
PXR Modulates the Prostate Cancer Cell Response to Afatinib by Regulating the Expression of the Monocarboxylate Transporter SLC16A1

 , , , , , , , ,
, , , , , , , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Drugs
2.3. Patients and Samples
2.4. Tissue Micro Arrays (TMA) and Immunohistochemistry
2.5. RT-qPCR
2.6. Western Blotting and Chromatin Immunoprecipitation
2.7. PXR Immunofluorescence Staining
2.8. Luciferase Assays
2.9. Cytotoxicity Assays
2.10. Determination of Intra- and Extra-Cellular Concentrations of Afatinib
3. Results
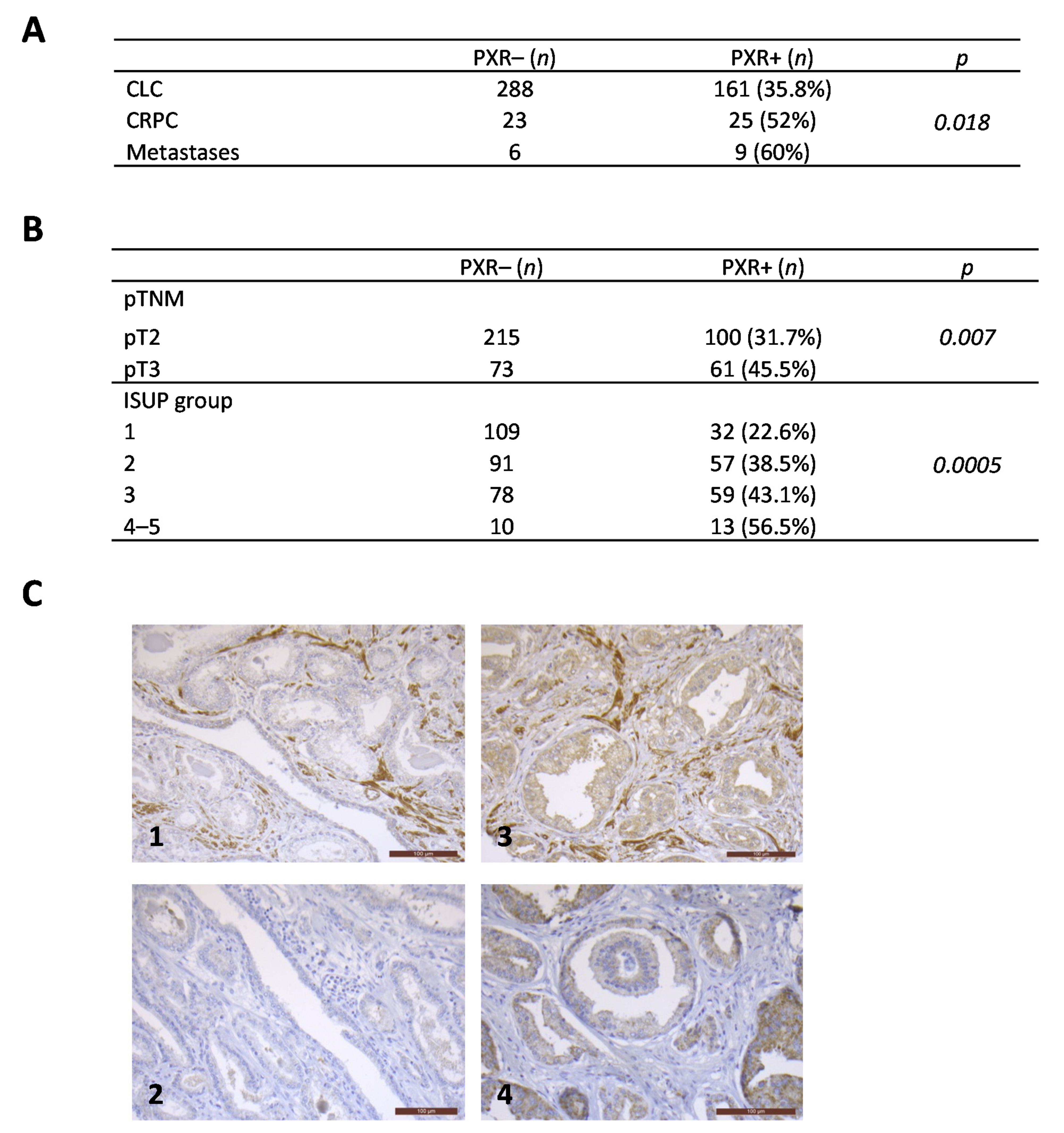
3.1. PXR Expression Is More Frequently Detected in Advanced Stages of Prostate Cancers
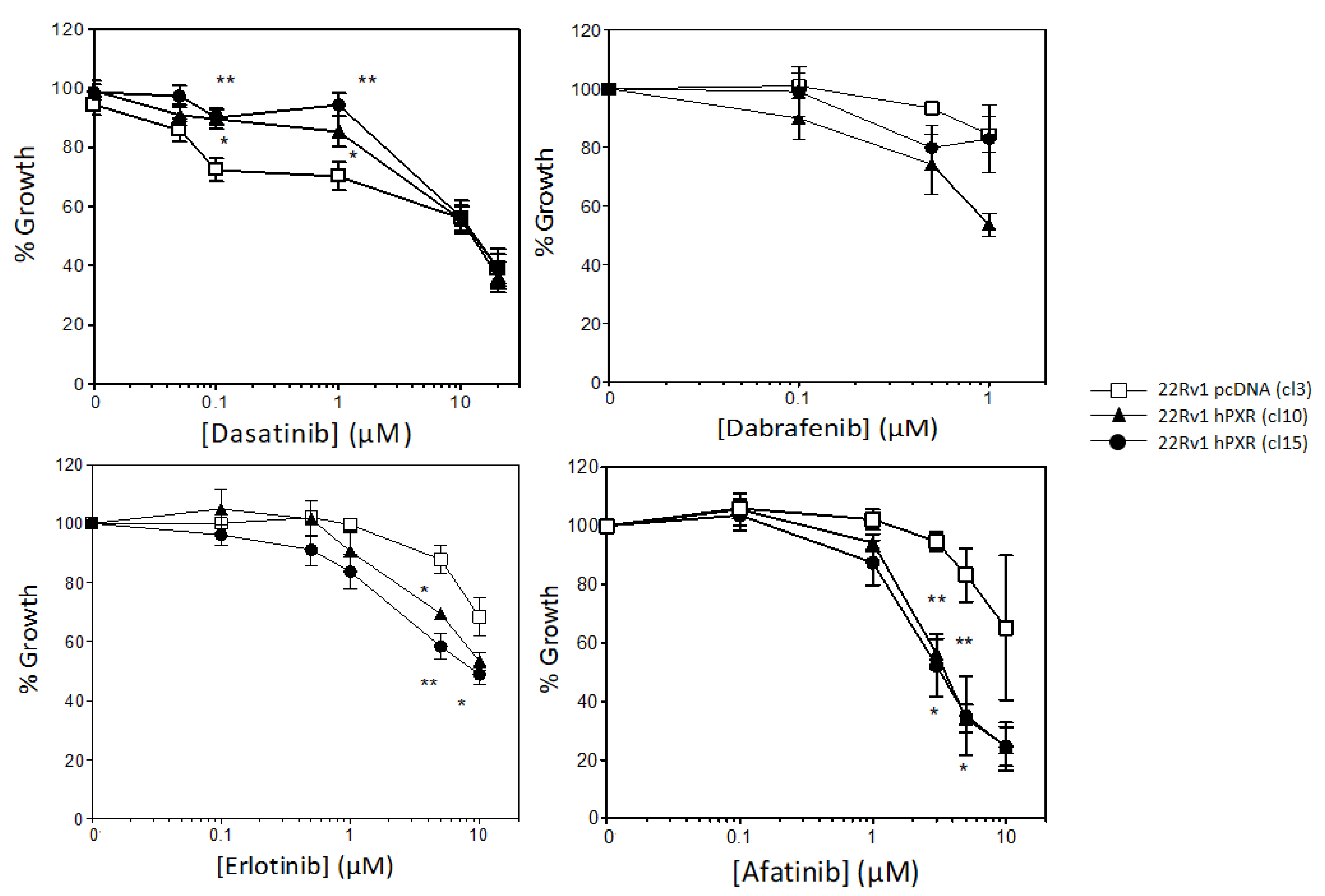
3.2. PXR Overexpression Sensitizes 22Rv1 Cells to Specific Kinase Inhibitors
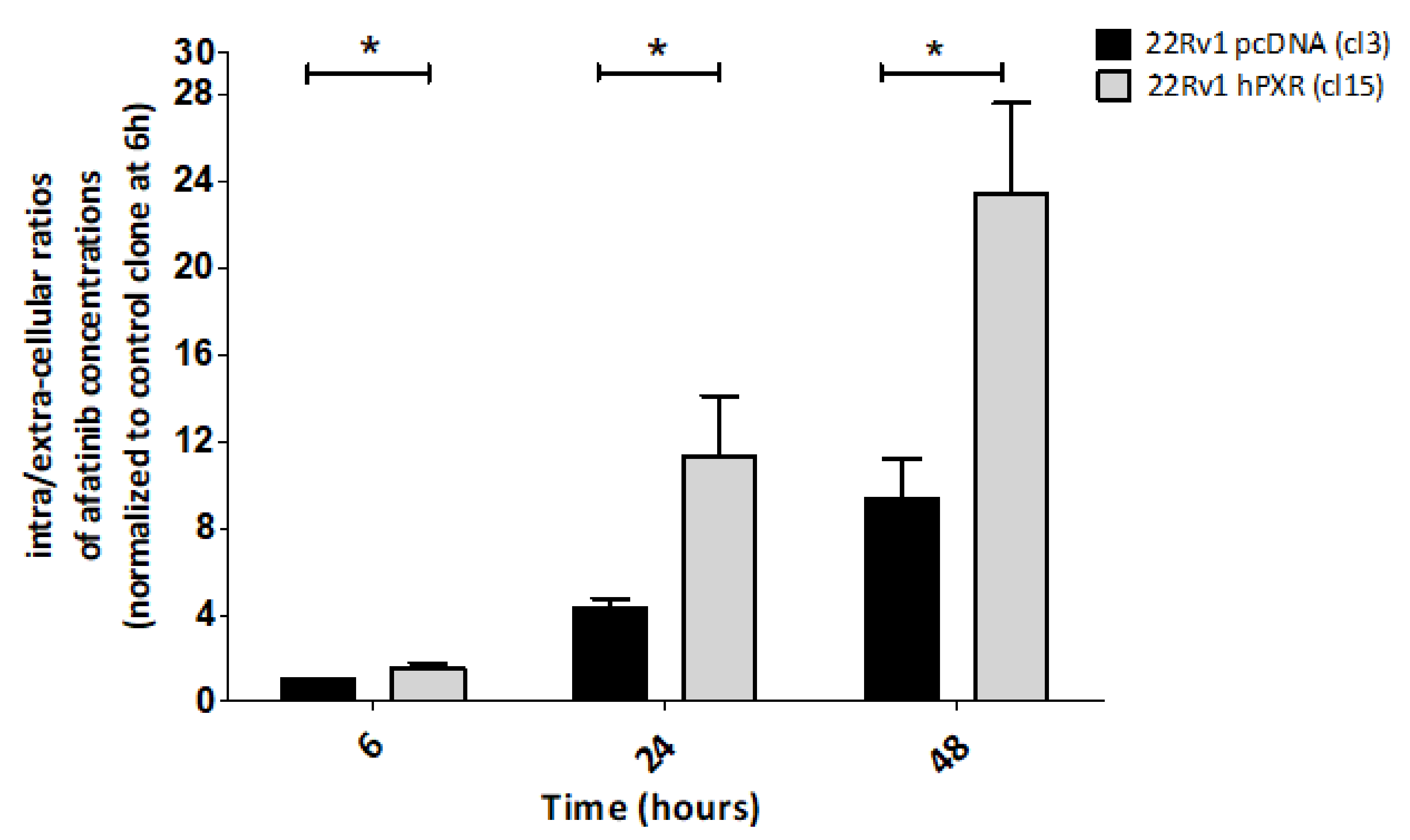
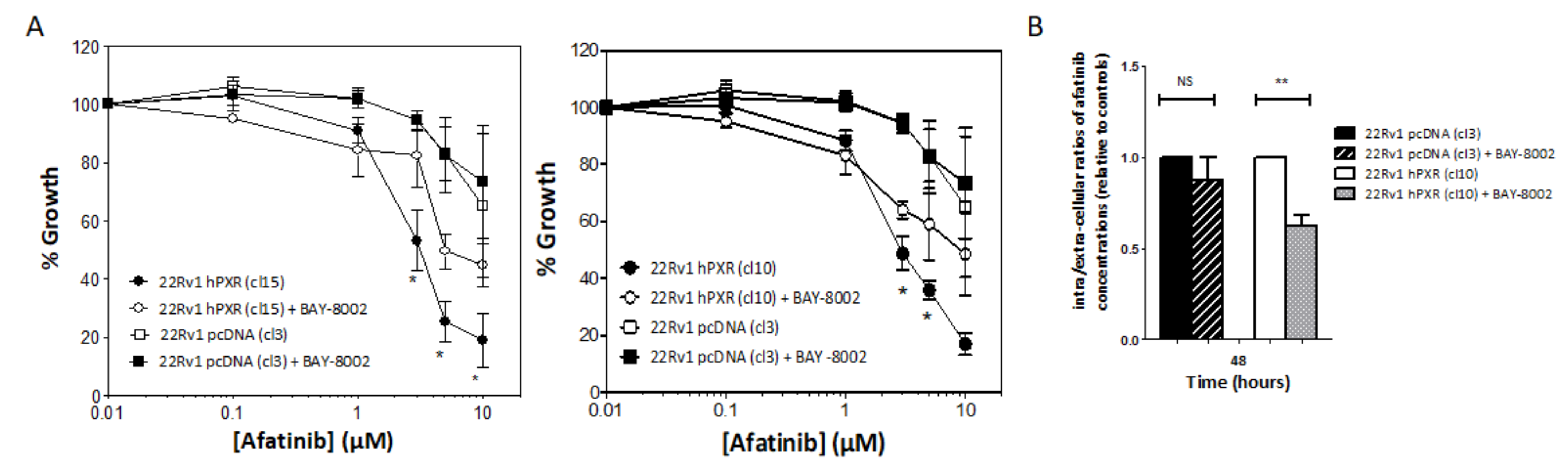
3.3. PXR-Mediated Increased Sensitivity to Afatinib of 22Rv1 Cells Is Associated with Enhanced Intracellular Concentration of the Drug
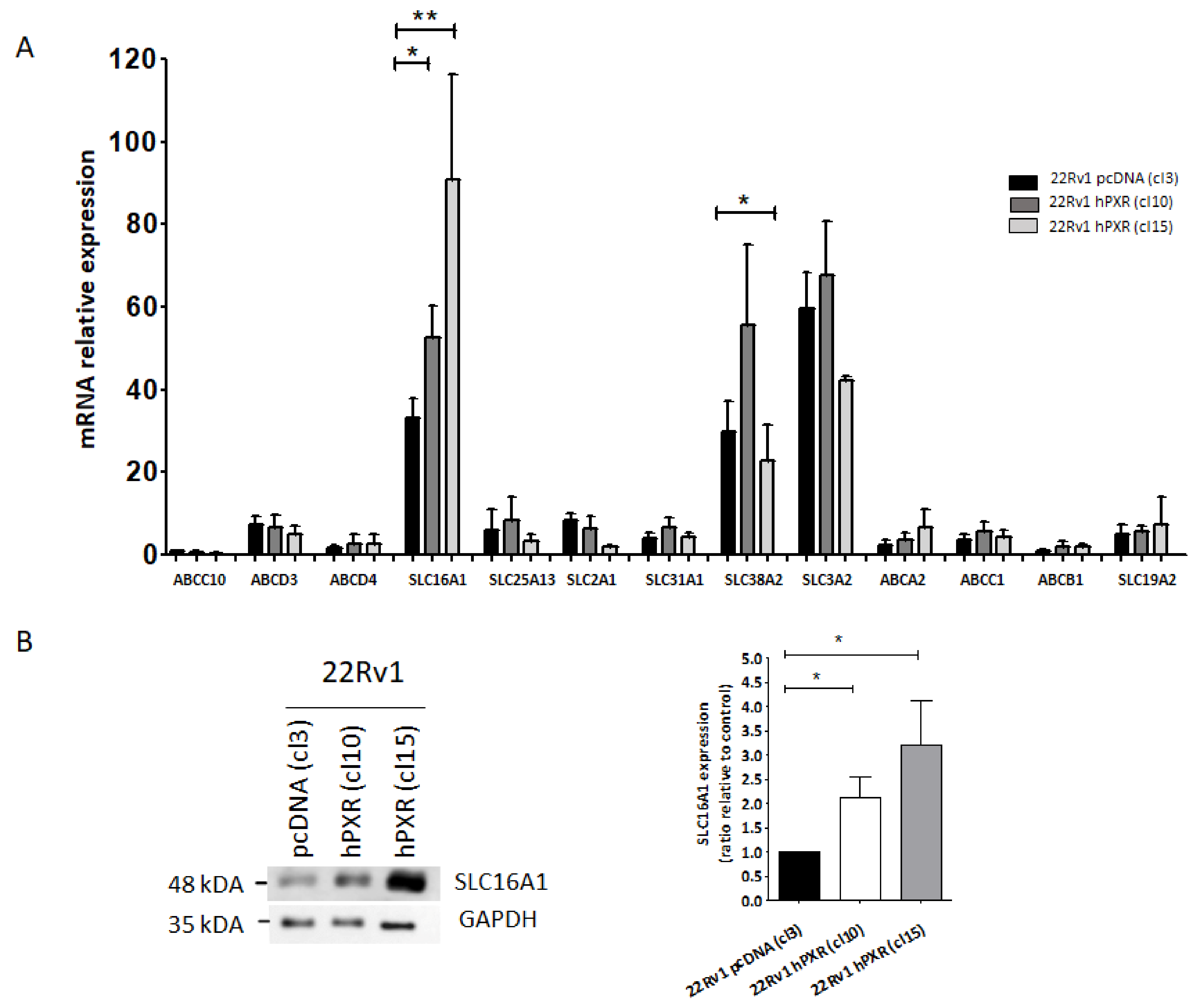
3.4. SLC16A1 Plays a Role in the Transport of Afatinib in 22Rv1 Cells Expressing PXR
3.5. SLC16A1 Inhibition Abrogates PXR-Mediated Sensitization of 22Rv1 hPXR Cells to Afatinib
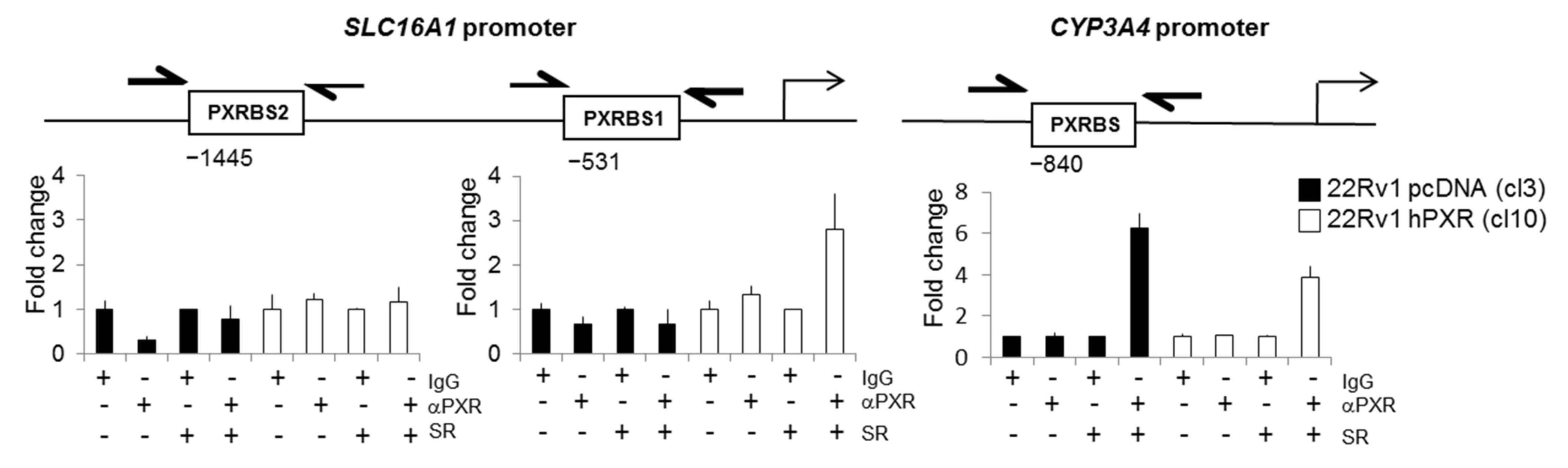
3.6. SLC16A1 Is a PXR Target Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate Cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of Resistance to Systemic Therapy in Metastatic Castration-Resistant Prostate Cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Wu, Z.; Ding, W.; Gao, S.; Gao, Y.; Xu, C. Mechanisms of Enzalutamide Resistance in Castration-Resistant Prostate Cancer and Therapeutic Strategies to Overcome It. