorigenic molecules, such as TNF-α and IL-9 [84]. The role of MC could therefore be cancer specific. Indeed, in thyroid, gastric, pancreatic and bladder cancers and in Merkel cell carcinomas, non-Hodgkin lymphomas and plasmacytoma, MC seems to be protumorigenic and associated with poor prognosis, while in others (e.g., breast cancer) they seem to have a protective role [85,86,87,88,89,90,91,92,93,94,95]. In cHL MC infiltration correlates with poor prognosis: Molin et al. reported a worse relapse-free survival in patients with higher mast cell density and Andersen et al. reported a significantly poorer outcome only in mixed cellularity CHL [96,97]. The latter also showed that the number of MC inversely correlated with the numbers of TAM and cytotoxic cells. Studies on the biological mechanisms underlying this adverse outcome are lacking: in the inflammatory background, MC were the predominant CD30L-positive cells and were able to activate HRSC in vitro through the CD30L–CD30 interaction. MC can however also promote the growth of Hodgkin’s tumours by modifying the tumour ME with the release of vascular endothelial growth factor-A and the induction of neovascularization [81,98]. There are increasing evidence supporting that targeting MC and/or their mediators represents a potential therapeutic target. Bortezomib inhibits degranulation and bortezomib-treated MC lose the ability to induce fibrosis and neoangiogenesis and are not able to promote the growth of the Hodgkin tumour in vivo [81]. Hansen et al. [98] showed that all Hodgkin cells release extracellular vesicles (EVs) containing CD30 (CD30EV) as a membrane protein. These EVs bind to CD30L on bystander cells and present additional membrane-associated CD30 sites for the binding of SGN-35. This study suggests that this mechanism allows a double targeting of both cancer and bystander cells along with MC and eosinophils.
7. The Myeloid Derived Suppressor Cells
The myeloid derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells produced by the bone marrow in neoplastic settings: when stimulated by chemical mediators (GM-CSF, G-CSF, M-CSF, VEGF, IL-1β, IL-4, Il-6, IL-10, IL-13 and IFNγ) they differentiate into monocytes and granulocytes, enter the bloodstream and reach the target organs where they perform an immunomodulatory function. In particular, they exert an immunosuppressive function both through the direct production of TGF-beta, ROS, IL-10 and peroxynitrite and through the indirect induction of T-reg differentiation in lymphocytes, thus promoting tumour growth and survival. In particular, MDSCs mostly exert the immune suppressive function against T-lymphocytes: this mostly derives from the high expression of arginase (Arg-1) that induces the depletion of tissue ME-arginine, which is an essential amino acid for the effective function of T-cell receptor (TCR) zeta chain assembly and downstream signalling. All this profoundly suppresses T cell immune responses [99,100,101,102,103]. The accumulation of MDSC has been reported in the ME of many cancer types where they can also differentiate into TAM and promote tumour angiogenesis and metastasis formation. In cHL tissue samples, a high number of Arg-1+ myeloid cells are significantly associated to shorter PFS in early stage patients and generally correlated with a worse OS [104]. However, the difficulty in defining the precise antigenic profile of MDSC, which overlaps with the granulocytic and monocytic phenotypes, hinders a thorough evaluation of their role in the disease. In the attempt to find a peripheral blood marker that could biologically mirror the dysregulated MDSC in tissue ME, Romano A et al. showed that the three main circulating MDSC derived subtypes (monocytic, granulocytic and CD34+ fractions) are increased in cHL and that CD34+ MDSCs predict short PFS, similarly to PET-2 [105]. The same group found that elevated serum Arg-1 (s-Arg-1) levels are associated with a shorter PFS and proposed the enzyme to be a novel potential biomarker for cHL prognosis [106]. Interestingly they also demonstrated that treatment with brentuximab vedotin reduced the absolute number of both the three MDSC subtypes and the s-Arg-1 levels [107]. Furthermore, patients with cHL experiencing objective responses (complete and partial responses) in a phase I trial using the PI3Kδ/γ inhibitor RP6530, managed to show a significant inhibition of circulating MDSC: these observations reinforce the possibility that MDSCs can serve as a therapeutic target [107].
8. Natural Killer Cells
Natural killer (NK) cells belong to group 1, innate lymphoid cells, and are a heterogeneous cell population with different functions and tissue distribution [108]. In peripheral blood two types of NK cells coexist: the CD56brightCD16– subset specialized in IFN secretion and the more cytotoxic subset CD56dimCD16+. NK cells are involved in tumour immune surveillance and recognize the transformed cells that downregulated MHC class I through their inhibitory receptors (KIRs) and NKG2x/CD94. NK cells can actually also eliminate cancers that retain MHC class I provided they express activating ligands, which engage activating receptors on NK cells (NKG2D, DNAM-1, NKp46, NKp30 and NKp44). Compromised NK cell activity was reported in many solid and haematological malignancies, which are able to alter NK cell maturation, to directly suppress their function, to enrich the ME of less cytotoxic CD56brightCD16– NK cells and to decrease or increase the levels of activating and inhibitory NK receptors, respectively. Tissue ME of cHL contains low levels of CD56-expressing NK cells and few studies indicated a dysregulation of NK function [109,110]. In particular, in the blood of cHL patients a new subset of mature NK cells was found lacking DNAM-1 expression, having limited killing activity and being poor INFγ producers [110]. These findings could have important clinical implications for the design of NK cell-targeting therapies.
