
Comparing Tumor Cell Invasion and Myeloid Cell Composition in Compatible Primary and Relapsing Glioblastoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Ganciclovir Treatment In Vitro
2.2. Animals
2.3. Tumor Implantation, Tamoxifen and GCV Treatment In Vivo
2.4. Mouse Brain Tissue Preparation
2.5. H&E Staining, Tumor Size Quantification and Scoring of Invasiveness
2.6. Immunofluorescence Staining and Quantification on Mouse Brain Sections
2.7. Statistical Analysis
3. Results
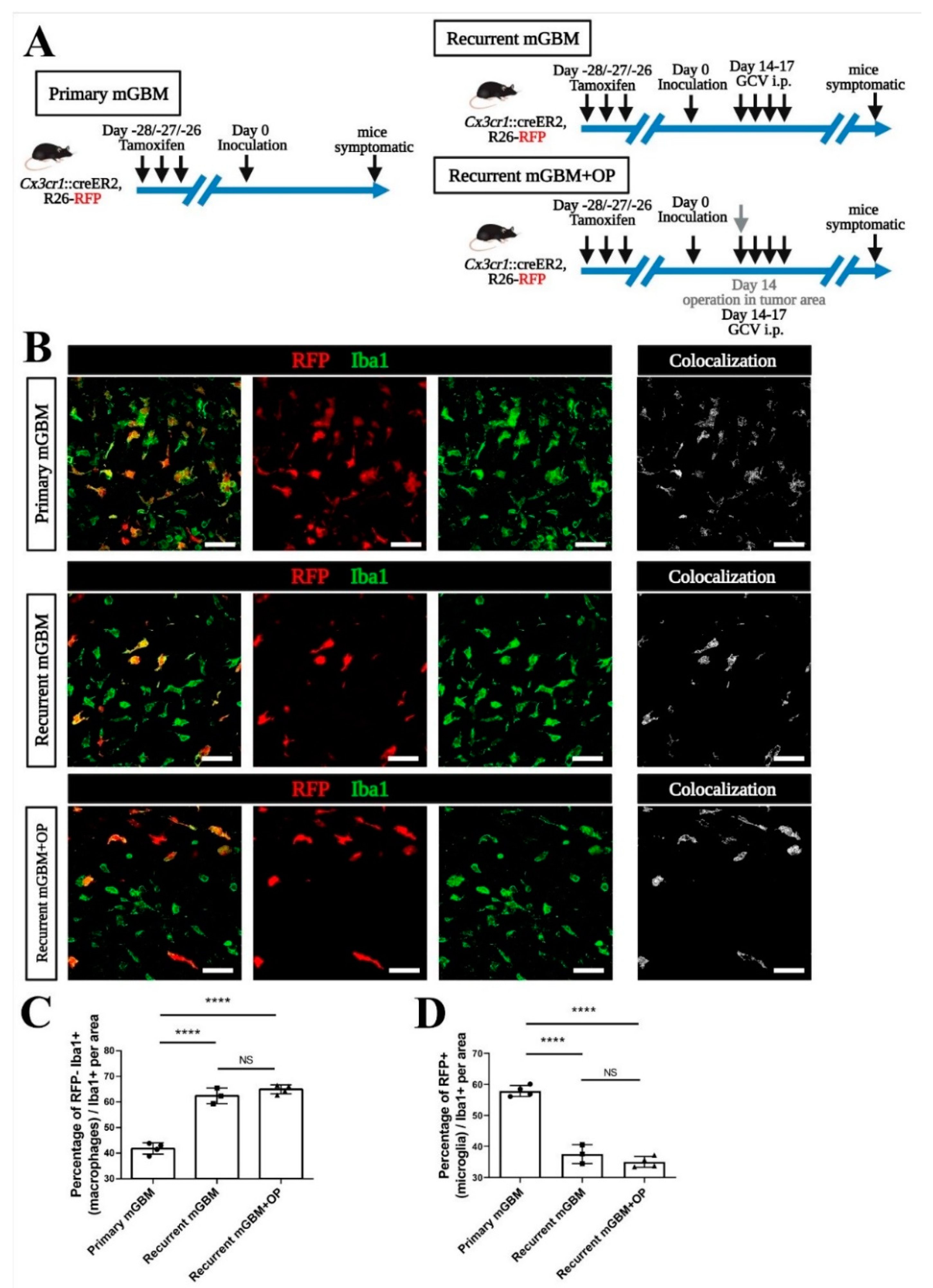
3.1. Establishing a Non-Invasive Mouse Model for GBM Recurrence
3.2. Increased Invasiveness in the Recurrent GBM Mouse Model
3.3. Monocyte-Derived Macrophages Make Up the Majority of TAM in Recurrent GBM
3.4. Signals from GBM Relapse and Not Surgical Intervention Itself Attract Peripheral Macrophages
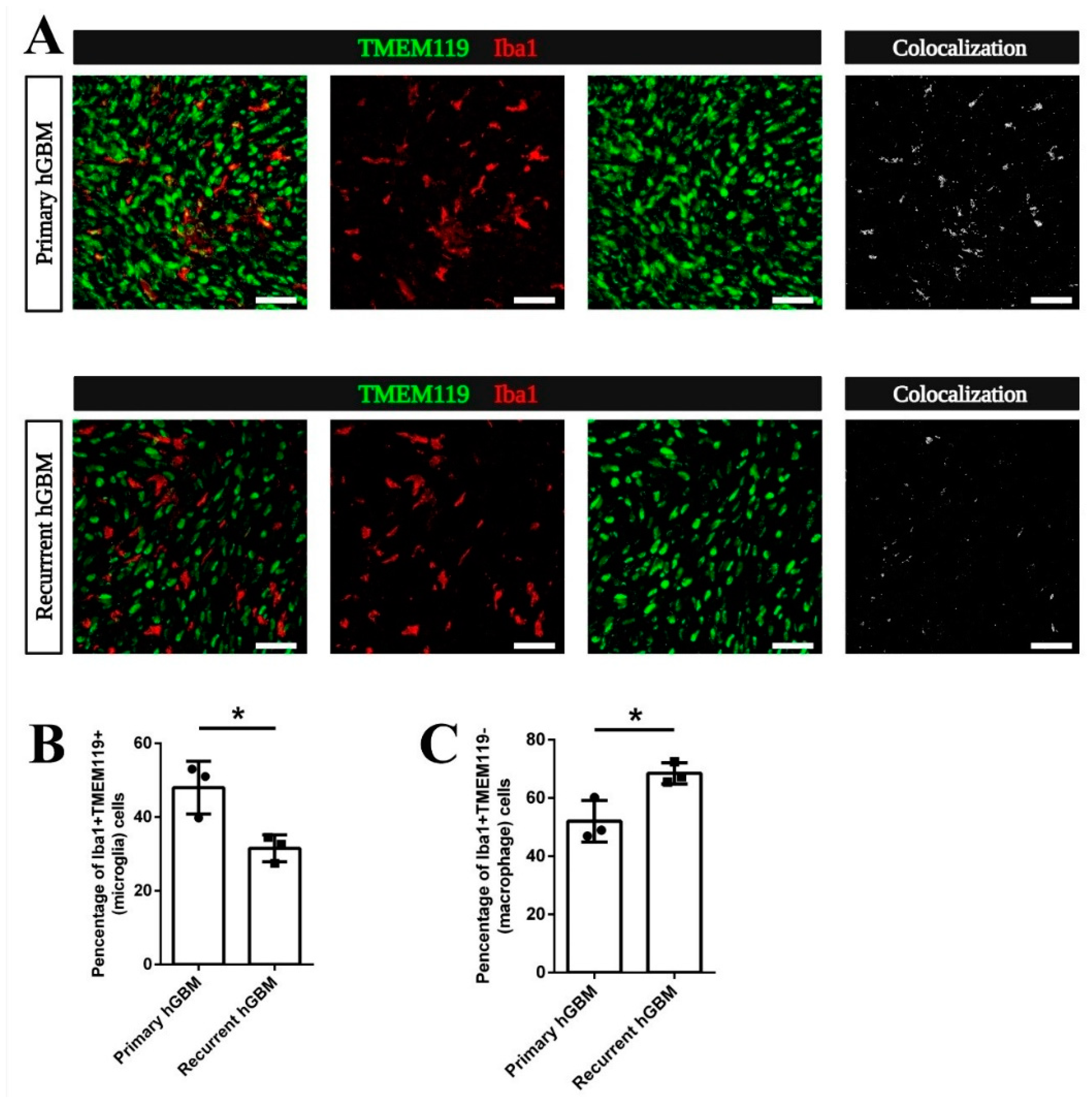
3.5. Increased Macrophage Attraction Is Preserved in a Patient-Derived Xenograft Model for Recurrent GBM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Bernstock, J.D.; Mooney, J.H.; Ilyas, A.; Chagoya, G.; Estevez-Ordonez, D.; Ibrahim, A.; Nakano, I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: Clinical and translational implications. J. Neurosurg. 2019, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Burger, P.C.; Dubois, P.J.; Schold, S.C., Jr.; Smith, K.R., Jr.; Odom, G.L.; Crafts, D.C.; Giangaspero, F. Computerized tomographic and pathologic studies of the untreated, quiescent, and recurrent glioblastoma multiforme. J. Neurosurg. 1983, 58, 159–169. [Google Scholar] [CrossRef]
- De Bonis, P.; Anile, C.; Pompucci, A.; Fiorentino, A.; Balducci, M.; Chiesa, S.; Lauriola, L.; Maira, G.; Mangiola, A. The influence of surgery on recurrence pattern of glioblastoma. Clin. Neurol. Neurosurg. 2013, 115, 37–43. [Google Scholar] [CrossRef]