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Culig, Z. Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr. Mol. Biol. Rep. 2017, 3, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Attard, G.; Antonarakis, E.S. Prostate Cancer: AR Aberrations and Resistance to Abiraterone or Enzalutamide. Nat. Rev. Urol. 2016, 13, 697–698. [Google Scholar] [CrossRef]
- Lombard, A.P.; Liu, C.; Armstrong, C.M.; Cucchiara, V.; Gu, X.; Lou, W.; Evans, C.P.; Gao, A.C. ABCB1 Mediates Cabazitaxel-Docetaxel Cross-Resistance in Advanced Prostate Cancer. Mol. Cancer Ther. 2017, 16, 2257–2266. [Google Scholar] [CrossRef] [Green Version]
- Sissung, T.M.; Baum, C.E.; Deeken, J.; Price, D.K.; Aragon-Ching, J.; Steinberg, S.M.; Dahut, W.; Sparreboom, A.; Figg, W.D. ABCB1 Genetic Variation Influences the Toxicity and Clinical Outcome of Patients with Androgen-Independent Prostate Cancer Treated with Docetaxel. Clin. Cancer Res. 2008, 14, 4543–4549. [Google Scholar] [CrossRef] [Green Version]
- Oprea-Lager, D.E.; Bijnsdorp, I.V.; VAN Moorselaar, R.J.A.; Van den Eertwegh, A.J.M.; Hoekstra, O.S.; Geldof, A.A. ABCC4 Decreases Docetaxel and Not Cabazitaxel Efficacy in Prostate Cancer Cells in Vitro. Anticancer Res. 2013, 33, 387–391. [Google Scholar] [PubMed]
- Kawanobe, T.; Kogure, S.; Nakamura, S.; Sato, M.; Katayama, K.; Mitsuhashi, J.; Noguchi, K.; Sugimoto, Y. Expression of Human ABCB5 Confers Resistance to Taxanes and Anthracyclines. Biochem. Biophys. Res. Commun. 2012, 418, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C. Investigational Serine/Threonine Kinase Inhibitors against Prostate Cancer Metastases. Exp. Opin. Investig. Drugs 2017, 26, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Cameron, M.D. Metabolism Considerations for Kinase Inhibitors in Cancer Treatment. Exp. Opin. Drug Metab. Toxicol. 