9. HRCs Escape the Immunosurveillance
In cHL, the inflammatory ME often accounts for more than 90% of the lesion; HRSCs must therefore utilize multiple strategies to evade the immune surveillance. We previously highlighted their ability to reprogram immune populations toward functional phenotypes that are not effective in the antitumour response. However, HRSC can also reduce or lose their antigen presenting function making themselves invisible to the immune system and trigger exhaustion programs in the ME lymphocytes and NK cells. β2M inactivating mutations disrupt the expression of the β2-microglobulin (β2M)/MHC class I dual protein complex at the HRSC cell surface and consistently only 20% of the cHL cases show positive membrane immunohistochemical staining for β2M or MHC class I proteins. Patients with decreased/absent β2M/MHC class I have shorter PFS, underlining the importance of MHC class I mediated antigen presentation by HRSC to cytotoxic T cells for an optimal response to standard therapy [111]. In addition, decreased/absent protein expression of also MHC class II is observed in about 70% of the cases; in 15% of cHL cases this is a functional consequence of the rearrangement of the major histocompatibility complex (MHC) class II transactivator (CIITA), which is a master regulator of HLA class II transcription [111,112]. Homozygous deletions within the CD58 gene were reported in cHL and lead to the loss of protein expression in a subset of relapsed disease. CD58 is involved in the immune recognition of the tumour cells via binding of the CD2 receptor expressed by most cytotoxic CD8+ T cells and NK cells: its downregulation further represent an immune escape mechanism of HL tumour cells, at least in the clinically aggressive disease [113,114]. Beside perturbing the process of antigen presentation, HRSC are also skilled in triggering exhaustion programs in T cells. Activation of T cells is mediated by the interaction of the T-cell receptor (TCR) with an MHC-bound antigen and by costimulation of coreceptors on the surface of the antigen presenting cells (APCs) and the T cells. However, in order to prevent the tissue damage during the immune reaction, signals that limit TCR activation are simultaneously triggered by coinhibitory coreceptors called negative immune checkpoints (ICPs). Persistent stimulation of the latter induces T-cell functional exhaustion, characterized by a hyporesponsive state, a decreased production of effector cytokines, an inhibition of proliferation and a reduced cytotoxic activity. Binding of PD-1 by its ligands PD-L1/PD-L2 is the major coinhibitory pathway regulating T-cell exhaustion, which is nevertheless a reversible process. PD-1 can be upregulated on activated T cells, NK cells, B lymphocytes and macrophages and was reported to be variably present in cHL ME also in T cells rosetting around HRSCs. PD-L1 was found to be expressed by TAM, APCs and by neoplastic cells of several cancers and hematologic malignancies [115,116]. In cHL the copy gain or amplification of the 9p24.1 locus that include PDL1/PD-L2/JAK-2 genes results in the constitutive expression of PD-L1 and PD-L2 in more than 85% of the patients [117]. A genetic base for the enhanced PD-L1/PD-1 signalling is extremely rare among neoplasms and could at least partially explain the impressive overall response rate (about 70%) to the PD-1 blockade by monoclonal anti-PD1 antibodies (nivolumab and pembrolizumab, Table 1) observed in relapsed and refractory cHL patients [14,15]. More recently, the combination of nivolumab with AVD was tested in patients with newly diagnosed, advanced-stage disease and was found to be highly effective, with an overall response rate of 87% and a complete response rate of 67% [118]. Furthermore, several trials are also evaluating the association of BV and nivolumab, showing that this combination could be an active and well-tolerated first salvage regimen in relapsed or refractory cHL, and being also active in previously untreated older patients with comorbidities [119,120]. In cHL other corepressors capable of inducing functional exhaustion/anergy of T cells are however present. Cytotoxic T-lymphocyte antigen 4 (CTLA-4) is a member of the immunoglobulin superfamily: it is expressed on activated CD4+ or CD8+ lymphocytes and competes with its homologCD28 receptor for the engagement of their cognate ligands, CD80(B7.1)/CD86(B7.2) on APC. CTLA-4 regulates the development and functioning of T-reg and is a key negative regulator of the T-cell response. Hodgkin ME is enriched of both CD4+/CTLA-4+/FOXP3+ regulatory cells and CTLA-4+ non-regulatory T lymphocytes, whereas CD86 was found to be expressed in TAM and HRSC [115]. These findings suggest that the CTLA-4:CD86 axis may play a role in the creation of an immune-tolerant ME and that patients with cHL may also benefit from the CTLA-4 blockade. Clinical trials evaluating the effects of the administration of the anti-CTLA-4 antibody (ipilimumab) in combination with nivolumab or nivolumab and BV are ongoing [117,121]. The lymphocyte activation gene-3 (LAG-3) molecule is on the cell surface of activated CD4+ and CD8+ effector T cells, CD4+FOXP3+ Treg, Tr1 cells, B cells, plasmacytoid dendritic cells and NK cells. It structurally resembles the CD4 coreceptor and binds to MHC class II with high affinity [122,123]. LAG-3 acts synergistically with PD-1 and/or CTLA-4 to negatively regulate T cell expansion and LAG-3+ T cells populate cHL ME in most of the cases [115,124,125,126]. As mentioned above, a distinct CD4+/LAG-3+/FOXP3− T cell subset, expressing IL-10 and TGFβ, was recently identified by Aoki et al [20].; it probably corresponds to a Th1-type T-regs population, able to reduce both proliferation and TNF production in LAG-3− T cells and previously demonstrated to suppress effector CD8+ T-cell function in cHL [38]. A clinical study investigating the preliminary efficacy of the anti-LAG-3 monoclonal antibody BMS-986016 in combination with nivolumab in relapsed or refractory cHL is currently ongoing (clinicaltrials.gov identifier: 02061761). Immune checkpoint regulator T cell immunoglobulin-3 (TIM-3) is a type I transmembrane protein that is expressed on T-regs, NK cells, monocytes, macrophages and dendritic cells. TIM-3 plays an important role in the induction of T cell tolerance and TIM-3+ T-regs were correlated to an exhausted CD8+ T phenotype in tumour ME [126]. TIM-3 is nearly always expressed in the ME of cHL and may be targeted in the treatment of relapsed/refractory HL [120]. Moreover, T cell immunoglobulin and the ITIM domain (TIGIT) is a coinhibitory transmembrane glycoprotein expressed on CD8+ cytotoxic T cells, CD4+ Th cells and FOXP3+ T-regs, which has a critical role in regulating exhausted CD8+ T cell responses in tumours [127,128] Li and coworkers found a variable proportion of TIGIT+/PD-1+ in cHL samples and this combination represents an attractive candidate for coblockade with the PD-1 pathway inhibitor [128].
10. Conclusions
cHL is a unique neoplasm because it is largely composed of a non-neoplastic inflammatory infiltrate. Due to the production of a large number of cytokines, HC and HRSC attract and favour the expansion of different immune cell populations, modifying their functional status in order to receive prosurvival stimuli and to turn off the antitumour immune response. To this purpose HRSC shape a biological niche by organizing the spatial distribution of cells in the ME.
Areas surrounding HRSC are enriched in CD4+ T lymphocytes and PD-L1+/CD86+ macrophages. The interaction of negative immune checkpoints expressed on T cells with their respective ligands on macrophages and HRSC induces a state of local immune anergy. The prevalence of the CD4+ T lymphocyte subsets PD-1+, CTLA-4+ or LAG-3+ and the absence of MHC class I on HRSC suggest that the action of ICP inhibitors may be mediated by CD4+ T cells rather than by cytotoxic CD8 lymphocytes, as occurring in several solid neoplasms. Interestingly, the tumour-reactive cytotoxic CD4+ T cells were demonstrated to play a role in immunity against human cancers and several studies indicate that their activity may be enhanced by the ICP inhibitor treatment [129]. In line with these findings, circulating CD4+ cytotoxic T cells (CD4+/GrB+/PD-1+ Th-1 effector memory cells) are more abundant in patients with relapsed refractory cHL and expression of MHC class II, but not MHC class I, on HRSC was associated with the response to nivolumab in the CheckMate 205 trial. Recently, the effectiveness of the treatment with ICP inhibitors was related to the activation of innate effectors: the abundance of activated NK cells was associated with more favourable responses to the PD-1 blockade, which reverses the immune evasion mediated by the interaction of PD-1+ NK cells and PD-L1+monocytes/macrophages [130,131]. Moreover, Cader et al. identified a new CD3−/CD68+/CD4+/GrB+ subset that was associated with the response to PD-1 inhibitors in the blood and tissue ME of cHL patients; these cells may represent a monocyte population that utilize granzyme B to destroy antibody-coated targets via antibody-dependent cellular cytotoxicity [132]. As CD3−CD68+CD4+GrB+ elements also express IRF4, pSTAT1 and pS6, indicating a prior exposure to IF-γ, it was speculated that the CD4+ T cell might modulate innate cytotoxic effectors.