- Van Nifterik, K.A.; Elkhuizen, P.H.; van Andel, R.J.; Stalpers, L.J.; Leenstra, S.; Lafleur, M.V.; Vandertop, W.P.; Slotman, B.J.; Hulsebos, T.J.; Sminia, P. Genetic profiling of a distant second glioblastoma multiforme after radiotherapy: Recurrence or second primary tumor? J. Neurosurg. 2006, 105, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Tully, P.A.; Gogos, A.J.; Love, C.; Liew, D.; Drummond, K.J.; Morokoff, A.P. Reoperation for Recurrent Glioblastoma and Its Association With Survival Benefit. Neurosurgery 2016, 79, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, A.; Conroy, S.; Glass, R.; Bhat, K.P.L. The Impact of the Tumor Microenvironment on the Properties of Glioma Stem-Like Cells. Front. Oncol. 2017, 7, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Wieghofer, P.; Knobeloch, K.P.; Prinz, M. Genetic targeting of microglia. Glia 2015, 63, 1–22. [Google Scholar] [CrossRef]
- Kälin, R.E.; Cai, L.; Li, Y.; Zhao, D.; Zhang, H.; Cheng, J.; Zhang, W.; Wu, Y.; Eisenhut, K.; Janssen, P.; et al. TAMEP are brain tumor parenchymal cells controlling neoplastic angiogenesis and progression. Cell Syst. 2021. [Google Scholar] [CrossRef]
- Glass, R.; Synowitz, M.; Kronenberg, G.; Walzlein, J.H.; Markovic, D.S.; Wang, L.P.; Gast, D.; Kiwit, J.; Kempermann, G.; Kettenmann, H. Glioblastoma-induced attraction of endogenous neural precursor cells is associated with improved survival. J. Neurosci. 2005, 25, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watters, J.J.; Schartner, J.M.; Badie, B. Microglia function in brain tumors. J. Neurosci. Res. 2005, 81, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical debulking promotes recruitment of macrophages and triggers glioblastoma phagocytosis in combination with CD47 blocking immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef] [Green Version]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Beltinger, C.; Fulda, S.; Kammertoens, T.; Meyer, E.; Uckert, W.; Debatin, K.M. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc. Natl. Acad. Sci. USA 1999, 96, 8699–8704. [Google Scholar] [CrossRef] [Green Version]
- Tomicic, M.T.; Thust, R.; Kaina, B. Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: Critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene 2002, 21, 2141–2153. [Google Scholar] [CrossRef] [Green Version]