2010, 6, 1175–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willson, T.M.; Kliewer, S.A. PXR, CAR and Drug Metabolism. Nat. Rev. Drug Discov. 2002, 1, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.C.; Cherian, M.T.; Wang, Y.-M.; Chen, T. Small-Molecule Modulators of PXR and CAR. Biochim. Biophys. Acta 2016, 1859, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-M.; Ong, S.S.; Chai, S.C.; Chen, T. Role of CAR and PXR in Xenobiotic Sensing and Metabolism. Exp. Opin. Drug Metab. Toxicol. 2012, 8, 803–817. [Google Scholar] [CrossRef] [Green Version]
- Oladimeji, P.O.; Chen, T. PXR: More Than Just a Master Xenobiotic Receptor. Mol. Pharmacol. 2018, 93, 119–127. [Google Scholar] [CrossRef]
- De Mattia, E.; Cecchin, E.; Roncato, R.; Toffoli, G. Pregnane X Receptor, Constitutive Androstane Receptor and Hepatocyte Nuclear Factors as Emerging Players in Cancer Precision Medicine. Pharmacogenomics 2016, 17, 1547–1571. [Google Scholar] [CrossRef]
- Hassen, W.; Kassambara, A.; Reme, T.; Sahota, S.; Seckinger, A.; Vincent, L.; Cartron, G.; Moreaux, J.; Hose, D.; Klein, B. Drug Metabolism and Clearance System in Tumor Cells of Patients with Multiple Myeloma. Oncotarget 2015, 6, 6431–6447. [Google Scholar] [CrossRef] [Green Version]
- Kong, Q.; Han, Z.; Zuo, X.; Wei, H.; Huang, W. Co-Expression of Pregnane X Receptor and ATP-Binding Cassette Sub-Family B Member 1 in Peripheral Blood: A Prospective Indicator for Drug Resistance Prediction in Non-Small Cell Lung Cancer. Oncol. Lett. 2016, 11, 3033–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmsen, S.; Meijerman, I.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H.M. PXR-Mediated P-Glycoprotein Induction by Small Molecule Tyrosine Kinase Inhibitors. Eur. J. Pharm. Sci. 2013, 48, 644–649. [Google Scholar] [CrossRef]