However, there is still much to discover about the microenvironment of cHL. Multistaining techniques, supported by image analysis systems and software, are useful in defining the phenotypic characteristics of immune subpopulations, but the structural and functional complexity of the tumour microenvironment will probably be further defined by the application of new technologies of digital spatial profiling. The latter being able to simultaneously detect, localize and quantify a large number of proteins and RNA molecules directly on tissue sections will allow one to further define the cellular composition and functional dynamics of the Hodgkin niche, providing a rationale for the appropriate use of ICPIs and new drugs that will target the microenvironment.
Author Contributions
Conceptualization: C.A., C.B., E.S.; writing—original draft preparation: C.A., C.B., E.S.; writing—review and editing: C.B., C.A.; Figure: C.A. All authors have read and agreed to the published version of the manuscript.
Funding
AIL Bologna: Associazione Italiana contro Leucemie linfomi e mieloma.
Institutional Review Board Statement
Ethical review and approval were waived for this study, due to it being a current literature review.
Informed Consent Statement
“Not applicable” (current literature review).
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hodgkin, T. On some Morbid Appearances of the Absorbent Glands and Spleen. Med. Chir. Trans. 1832, 17, 68–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, H.; Delsol, G.; Pileri, S.A.; Weiss, L.; Poppema, S.; Jaffe, E.S. Classical Hodgkin lymphoma. In Who Classification of Tumors of Haematopoietic and Lymphoid Tissues; Swerdlow, S., Campo, E., Harris, N., Eds.; IARC: Lyon, France, 2017; pp. 423–441. [Google Scholar]
- Stein, H.; Marafioti, T.; Foss, H.-D.; Laumen, H.; Hummel, M.; Anagnostopoulos, I.; Wirth, T.; Demel, G.; Falini, B. Down-regulation of BOB.1/OBF.1 and Oct2 in classical Hodgkin disease but not in lymphocyte predominant Hodgkin disease correlates with immunoglobulin transcription. Blood 2001, 97, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostinelli, C.; Sabattini, E.; Gjørret, J.O.; Righi, S.; Rossi, M.; Mancini, M.; Piccaluga, P.P.; Bacci, F.; Marafioti, T.; Bettini, G.; et al. Characterization of a new monoclonal antibody against PAX5/BASP in 1525 paraffin-embedded human and animal tissue samples. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 561–572. [Google Scholar] [CrossRef]
- Mathas, S.; Jöhrens, K.; Joos, S.; Lietz, A.; Hummel, F.; Janz, M.; Jundt, F.; Anagnostopoulos, I.; Bommert, K.; Lichter, P.; et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood 2005, 106, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Szymanowska, N.; Klapper, W.; Gesk, S.; Kuppers, R.; Martin-Subero, J.I.; Siebert, R. BCL2 and BCL3 are recurrent translocation partners of the IGH locus. Cancer Genet. Cytogenet. 2008, 186, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef]
- Schmidt, A.; Schmitz, R.; Giefing, M.; Martin-Subero, J.I.; Gesk, S.; Vater, I.; Massow, A.; Maggio, E.; Schneider, M.; Hansmann, M.-L.; et al. Rare occurrence of biallelic CYLD gene mutations in classical Hodgkin lymphoma. Genes Chromosomes Cancer 2002, 49, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.-L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Canellos, G.P.; Niedzwiecki, D.; Johnson, J.L. Long-term follow-up of survival in Hodgkin’s lymphoma. N. Engl. J. Med. 2009, 361, 2390–2391. [Google Scholar] [CrossRef]
- Alperovich, A.; Younes, A. Targeting CD30 Using Brentuximab Vedotin in the Treatment of Hodgkin Lymphoma. Cancer J. 2016, 22, 23–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, C.H.; Ribrag, V.; Michot, J.M. PD-1 Blockade with the Monoclonal Antibody Pembrolizumab (MK-3475) in Patients with Classical Hodgkin Lymphoma after Brentuximab Vedotin Failure: Preliminary Results from a Phase 1b Study (KEYNOTE-013). Blood 2014, 124, 290. [Google Scholar] [CrossRef]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Pinto, A.; Gloghini, A.; Carbone, A. Chemokine receptors as therapeutic tools in Hodgkin lymphoma: CCR4 and beyond. Blood 2010, 115, 746–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, M.; Carreras, J.; Higuchi, H.; Kotaki, R.; Hoshina, T.; Okuyama, K.; Suzuki, N.; Kakizaki, M.; Miyatake, Y.; Ando, K.; et al. PD-L1/L2 protein levels rapidly increase on monocytes via trogocytosis from tumor cells in classical Hodgkin lymphoma. Leukemia 2020, 34, 2405–2417. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, M.H.M.; Segges, P.; Vera-Lozada, G.; Hassan, R.; Niedobitek, G. Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PLoS ONE 2015, 10, e0124531. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.; Xu, W.; He, B.; Dillon, S.R.; Gross, J.A.; Sievers, E.; Qiao, X.; Santini, P.; Hyjek, E.; Lee, J.-W.; et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 2007, 109, 729–739. [Google Scholar] [CrossRef]
- Piccaluga, P.P.; Agostinelli, C.; Gazzola, A.; Tripodo, C.; Bacci, F.; Sabattini, E.; Sista, M.T.; Mannu, C.; Sapienza, M.R.; Rossi, M.; et al. Pathobiology of hodgkin lymphoma. Adv. Hematol. 2011, 2011, 920898. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.G.M.; Vera-Lozada, G.; Soares, F.A.; Niedobitek, G.; Hassan, R. Tumor microenvironment composition in pediatric classical Hodgkin lymphoma is modulated by age and Epstein-Barr virus infection. Int. J. Cancer 2012, 131, 1142–1152. [Google Scholar] [CrossRef]
- Duffield, A.S.; Ascierto, M.L.; Anders, R.A.; Taube, J.M.; Meeker, A.K.; Chen, S.; McMiller, T.L.; Phillips, N.A.; Xu, H.; Ogurtsova, A.; et al. Th17 immune microenvironment in Epstein-Barr virus-negative Hodgkin lymphoma: Implications for immunotherapy. Blood Adv. 2017, 1, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Halabi, L.; Adam, J.; Gravelle, P.; Marty, V.; Danu, A.; Lazarovici, J.; Ribrag, V.; Bosq, J.; Camara-Clayette, V.; Laurent, C.; et al. Expression of the Immune Checkpoint Regulators LAG-3 and TIM-3 in Classical Hodgkin Lymphoma. Clin. Lymphoma Myeloma Leuk. 2020, 12, S2152–S2650. [Google Scholar] [CrossRef] [PubMed]
- Sabatos, C.; Chakravarti, S.; Cha, E.; Schubart, A.; Sánchez-Fueyo, A.; Zheng, X.X.; Coyle, A.J.; Strom, T.B.; Freeman, G.J.; Kuchroo, V.K. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003, 4, 1102–1110. [Google Scholar] [CrossRef]