- Bello, L.; Giussani, C.; Carrabba, G.; Pluderi, M.; Lucini, V.; Pannacci, M.; Caronzolo, D.; Tomei, G.; Villani, R.; Scaglione, F.; et al. Suppression of malignant glioma recurrence in a newly developed animal model by endogenous inhibitors. Clin. Cancer Res. 2002, 8, 3539–3548. [Google Scholar]
- Kauer, T.M.; Figueiredo, J.L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011, 15, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Hingtgen, S.; Figueiredo, J.L.; Farrar, C.; Duebgen, M.; Martinez-Quintanilla, J.; Bhere, D.; Shah, K. Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J. Neuro Oncol. 2013, 111, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.S.; Holland, E.C. Standard of care therapy for malignant glioma and its effect on tumor and stromal cells. Oncogene 2012, 31, 1995–2006. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.V.; Bergers, G. Mechanisms of evasive resistance to anti-VEGF therapy in glioblastoma. CNS Oncol. 2013, 2, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Hossain, J.A.; Latif, M.A.; Ystaas, L.A.R.; Ninzima, S.; Riecken, K.; Muller, A.; Azuaje, F.; Joseph, J.V.; Talasila, K.M.; Ghimire, J.; et al. Long-term treatment with valganciclovir improves lentiviral suicide gene therapy of glioblastoma. Neuro Oncol. 2019, 21, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Drachsler, M.; Kleber, S.; Mateos, A.; Volk, K.; Mohr, N.; Chen, S.; Cirovic, B.; Tuttenberg, J.; Gieffers, C.; Sykora, J.; et al. CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Dis. 2016, 7, e2209. [Google Scholar] [CrossRef]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastrella, G.; Hou, M.; Li, M.; Stoecklein, V.M.; Zdouc, N.; Volmar, M.N.M.; Miletic, H.; Reinhard, S.; Herold-Mende, C.C.; Kleber, S.; et al. Targeting APLN/APLNR Improves Antiangiogenic Efficiency and Blunts Proinvasive Side Effects of VEGFA/VEGFR2 Blockade in Glioblastoma. Cancer Res. 2019, 79, 2298–2313. [Google Scholar] [CrossRef] [Green Version]