- MacLeod, A.K.; McLaughlin, L.A.; Henderson, C.J.; Wolf, C.R. Activation Status of the Pregnane X Receptor Influences Vemurafenib Availability in Humanized Mouse Models. Cancer Res. 2015, 75, 4573–4581. [Google Scholar] [CrossRef] [Green Version]
- Creusot, N.; Gassiot, M.; Alaterre, E.; Chiavarina, B.; Grimaldi, M.; Boulahtouf, A.; Toporova, L.; Gerbal-Chaloin, S.; Daujat-Chavanieu, M.; Matheux, A.; et al. The Anti-Cancer Drug Dabrafenib Is a Potent Activator of the Human Pregnane X Receptor. Cells 2020, 9, 1641. [Google Scholar] [CrossRef]
- Rigalli, J.P.; Ciriaci, N.; Arias, A.; Ceballos, M.P.; Villanueva, S.S.M.; Luquita, M.G.; Mottino, A.D.; Ghanem, C.I.; Catania, V.A.; Ruiz, M.L. Regulation of Multidrug Resistance Proteins by Genistein in a Hepatocarcinoma Cell Line: Impact on Sorafenib Cytotoxicity. PLoS ONE 2015, 10, e0119502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, F.; Jiang, Q.; Cao, S.; Cao, Y.; Li, R.; Shen, L.; Zhu, H.; Wang, T.; Sun, L.; Liang, E.; et al. Pregnane X Receptor Mediates Sorafenib Resistance in Advanced Hepatocellular Carcinoma. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Noll, E.M.; Eisen, C.; Stenzinger, A.; Espinet, E.; Muckenhuber, A.; Klein, C.; Vogel, V.; Klaus, B.; Nadler, W.; Rösli, C.; et al. CYP3A5 Mediates Basal and Acquired Therapy Resistance in Different Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Med. 2016, 22, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tang, Y.; Wang, M.