- Sakuishi, K.; Ngiow, S.F.; Sullivan, J.M.; Teng, M.W.L.; Kuchroo, V.K.; Smyth, M.J.; Anderson, A.C. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. OncoImmunology 2013, 2, e23849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blessin, N.C.; Simon, R.; Kluth, M.; Fischer, K.; Hube-Magg, C.; Li, W.; Makrypidi-Fraune, G.; Wellge, B.; Mandelkow, T.; Debatin, N.F.; et al. Patterns of TIGIT Expression in Lymphatic Tissue, Inflammation, and Cancer. Dis. Markers 2019, 2019, 5160565. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.V.; Jones, D.B.; Wright, D.H. Reed-Sternberg-cell/lymphocyte interaction. Lancet 1977, 2, 768–769. [Google Scholar] [CrossRef]
- Stuart, A.E.; Williams, A.R.; Habeshaw, J.A. Rosetting and other reactions of the Reed-Sternberg cell. J. Pathol. 1977, 122, 81–90. [Google Scholar] [CrossRef]
- Steidl, C.; Connors, J.M.; Gascoyne, R.D. Molecular pathogenesis of Hodgkin’s lymphoma: Increasing evidence of the importance of the microenvironment. J. Clin. Oncol. 2011, 29, 1812–1826. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.A.; Christie, L.E.; Munro, L.R.; Culligan, D.J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 2004, 103, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinnider, B.F.; Mak, T.W. The role of cytokines in classical Hodgkin lymphoma. Blood 2002, 99, 4283–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Juszczyński, P.; Ouyang, J.; Monti, S.; Rodig, S.J.; Takeyama, K.; Abramson, J.; Chen, W.; Kutok, J.L.; Rabinovich, G.A.; Shipp, M.A. The AP1-dependent secretion of galectin-1 by Reed–Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2007, 104, 13134–13139. [Google Scholar] [CrossRef] [Green Version]
- Greaves, P.; Clear, A.; Owen, A.; Iqbal, S.; Lee, A.; Matthews, J.; Wilson, A.; Calaminici, M.; Gribben, J.G. Defining characteristics of classical Hodgkin lymphoma microenvironment T-helper cells. Blood 2013, 122, 2856–2863. [Google Scholar] [CrossRef]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4 + regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Ferrarini, I.; Rigo, A.; Zamò, A.; Vinante, F.; Ferrarini, I.; Rigo, A.; Zamò, A.; Vinante, F. Classical Hodgkin lymphoma cells may promote an IL-17-enriched microenvironment. Leuk. Lymphoma 2019, 60, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Le, K.-S.; Amé-Thomas, P.; Tarte, K.; Gondois-Rey, F.; Granjeaud, S.; Orlanducci, F.; Foucher, E.D.; Broussais, F.; Bouabdallah, R.; Fest, T.; et al. CXCR5 and ICOS expression identifies a CD8 T-cell subset with T FH features in Hodgkin lymphomas. Blood Adv. 2018, 2, 1889–1900. [Google Scholar] [CrossRef]
- Ree, H.J.; Kadin, M.E. Macrophage-histiocytes in Hodgkin’s disease. The relationship of peanut-agglutinin-binding macrophage-histiocytes to clinicopathologic presentation and course of disease. Cancer 1985, 56, 2. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In Vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Aldinucci, D.; Celegato, M.; Casagrande, N. Microenvironmental interactions in classical Hodgkin lymphoma and their role in promoting tumor growth, immune escape and drug resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits Hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2019, 104, 564–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 62, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.L.; Scott, D.W.; Hong, F.; Kahl, B.S.; Fisher, R.I.; Bartlett, N.; Advani, R.; Buckstein, R.; Rimsza, L.M.; Connors, J.M.; et al. Tumor-associated macrophages predict inferior outcomes in classic Hodgkin lymphoma: A correlative study from the E2496 Intergroup trial. Blood 2012, 120, 3280–3287. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Cen, H.; Tan, X.; Ke, Q. Meta-analysis of the prognostic and clinical value of tumor-associated macrophages in adult classical Hodgkin lymphoma. BMC Med. 2016, 14, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzankov, A.; Matter, M.S.; Dirnhofer, S. Refined prognostic role of CD68-positive tumor macrophages in the context of the cellular micromilieu of classical Hodgkin lymphoma. Pathobiology 2010, 77, 6. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.A.A.; Wada, N.; Ikeda, J.; Shibayama, H.; Hashimoto, K.; Yamagami, T.; Tatsumi, Y.; Tsukaguchi, M.; Take, H.; Tsudo, M.; et al. Prognostic implication of types of tumor-associated macrophages in Hodgkin lymphoma. Virchows. Arch. 2011, 459, 4. [Google Scholar] [CrossRef] [PubMed]
- Kamper, P.; Bendix, K.; Hamilton-Dutoit, S.; Honore, B.; Nyengaard, J.R.; d’Amore, F. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica 2011, 96, 269–276. [Google Scholar] [CrossRef]
- Jakovic, L.R.; Mihaljevic, B.S.; Jovanovic, M.D.; Bogdanovic, A.D.; Andjelic, B.M.; Bumbasirevic, V.Z. Prognostic significance of Bcl-2, tumor-associated macrophages, and total neoplastic and inflammatory lymph node involvement in advanced stage classical Hodgkin’s lymphoma. Onkologie 2012, 35, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Deau, B.; Bachy, E.; Ribrag, V.; Delarue, R.; Rubio, M.T.; Bosq, J.; Varet, B.; Brousse, N.; Hermine, O.; Canioni, D. Macrophage, mast cell and T lymphocyte infiltrations are independent predictive biomarkers of primary refractoriness or early relapse in classical Hodgkin lymphoma. Leuk. Lymphoma 2013, 54, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jachimowicz, R.D.; Pieper, L.; Reinke, S.; Gontarewicz, A.; Plütschow, A.; Haverkamp, H.; Frauenfeld, L.; Fend, F.; Overkamp, M.; Jochims, F.; et al. Analysis of the tumor microenvironment by whole-slide image analysis identifies low B cell content as a predictor of adverse outcome in advanced-stage classical Hodgkin lymphoma treated with BEACOPP. Haematologica 2020, 106, 1684–1692. [Google Scholar] [CrossRef]