- Frisch, A.; Kälin, S.; Monk, R.; Radke, J.; Heppner, F.L.; Kälin, R.E. Apelin Controls Angiogenesis-Dependent Glioblastoma Growth. Int. J. Mol. Sci. 2020, 21, 4179. [Google Scholar] [CrossRef]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neuro Oncol. 2011, 101, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Heath, R.N.; Shah, A.H.; Sanjurjo, A.D.; Eichberg, D.G.; Luther, E.M.; de la Fuente, M.I.; Komotar, R.J.; Ivan, M.E. Resection versus biopsy in the treatment of multifocal glioblastoma: A weighted survival analysis. J. Neuro Oncol. 2020, 148, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Stupp, R.; Beule, A.C.; Bromberg, J.; Wick, A.; Ernemann, U.; Platten, M.; Marosi, C.; Mason, W.P.; van den Bent, M.; et al. A novel tool to analyze MRI recurrence patterns in glioblastoma. Neuro Oncol. 2008, 10, 1019–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Volmar, M.N.M.; Cheng, J.; Alenezi, H.; Richter, S.; Haug, A.; Hassan, Z.; Goldberg, M.; Li, Y.; Hou, M.; Herold-Mende, C.; et al. Cannabidiol converts NFkappaB into a tumor suppressor in glioblastoma with defined antioxidative properties. Neuro Oncol. 2021. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Woodroffe, R.W.; Zanaty, M.; Soni, N.; Mott, S.L.; Helland, L.C.; Pasha, A.; Maley, J.; Dhungana, N.; Jones, K.A.; Monga, V.; et al. Survival after reoperation for recurrent glioblastoma. J. Clin. Neurosci. 2020, 73, 118–124. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Aizer, A.A.; Ancukiewicz, M.; Nguyen, P.L.; Shih, H.A.; Loeffler, J.S.; Oh, K.S. Underutilization of radiation therapy in patients with glioblastoma: Predictive factors and outcomes. Cancer 2014, 120, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Sulman, E.P.; Ismaila, N.; Armstrong, T.S.; Tsien, C.; Batchelor, T.T.; Cloughesy, T.; Galanis, E.; Gilbert, M.; Gondi, V.; Lovely, M.; et al. Radiation Therapy for