-T.; Zeng, S.; Nie, D. Human Pregnane X Receptor and Resistance to Chemotherapy in Prostate Cancer. Cancer Res. 2007, 67, 10361–10367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, T.; Takahashi, S.; Urano, T.; Tanaka, T.; Zhang, W.; Azuma, K.; Takayama, K.; Obinata, D.; Murata, T.; Horie-Inoue, K.; et al. Clinical Significance of Steroid and Xenobiotic Receptor and Its Targeted Gene CYP3A4 in Human Prostate Cancer. Cancer Sci. 2012, 103, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Sramkoski, R.M.; Pretlow, T.G.; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A New Human Prostate Carcinoma Cell Line, 22Rv. In Vitro Cell. Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Van Hoppe, S.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast Cancer Resistance Protein (BCRP/ABCG2) and P-Glycoprotein (P-Gp/ABCB1) Transport Afatinib and Restrict Its Oral Availability and Brain Accumulation. Pharmacol. Res. 2017, 120, 43–50. [Google Scholar] [CrossRef]
- Stopfer, P.; Marzin, K.; Narjes, H.; Gansser, D.; Shahidi, M.; Uttereuther-Fischer, M.; Ebner, T. Afatinib Pharmacokinetics and Metabolism after Oral Administration to Healthy Male Volunteers. Cancer Chemother. Pharmacol. 2012, 69, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Bender, E.; Kopitz, C.; Grünewald, S.; Schlicker, A.; Schwede, W.; Eheim, A.; Toschi, L.; Neuhaus, R.; Richter, C.; et al. Preclinical Efficacy of the Novel Monocarboxylate Transporter 1 Inhibitor BAY-8002 and Associated Markers of Resistance. Mol. Cancer Ther. 2018, 17, 2285–2296. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Cheng, Q.; Ou, Z.; Lee, J.H.; Xu, M.; Kochhar, U.; Ren, S.; Huang, M.; Pflug, B.R.; Xie, W. Pregnane X Receptor as a Therapeutic Target to Inhibit Androgen Activity. Endocrinology 2010, 151, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Hiwase, D.K.; Saunders, V.; Hewett, D.; Frede, A.; Zrim, S.; Dang, P.; Eadie, L.; To, L.B.; Melo, J.; Kumar, S.; et al. Dasatinib Cellular Uptake and Efflux in Chronic Myeloid Leukemia Cells: Therapeutic Implications. Clin. Cancer Res. 2008, 14, 3881–3888. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, M.; He, P.; Hidalgo, M.; Baker, S.D. Differential Metabolism of Gefitinib and Erlotinib by Human Cytochrome P450 Enzymes. Clin. Cancer Res. 2007, 13, 3731–3737. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, H.-T.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.; et al. Dose Selection, Pharmacokinetics, and Pharmacodynamics of BRAF Inhibitor Dabrafenib (GSK2118436). Clin. Cancer Res. 2014, 20, 4449–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, S.; de Vries, N.A.; Buckle, T.; Bolijn, M.J.; van Eijndhoven, M.A.J.; Beijnen, J.H.; Mazzanti, R.; van Tellingen, O.; Schellens, J.H.M. Effect of the ATP-Binding Cassette Drug Transporters ABCB1, ABCG2, and ABCC2 on Erlotinib Hydrochloride (Tarceva) Disposition in in Vitro and in Vivo Pharmacokinetic Studies Employing Bcrp1-/-/Mdr1a/1b-/- (Triple-Knockout) and Wild-Type Mice. Mol. Cancer Ther. 2008, 7, 2280–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellens, H.; Johnson, M.; Lawrence, S.K.; Watson, C.; Chen, L.; Richards-Peterson, L.E. Prediction of the Transporter-Mediated Drug-Drug Interaction Potential of Dabrafenib and Its Major Circulating Metabolites. Drug Metab. Dispos. Biol. Fate Chem. 2017, 45, 646–656. [Google Scholar] [CrossRef]
- CHMP Assessment Report Giotrif. Available online: https://www.ema.europa.eu/en/documents/assessment-report/giotrif-epar-public-assessment-report_en.pdf (accessed on 1 February 2021).