- Barros, M.H.M.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage polarisation: An immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [Green Version]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I.; et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Werner, L.; Dreyer, J.H.; Hartmann, D.; Barros, M.H.M.; Büttner-Herold, M.; Grittner, U.; Niedobitek, G. Tumor-associated macrophages in classical Hodgkin lymphoma: Hormetic relationship to outcome. Sci. Rep. 2020, 10, 9410. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Von Bonin, F.; Rehberg, T.; Perez-Rubio, P.; Engelmann, J.C.; Limm, K.; Reinke, S.; Dullin, C.; Sun, X.; Specht, R.; et al. High CD206 levels in Hodgkin lymphoma-educated macrophages are linked to matrix-remodeling and lymphoma dissemination. Mol. Oncol. 2020, 14, 571–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casagrande, N.; Borghese, C.; Favero, A.; Vicenzetto, C.; Aldinucci, D. Trabectedin overcomes doxorubicin-resistance, counteracts tumor-immunosuppressive reprogramming of monocytes and decreases xenograft growth in Hodgkin lymphoma. Cancer Lett. 2021, 500, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.; Careddu, G.; Serio, S.; Consonni, F.M.; Maeda, A.; Viswanadha, S.; Castagna, L.; Santoro, A.; Allavena, P.; Sica, A.; et al. Targeting cancer cells and tumor microenvironment in preclinical and clinical models of hodgkin lymphoma using the dual PI3Kδ/γ inhibitor RP6530. Clin. Cancer Res. 2019, 25, 1098–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Advani, R.H.; Horning, S.J.; Hoppe, R.T.; Daadi, S.; Allen, J.; Natkunam, Y.; Bartlett, N.L. Mature results of a phase II study of rituximab therapy for nodular lymphocyte-predominant Hodgkin lymphoma. J. Clin. Oncol. 2014, 32, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Romaguera, J.; Hagemeister, F.; McLaughlin, P.; Rodriguez, M.A.; Fiumara, P.; Goy, A.; Jeha, S.; Manning, J.T.; Jones, D.; et al. A pilot study of rituximab in patients with recurrent, classic Hodgkin disease. Cancer 2003, 98, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Oki, Y.; McLaughlin, P.; Copeland, A.R.; Goy, A.; Pro, B.; Feng, L.; Yuan, Y.; Chuang, H.H.; Macapinlac, H.A.; et al. Phase 2 study of rituximab plus ABVD in patients with newly diagnosed classical Hodgkin lymphoma. Blood 2012, 119, 4123–4128. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Clear, A.; Coutinho, R.; Wilson, A.; Matthews, J.; Owen, A.; Shanyinde, M.; Lister, T.A.; Calaminici, M.; Gribben, J.G. Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of classical Hodgkin lymphoma is predictive of outcome. J. Clin. Oncol. 2013, 31, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Panico, L.; Tenneriello, V.; Ronconi, F.; Lepore, M.; Cantore, N.; Dell’Angelo, A.C.; Ferbo, L.; Ferrara, F. High CD20+ background cells predict a favorable outcome in classical Hodgkin lymphoma and antagonize CD68+ macrophages. Leuk. Lymphoma 2015, 56, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Chetaille, B.; Bertucci, F.; Finetti, P.; Esterni, B.; Stamatoullas, A.; Picquenot, J.M.; Copin, M.C.; Morschhauser, F.; Casasnovas, R.-O.; Petrella, T.; et al. Molecular profiling of classical Hodgkin lymphoma tissues uncovers variations in the tumor microenvironment and correlations with EBV infection and outcome. Blood 2009, 113, 2765–3775. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Leitner, W.W.; Golding, B.; Scott, D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006, 66, 7741–7747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, K.E.; Korets, L.V.; Coussens, L.M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005, 7, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigami, E.; Sakakibara, M.; Sakakibara, J.; Masuda, T.; Fujimoto, H.; Hayama, S.; Nagashima, T.; Sangai, T.; Nakagawa, A.; Nakatani, Y.; et al. Coexistence of regulatory B cells and regulatory T cells in tumor-infiltrating lymphocyte aggregates is a prognostic factor in patients with breast cancer. Breast Cancer 2019, 26, 180–189. [Google Scholar] [CrossRef]
- Mohammed, Z.M.A.; Going, J.J.; Edwards, J.; Elsberger, B.; McMillan, D.C. The relationship between lymphocyte subsets and clinico-pathological determinants of survival in patients with primary operable invasive ductal breast cancer. J. Cancer 2013, 109, 1676–1684. [Google Scholar] [CrossRef] [Green Version]
- Berntsson, J.; Nodin, B.; Eberhard, J.; Micke, P.; Jirström, K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int. J. Cancer 2016, 139, 1129–1139. [Google Scholar] [CrossRef]
- Lundgren, S.; Berntsson, J.; Nodin, B.; Micke, P.; Jirström, K. Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer. J. Ovarian. Res. 2016, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Lohr, M.; Edlund, K.; Botling, J.; Hammad, S.; Hellwig, B.; Othman, A.; Berglund, A.; Lambe, M.; Holmberg, L.; Ekman, S.; et al. The prognostic relevance of tumour-infiltrating plasma cells and immunoglobulin kappa C indicates an important role of the humoral immune response in non-small cell lung cancer. Cancer Lett. 2013, 333, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Gholiha, A.R.; Hollander, P.; Hedstrom, G.; Sundstrom, C.; Molin, D.; Smedby, K.E.; Hjalgrim, H.; Glimelius, I.; Amini, R.-M.; Enblad, G. High tumour plasma cell infiltration reflects an important microenvironmental component in classic Hodgkin lymphoma linked to presence of B-symptoms. Br. J. Haematol. 2019, 184, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.A.; Maurer, M.J.; Cerhan, J.; Katzmann, J.A.; Ansell, S.M.; Habermann, T.M.; Macon, W.R.; Weiner, G.; Link, B.; Witzig, T.E. Elevated serum free light chains are associated with inferior event free and overall survival in Hodgkin lymphoma. Am. J. Hematol. 2011, 86, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Affara, N.I.; Ruffell, B.; Medler, T.R.; Gunderson, A.J.; Johansson, M.; Bornstein, S.; Bergsland, E.; Steinhoff, M.; Li, Y.; Gong, Q.; et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 2014, 25, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Crocker, J.; Smith, P.J. A quantitative study of mast cells in Hodgkin’s disease. J. Clin. Pathol. 1984, 37, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, S.; Yokote, T.; Hiraoka, N.; Nishiwaki, U.; Hanafusa, T.; Nishimura, Y.; Tsuji, M. Role of mast cells in fibrosis of classical Hodgkin lymphoma. J. Immunopathol. Pharmacol. 2016, 29, 603–611. [Google Scholar] [CrossRef]