Glioblastoma: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, J.A.; Ystaas, L.R.; Mrdalj, J.; Valk, K.; Riecken, K.; Fehse, B.; Bjerkvig, R.; Gronli, J.; Miletic, H. Lentiviral HSV-Tk.007-mediated suicide gene therapy is not toxic for normal brain cells. J. Gene Med. 2016, 18, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Grathwohl, S.A.; Kälin, R.E.; Bolmont, T.; Prokop, S.; Winkelmann, G.; Kaeser, S.A.; Odenthal, J.; Radde, R.; Eldh, T.; Gandy, S.; et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 2009, 12, 1361–1363. [Google Scholar] [CrossRef] [PubMed]
- Huszthy, P.C.; Giroglou, T.; Tsinkalovsky, O.; Euskirchen, P.; Skaftnesmo, K.O.; Bjerkvig, R.; von Laer, D.; Miletic, H. Remission of invasive, cancer stem-like glioblastoma xenografts using lentiviral vector-mediated suicide gene therapy. PLoS ONE 2009, 4, e6314. [Google Scholar] [CrossRef]
- Le Reste, P.J.; Pineau, R.; Voutetakis, K.; Samal, J.; Jegou, G.; Lhomond, S.; Gorman, A.M.; Samali, A.; Patterson, J.B.; Zeng, Q.; et al. Local intracerebral inhibition of IRE1 by MKC8866 sensitizes glioblastoma to irradiation/chemotherapy in vivo. Cancer Lett. 2020, 494, 73–83. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.; Zhang, H.; Uyar, R.; Hossain, J.A.; Miletic, H.; Tonn, J.-C.; Glass, R.; Kälin, R.E. Comparing Tumor Cell Invasion and Myeloid Cell Composition in Compatible Primary and Relapsing Glioblastoma. Cancers 2021, 13, 3636. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143636
Zhao D, Zhang H, Uyar R, Hossain JA, Miletic H, Tonn J-C, Glass R, Kälin RE. Comparing Tumor Cell Invasion and Myeloid Cell Composition in Compatible Primary and Relapsing Glioblastoma. Cancers. 2021; 13(14):3636. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143636
Chicago/Turabian StyleZhao, Dongxu, Huabin Zhang, Ramazan Uyar, Jubayer A. Hossain, Hrvoje Miletic, Jörg-Christian Tonn, Rainer Glass, and Roland E. Kälin. 2021. "Comparing Tumor Cell Invasion and Myeloid Cell Composition in Compatible Primary and Relapsing Glioblastoma" Cancers 13, no. 14: 3636. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143636