- Sun, X.; Wang, M.; Wang, M.; Yao, L.; Li, X.; Dong, H.; Li, M.; Sun, T.; Liu, X.; Liu, Y.; et al. Role of Proton-Coupled Monocarboxylate Transporters in Cancer: From Metabolic Crosstalk to Therapeutic Potential. Front. Cell Dev. Biol. 2020, 8, 651. [Google Scholar] [CrossRef] [PubMed]
- Marchiq, I.; Pouysségur, J. Hypoxia, Cancer Metabolism and the Therapeutic Benefit of Targeting Lactate/H(+) Symporters. J. Mol. Med. 2016, 94, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Fisel, P.; Schaeffeler, E.; Schwab, M. Clinical and Functional Relevance of the Monocarboxylate Transporter Family in Disease Pathophysiology and Drug Therapy. Clin. Transl. Sci. 2018, 11, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Felisbino, S.; Massie, C.E.; Vizcaino, J.R.; Coelho, R.; Sandi, C.; Simoes-Sousa, S.; Jurmeister, S.; Ramos-Montoya, A.; Asim, M.; et al. A Glycolytic Phenotype Is Associated with Prostate Cancer Progression and Aggressiveness: A Role for Monocarboxylate Transporters as Metabolic Targets for Therapy. J. Pathol. 2015, 236, 517–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cundy, K.C.; Annamalai, T.; Bu, L.; De Vera, J.; Estrela, J.; Luo, W.; Shirsat, P.; Torneros, A.; Yao, F.; Zou, J.; et al. XP13512 [(+/−)-1-([(Alpha-Isobutanoyloxyethoxy)Carbonyl] Aminomethyl)-1-Cyclohexane Acetic Acid], a Novel Gabapentin Prodrug: II. Improved Oral Bioavailability, Dose Proportionality, and Colonic Absorption Compared with Gabapentin in Rats and Monkeys. J. Pharmacol. Exp. Ther. 2004, 311, 324–333. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Possemato, R.; Yilmaz, O.H.; Koch, C.E.; Chen, W.W.; Hutchins, A.W.; Gultekin, Y.; Peterson, T.R.; Carette, J.E.; et al. MCT1-Mediated Transport of a Toxic Molecule Is an Effective Strategy for Targeting Glycolytic Tumors. Nat. Genet. 2013, 45, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Coelho, F.; Nunes, C.; Gouveia-Fernandes, S.; Rosas, R.; Silva, F.; Gameiro, P.; Carvalho, T.; Gomes da Silva, M.; Cabeçadas, J.; Dias, S.; et al. Monocarboxylate Transporter 1 (MCT1), a Tool to Stratify Acute Myeloid Leukemia (AML) Patients and a Vehicle to Kill Cancer Cells. Oncotarget 2017, 8, 82803–82823. [Google Scholar] [CrossRef]
- Hamada, A.; Sissung, T.; Price, D.K.; Danesi, R.; Chau, C.H.; Sharifi, N.; Venzon, D.; Maeda, K.; Nagao, K.; Sparreboom, A.; et al. Effect of SLCO1B3 Haplotype on Testosterone Transport and Clinical Outcome in Caucasian Patients with Androgen-Independent Prostatic Cancer. Clin. Cancer Res. 2008, 14, 3312–3318. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, C.-Y.; Duan, Y.-J.; Huo, X.-K.; Meng, Q.; Liu, Z.-H.; Sun, H.-J.; Ma, X.-D.; Liu, K.-X. Afatinib Decreases P-Glycoprotein Expression to Promote Adriamycin Toxicity of A549T Cells. J. Cell. Biochem. 2018, 119, 414–423. [Google Scholar] [CrossRef]
- Toporova, L.; Grimaldi, M.; Boulahtouf, A.; Balaguer, P. Assessing the Selectivity of FXR, LXRs, CAR, and RORγ Pharmaceutical Ligands with Reporter Cell Lines. Front. Pharmacol. 2020, 11, 1122. [Google Scholar] [CrossRef]
- Bailey, I.; Gibson, G.G.; Plant, K.; Graham, M.; Plant, N. A PXR-Mediated Negative Feedback Loop Attenuates the Expression of CYP3A in Response to the PXR Agonist Pregnenalone-16α-Carbonitrile. PLoS ONE 2011, 6, e16703. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.K.; Daujat-Chavanieu, M.; Gerbal-Chaloin, S. Activation of the Aryl Hydrocarbon Receptor Decreases Rifampicin-Induced CYP3A4 Expression in Primary Human Hepatocytes and HepaRG. Toxicol. Lett. 2017, 277, 1–8. [Google Scholar] [CrossRef]
- Lee, C.; Ding, X.; Riddick, D.S. Downregulation of Mouse Hepatic CYP3A Protein by 3-Methylcholanthrene Does Not Require Cytochrome P450-Dependent Metabolism. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 1782–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Riddick, D.S. Aryl Hydrocarbon Receptor-Dependence of Dioxin’s Effects on Constitutive Mouse Hepatic Cytochromes P450 and Growth Hormone Signaling Components. Can. J. Physiol. Pharmacol. 2012, 90, 1354–1363. [Google Scholar] [CrossRef] [Green Version]