- Mizuno, H.; Nakayama, T.; Miyata, Y.; Saito, S.; Nishiwaki, S.; Nakao, N.; Takeshita, K.; Naoe, T. Mast cells promote the growth of Hodgkin’s lymphoma cell tumor by modifying the tumor microenvironment that can be perturbed by bortezomib. Leukemia 2012, 26, 2269–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, K.; He, W.; Gao, Y.; Huang, W.; Lin, X.; Cai, L.; Fang, Z.; Zhou, Q.; Luo, Z.; et al. Transforming growth factor beta 1 plays an important role in inducing CD4(+)CD25(+)forhead box P3(+) regulatory T cells by mast cells. Clin. Exp. Immunol. 2010, 161, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Melillo, R.M.; Guarino, V.; Avilla, E.; Galdiero, M.R.; Liotti, F.; Prevete, N.; Rossi, F.W.; Basolo, F.; Ugolini, C.; De Paulis, A.; et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 2010, 29, 6203–6215. [Google Scholar] [CrossRef] [Green Version]
- Ammendola, M.; Sacco, R.; Donato, G.; Zuccalà, V.; Russo, E.; Luposella, M.; Vescio, G.; Rizzuto, A.; Patruno, R.; De Sarro, G.; et al. Mast cell positivity to tryptase correlates with metastatic lymph nodes in gastrointestinal cancer patients treated surgically. Oncology 2013, 85, 111–116. [Google Scholar] [CrossRef]
- Ammendola, M.; Sacco, R.; Sammarco, G.; Donato, G.; Zuccalà, V.; Romano, R.; Luposella, M.; Patruno, R.; Vallicelli, C.; Verdecchia, G.M.; et al. Mast Cells Positive to Tryptase and c-Kit Receptor Expressing Cells Correlates with Angiogenesis in Gastric Cancer Patients Surgically Treated. Gastroenterol. Res. Pract. 2013, 2013, 703163. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Brown Swigart, L.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Rao, Q.; YChen, Y.; Yeh, C.R.; Ding, J.; Li, L.; Chang, C.; Yeh, S. Recruited mast cells in the tumor microenvironment enhance bladder cancer metastasis via modulation of ERβ/CCL2/CCR2 EMT/MMP9 signals. Oncotarget 2016, 7, 7842–7855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.W.; Ng, L.B.; Murray, K. Mast cells have prognostic value in Merkel cell carcinoma. Am. J. Dermatopathol. 2008, 30, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, A.; Schlaak, M.; Heukamp, L.C.; Förster, A.; Theurich, S.; Von Bergwelt-Baildon, M.; Büttner, R.; Kurschat, P.; Mauch, C.; Roers, A.; et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood 2012, 120, 2042–2054. [Google Scholar] [CrossRef] [Green Version]
- Tripodo, C.; Gri, G.; Piccaluga, P.P.; Frossi, B.; Guarnotta, C.; Piconese, S.; Franco, V.; Vetri, V.; Pucillo, C.; Florena, A.M.; et al. Mast cells and Th17 cells contribute to the lymphoma-associated pro-inflammatory microenvironment of angioimmunoblastic T-cell lymphoma. Am. J. Pathol. 2010, 177, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Franco, G.; Guarnotta, C.; Frossi, B.; Piccaluga, P.P.; Boveri, E.; Gulino, A.; Fuligni, F.; Rigoni, A.; Porcasi, R.; Buffa, S.; et al. Bone marrow stroma CD40 expression correlates with inflammatory mast cell infiltration and disease progression in splenic marginal zone lymphoma. Blood 2014, 123, 1836–1849. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Vacca, A.; Nico, B.; Quondamatteo, F.; Ria, R.; Minischetti, M.; Marzullo, A.; Herken, R.; Roncali, L.; Dammacco, F. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. Br. J. Cancer 1999, 79, 451–455. [Google Scholar] [CrossRef] [Green Version]
- Rajput, A.B.; Turbin, D.A.; Cheang, M.C.U.; Voduc, D.K.; Leung, S.; Gelmon, K.A.; Gilks, C.B.; Huntsman, D.G. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: A study of 4444 cases. Breast Cancer Res. Treat. 2008, 107, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.D.; Kamper, P.; Nielsen, P.S.; Bendix, K.; Riber-Hansen, R.; Steiniche, T.; Hamilton-Dutoit, S.; Clausen, M.R.; D’Amore, F. Tumour-associated mast cells in classical Hodgkin’s lymphoma: Correlation with histological subtype, other tumour-infiltrating inflammatory cell subsets and outcome. Eur. J. Haematol. 2016, 96, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Gruss, H.J.; Boiani, N.; Williams, D.E.; Armitage, R.J.; Smith, C.A.; Goodwin, R.G. Pleiotropic effects of the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood 1994, 83, 2045–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molin, D.; Edström, A.; Glimelius, I.; Glimelius, B.; Nilsson, G.; Sundström, C.; Enblad, G. Mast cell infiltration correlates with poor prognosis in Hodgkin’s lymphoma. Br. J. Haematol. 2002, 119, 122–124. [Google Scholar] [CrossRef]
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schön, G.; Grenzi, P.; Bachurski, D.; Aquino, B.; et al. CD30 on extracellular vesicles from malignant Hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with Brentuximab-Vedotin, In Vitro. Oncotarget 2016, 7, 30523–30535. [Google Scholar] [CrossRef] [Green Version]
- Darcy, C.J.; Woodberry, T.; Davis, J.S.; Piera, K.A.; McNeil, Y.R.; Chen, Y.; Yeo, T.W.; Weinberg, J.B.; Anstey, N.M. Increased plasma arginase activity in human sepsis: Association with increased circulating neutrophils. Clin. Chem. Lab. Med. 2014, 52, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.G.; Megiel, C.; Russell, S.M.; Bingham, B.; Arger, N.; Woo, T.; Epstein, A.L. Functional characterization of human Cd33+ and Cd11b+ myeloid-derived suppressor cell subsets induced from peripheral blood mononuclear cells co-cultured with a diverse set of human tumor cell lines. J. Transl. Med. 2011, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Raber, P.; Ochoa, A.C.; Rodriguez, P.C. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: Mechanisms of T cell suppression and therapeutic perspectives. Immunol. Invest. 2012, 41, 614–634. [Google Scholar] [CrossRef]
- Popovic, P.J.; Zeh 3rd, H.J.; Ochoa, J.B. Arginine and immunity. J. Nutr. 2007, 137, 1681s–1686s. [Google Scholar] [CrossRef] [Green Version]
- Gallamini, A.; Agostinelli, C.; Tripodo, C.; Starcqualursi, L.; Fuligni, F.; Fiore, F. Analysis of myeloid suppressor marker arginase identifies CD68+/arginase+ myeloid/monocytic subsets and exerts stronger prognostic influence than macrophage quantification in classical Hodgkin lymphoma. Haematologica 2013, 98, 1–64. [Google Scholar]
- Romano, A.; Parrinello, N.L.; Vetro, C.; Forte, S.; Chiarenza, A.; Figuera, A.; Motta, G.; Palumbo, G.A.M.; Ippolito, M.; Consoli, U.; et al. Circulating myeloid-derived suppressor cells correlate with clinical outcome in Hodgkin Lymphoma patients treated up-front with a risk-adapted strategy. Br. J. Haematol. 2015, 168, 689–700. [Google Scholar] [CrossRef]
- Romano, A.; Parrinello, N.; Vetro, C.; Tibullo, D.; Giallongo, C.; La Cava, P.; Chiarenza, A.; Motta, G.; Caruso, A.; Villari, L.; et al. The prognostic value of the myeloid-mediated immunosuppression marker Arginase-1 in classic Hodgkin lymphoma. Oncotarget 2016, 7, 67333–67346. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Parrinello, N.L.; Chiarenza, A.; Motta, G.; Tibullo, D.; Giallongo, C.; La Cava, P.; Camiolo, G.; Puglisi, F.; Palumbo, G.A.; et al. Immune off-target effects of Brentuximab Vedotin in relapsed/refractory Hodgkin Lymphoma. Br. J. Haematol. 2019, 185, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Cortez, V.S.; Colonna, M. Killing the Invaders: NK Cell Impact in Tumors and Anti-Tumor Therapy. Cancers 2021, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Koulis, A.; Trivedi, P.; Ibrahim, H.; Bower, M.; Naresh, K.N. The role of the microenvironment in human immunodeficiency virus-associated classical Hodgkin lymphoma. Histopathology 2014, 65, 749–756. [Google Scholar] [CrossRef]
- Stannard, K.A.; Lemoine, S.; Waterhouse, N.J.; Vari, F.; Chatenoud, L.; Gandhi, M.K.; Martinet, L.; Smyth, M.J.; Guillerey, C. Human peripheral blood DNAM-1 neg NK cells are a terminally differentiated subset with limited effector functions. Blood Adv. 2019, 3, 1681–1694. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [Green Version]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Ben Neriah, S.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Razak, F.R.A.; Diepstra, A.; Visser, L.; van den Berg, A. CD58 mutations are common in Hodgkin lymphoma cell lines and loss of CD58 expression in tumor cells occurs in Hodgkin lymphoma patients who relapse. Genes. Immun. 2016, 17, 363–366. [Google Scholar] [CrossRef]
- Schneider, M.; Schneider, S.; Zühlke-Jenisch, R.; Klapper, W.; Sundström, C.; Hartmann, S.; Hansmann, M.-L.; Siebert, R.; Küppers, R.; Giefing, M. Alterations of the CD58 gene in classical Hodgkin lymphoma. Genes Chromosomes Cancer 2015, 54, 638–645. [Google Scholar] [CrossRef]
- Patel, S.S.; Weirather, J.L.; Lipschitz, M.; Lako, A.; Chen, P.-H.; Griffin, G.K.; Armand, P.; Shipp, M.A.; Rodig, S.J. The microenvironmental niche in classic Hodgkin lymphoma is enriched for CTLA-4-positive T cells that are PD-1-negative. Blood 2019, 134, 2059–2069. [Google Scholar] [CrossRef]
- Agostinelli, C.; Gallamini, A.; Stracqualursi, L.; Agati, P.; Tripodo, C.; Fuligni, F.; Sista, M.T.; Fanti, S.; Biggi, A.; Vitolo, U.; et al. The combined role of biomarkers and interim PET scan in prediction of treatment outcome in classical Hodgkin’s lymphoma: A retrospective, European, multicentre cohort study. Lancet. Haematol. 2016, 3, e467–e479. [Google Scholar] [CrossRef]
- Herrera, A.F.; Moskowitz, A.J.; Bartlett, N.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Ansell, S.M.; Moskowitz, C.H.; Fenton, K.; et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2018, 131, 1183–1194. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.M.; Piccioni, D.; Kato, S.; Boichard, A.; Wang, H.-Y.; Frampton, G.; Lippman, S.M.; Connelly, C.; Fabrizio, D.; Miller, V.; et al. Prevalence of PDL1 Amplification and Preliminary Response to Immune Checkpoint Blockade in Solid Tumors. JAMA Oncol. 2018, 4, 1237–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramchandren, R.; Domenech, E.D.; Rueda, A.; Trněný, M.; Feldman, T.A.; Lee, H.J.; Provencio, M.; Sillaber, C.; Cohen, J.B.; Savage, K.J.; et al. Nivolumab for Newly Diagnosed Advanced-Stage Classic Hodgkin Lymphoma: Safety and Efficacy in the Phase II CheckMate 205 Study. J. Clin. Oncol. 2019, 37, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Bartlett, N.L.; LaPlant, B.; Lee, H.J.; Advani, R.J.; Christian, B.; Diefenbach, C.S.; Feldman, T.A.; Ansell, S.M. Brentuximab vedotin plus nivolumab as first-line therapy in older or chemotherapy-ineligible patients with Hodgkin lymphoma (ACCRU): A multicentre, single-arm, phase 2 trial. Lancet. Haematol. 2020, 7, e808–e815. [Google Scholar] [CrossRef]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; McKiver, M.P.; Ansell, S.M.; Armand, P.; Lesokhin, A.; et al. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2021, 35, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, C.S.; Hong, F.; Ambinder, R.F.; Cohen, J.B.; Robertson, M.J.; David, K.A.; Advani, R.H.; Fenske, T.S.; Barta, S.K.; Palmisiano, N.D.; et al. Ipilimumab, nivolumab, and brentuximab vedotin combination therapies in patients with relapsed or refractory Hodgkin lymphoma: Phase 1 results of an open-label, multicentre, phase 1/2 trial. Lancet. Haematol. 2020, 7, e660–e670. [Google Scholar] [CrossRef]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Okazaki, I.-M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation In Vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell. 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Blessin, N.C.; Simon, R.; Kluth, M.; Fischer, K.; Hube-Magg, C.; Makrypidi-Fraune, G.; Wellge, B.; Mandelkow, T.; Debatin, N.F.; et al. Expression of the immune checkpoint receptor TIGIT in Hodgkin’s lymphoma. MC Cancer 2018, 18, 1209. [Google Scholar] [CrossRef]
- Renné, C.; Willenbrock, K.; Küppers, R.; Hansmann, M.L.; Bräuninger, A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood 2005, 105, 4051–4059. [Google Scholar] [CrossRef] [Green Version]
- Koh, Y.W.; Yoon, D.H.; Suh, C.; Cha, H.J.; Huh, J. Insulin-like growth factor-1 receptor is associated with better prognosis in classical Hodgkin’s lymphoma: Correlation with MET expression. Exp. Pathol. 2015, 96, 232–239. [Google Scholar] [CrossRef]
- Xu, C.; Plattel, W.; Berg, A.V.D.; Rüther, N.; Huang, X.; Wang, M.; De Jong, D.; Vos, H.; Van Imhoff, G.; Viardot, A.; et al. Expression of the c-Met oncogene by tumor cells predicts a favorable outcome in classical Hodgkin’s lymphoma. Haematologica 2012, 97, 572–578. [Google Scholar] [CrossRef] [Green Version]
- Cader, F.Z.; Hu, X.; Goh, W.L.; Wienand, K.; Ouyang, J.; Mandato, E.; Redd, R.; Lawton, L.N.; Chen, P.-H.; Weirather, J.L.; et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat. Med. 2020, 26, 1468–1479. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representation of the Hodgkinian niche with CD30+ HRSC surrounded by several subsets of Th-1 and T-reg CD4+ lymphocytes, CD8+ cytotoxic T-cell, tumour associated macrophages (TAMs), granulocytes (GRs) and mast cells (MCs).