- Sugatani, J.; Mizushima, K.; Osabe, M.; Yamakawa, K.; Kakizaki, S.; Takagi, H.; Mori, M.; Ikari, A.; Miwa, M. Transcriptional Regulation of Human UGT1A1 Gene Expression through Distal and Proximal Promoter Motifs: Implication of Defects in the UGT1A1 Gene Promoter. Arch. Pharmacol. 2008, 377, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.G.; Mackenzie, P.I.; Nair, P.C.; McKinnon, R.A.; Meech, R. The Expression Profiles of ADME Genes in Human Cancers and Their Associations with Clinical Outcomes. Cancers 2020, 12, 3369. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | CLC (n = 449) | CRPC (n = 48) | Metastases (n = 28) Bone n = 15 Lymph Node n = 13 |
|---|---|---|---|
| Age y, median (range) | 63 (46–74) | 72 (56–86) | 66 (51–77) |
| PSA (ng/mL) | 10.1 (1.5–23) | 12.5 (0.2–285) | 12.4 (3.1–4894) |
| pTNM | |||
| pT2 | 315 | NA | NA |
| pT3 | 134 | ||
| ISUP group | |||
| 1 | 141 | NA | NA |
| 2 | 148 | ||
| 3 | 137 | ||
| 4–5 | 23 |
| ABCA12 | sense 5′- AGAACAGGCACTGCAAATGAAT antisense 5′-ATGGGAATGGCCAGCAACTT-3′; |
| ABCA2 | sense 5′-AGAACGTGACGCTCAAACGC, antisense 5′-GCGATTGCATGACAGGCAGG-3′; |
| ABCA5 | sense 5′-TGGAATGGACCCCTGTTCTCG, antisense 5′-CGGTAGCCGATCCCCCATTTA-3′; |
| ABCA9 | sense 5′-TGTTTTCTTATGATGCAAGGGCA, antisense 5′-AGGTCTTTCACACAACTTCCTTCA-3′; |
| ABCB1 | sense 5′-GTCGGACCACCATTGTGATAG-3′; antisense 5′-CATTTCCTGCTGTCTGCATTGTG-3′, |
| ABCC1 | sense 5′-GAATGGCATGCTGGTGACGG-3′; antisense 5′-AGCTTGACCTGCCCTGTCTG-3′; |
| ABCC10 | sense 5′-TGCCACCATCCGAGACAACA-3′; antisense 5′-CCTGGTAGACAGCACGAGCA-3′; |
| ABCD3 | sense 5′-TCGGCCTGCACGGTAAGAAA-3′; antisense 5′-TCGAGACACCAGCATAACAGCA-3′; |
| ABCD4 | sense 5′-CGGAGCCTGCTGCTTTCTAC-3′; antisense 5′-GACCCCGCTGAAAATGGGGA-3′; |
| ABCF1 | sense 5′-TCAGGATCAGAGTGAGGAAGAGGA-3′; antisense 5′-AGAGCAGCGAATTTATTTTGAGGC-3′; |
| CYP3A4 | sense 5′-GCCTGGTGCTCCTCTATCTA-3′; antisense 5′-GGCTGTTGACCATCATAAAAG-3′; |
| GAPDH | Sense 5′-AATTGAGCCCGCAGCCTCCC-3′; antisense 5′-CCAGGCGCCCAATACGACCA-3′ |
| HPRT | sense 5′-CTGACCTGCTGGATTACA-3′; antisense 5′-GCGACCTTGACCATCTTT-3′ |
| RPL0 | sense 5′-CAGATGGATCAGCCAAGAAGG-3′; antisense 5′-ATCAACGGGTACAAACGAGTC-3′ |
| SLC15A2 | sense 5′-AGTCCTTGGGTGCCTTACCA-3′; antisense 5′-AGCTCCCTGCATTGATGGACA-3′; |
| SLC16A1 | sense 5′-TTAAGGCGGCCCTGTTGAGA-3′; antisense 5′-TCCAATTACCACTGCCCAGC-3′ |
| SLC16A3 | sense 5′-GGAGGTGAGGCGGAACCAAC-3′; antisense 5′-TCCAGGCTGTGTCGCTGTAG-3′; |
| SLC19A2 | sense 5′-TTGCTGCAAACCTCAGCATGG-3′; antisense 5′-CCACTGGCCAGGAAAACCAC-3′; |
| SLC19A3 | sense 5′-CGGCAAGTGAGCGATTTGGT-3′; antisense 5′-TCTCTGCACTGGTCAGGTTTTT-3′; |
| SLC25A13 | sense 5′-GGCACCAGGAAAGATGTTGAAGT-3′; antisense 5′-GCCTCAGCCAAGTTAAAGGGC-3′; |
| SLC2A1 | sense 5′-CATGGCGGGTTGTGCCATAC-3′; antisense 5′-AGAAGCCTGCAACGGCAATG, |
| SLC31A1 | sense 5′-CAAGTGGCCAAAACCCCTGT, antisense 5′-CTCTGCCCTGAGGCACAACT-3′; |
| SLC38A2 | sense 5′-TCCTACCCCACCAAGCAAGC-3′; antisense 5′-ACCCTCCTTCATTGGCAGTCT-3′; |
| SLC3A2 | sense 5′-TCCTTCTTGCCGGCTCAACT-3′; antisense 5′-TTGGCACTTACAGCCCCTGG-3′ |
| SLC7A11 | sense 5′-CTCTGACTGGAGTCCCTGCG-3′; antisense 5′-TGTCTCCCCTTGGGCAGATTG-3′; |
| SLC7A6 | sense 5′-TTCATCCGCCTGTGGGTCTC-3′; antisense 5′-TGCCCCACTTGACATAGGCA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matheux, A.; Gassiot, M.; Fromont, G.; Leenhardt, F.; Boulahtouf, A.; Fabbrizio, E.; Marchive, C.; Garcin, A.; Agherbi, H.; Combès, E.; et al. PXR Modulates the Prostate Cancer Cell Response to Afatinib by Regulating the Expression of the Monocarboxylate Transporter SLC16A1. Cancers 2021, 13, 3635. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143635
Matheux A, Gassiot M, Fromont G, Leenhardt F, Boulahtouf A, Fabbrizio E, Marchive C, Garcin A, Agherbi H, Combès E, et al. PXR Modulates the Prostate Cancer Cell Response to Afatinib by Regulating the Expression of the Monocarboxylate Transporter SLC16A1. Cancers. 2021; 13(14):3635. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143635
Chicago/Turabian StyleMatheux, Alice, Matthieu Gassiot, Gaëlle Fromont, Fanny Leenhardt, Abdelhay Boulahtouf, Eric Fabbrizio, Candice Marchive, Aurélie Garcin, Hanane Agherbi, Eve Combès, and et al. 2021. "PXR Modulates the Prostate Cancer Cell Response to Afatinib by Regulating the Expression of the Monocarboxylate Transporter SLC16A1" Cancers 13, no. 14: 3635. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143635