Figure 1.
Representation of the Hodgkinian niche with CD30+ HRSC surrounded by several subsets of Th-1 and T-reg CD4+ lymphocytes, CD8+ cytotoxic T-cell, tumour associated macrophages (TAMs), granulocytes (GRs) and mast cells (MCs).


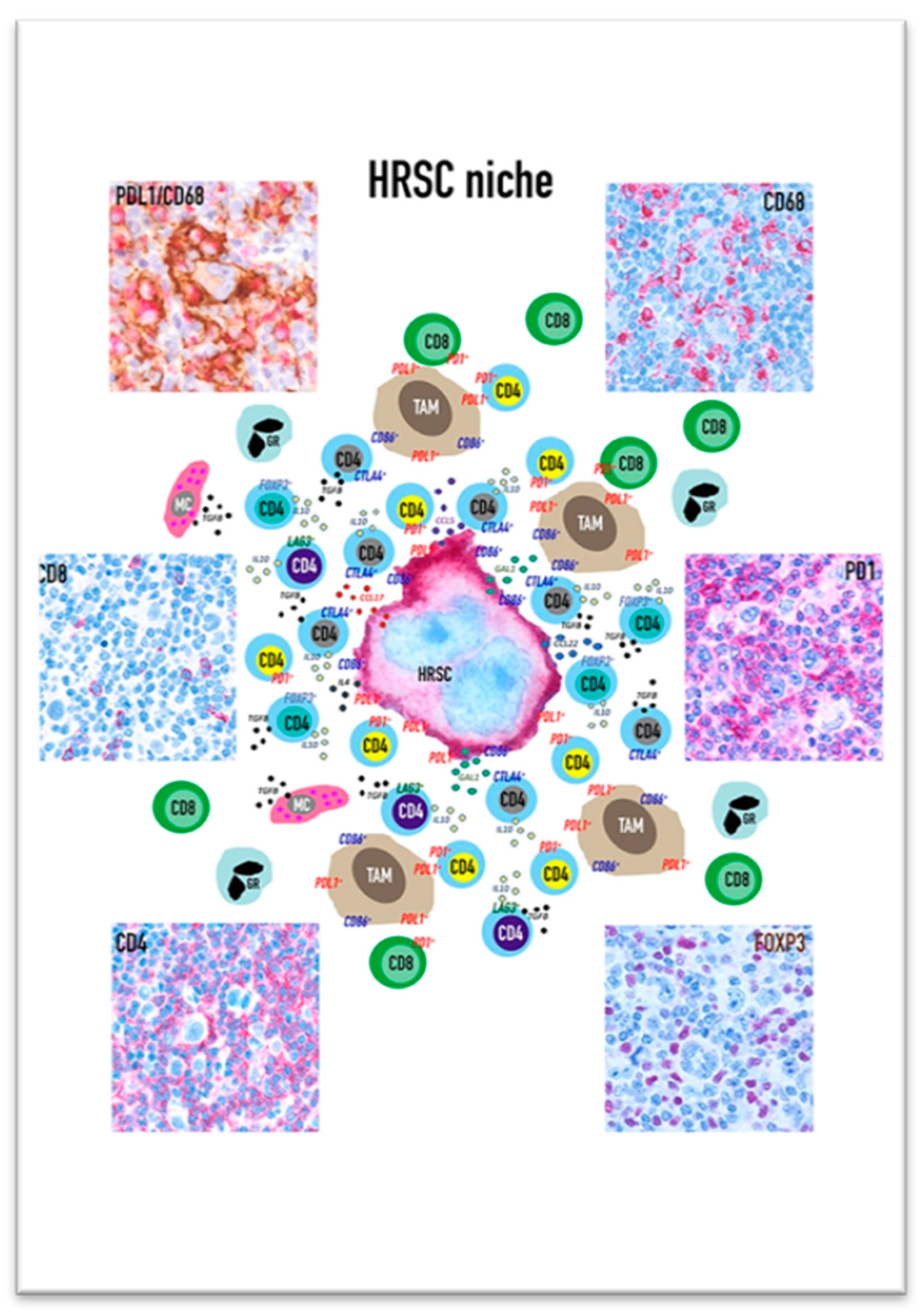{kind=link}
Table 1.
Immune checkpoint molecules.
| Function | Name | Uses |
|---|---|---|
| anti-PD1 | Nivolumab; Pembrolizumab | approved for metastatic melanoma, non-small cell lung cancer; refractory Hodgkin Lymphoma, advanced renal cell carcinoma, urothelial carcinoma |
| anti-PDL1 | Atezolizumab, Durvalumab | approved for bladder cancer, non-small cell lung cancer, Merkel cell carcinoma |
| anti-CTLA4 | Ipilimumab; Tremelimumab | approved for kin melanoma |
| anti-Lag 3 | BMS986016 | in study for cervical cancer, bladder cancer, colonic cancer, melanoma, noon small cell lung cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bertuzzi, C.; Sabattini, E.; Agostinelli, C. Immune Microenvironment Features and Dynamics in Hodgkin Lymphoma. Cancers 2021, 13, 3634. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143634
AMA Style
Bertuzzi C, Sabattini E, Agostinelli C. Immune Microenvironment Features and Dynamics in Hodgkin Lymphoma. Cancers. 2021; 13(14):3634. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143634
Chicago/Turabian StyleBertuzzi, Clara, Elena Sabattini, and Claudio Agostinelli. 2021. "Immune Microenvironment Features and Dynamics in Hodgkin Lymphoma" Cancers 13, no. 14: 3634. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143634
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.