
Geospatial Assessments of DNA Adducts in the Human Stomach: A Model of Field Cancerization

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Gastric Cancer Risk
3. Mutation Spectrum and Mutational Signature of Gastric Cancer
3.1. Mutation Spectrum
3.2. Mutational Signature
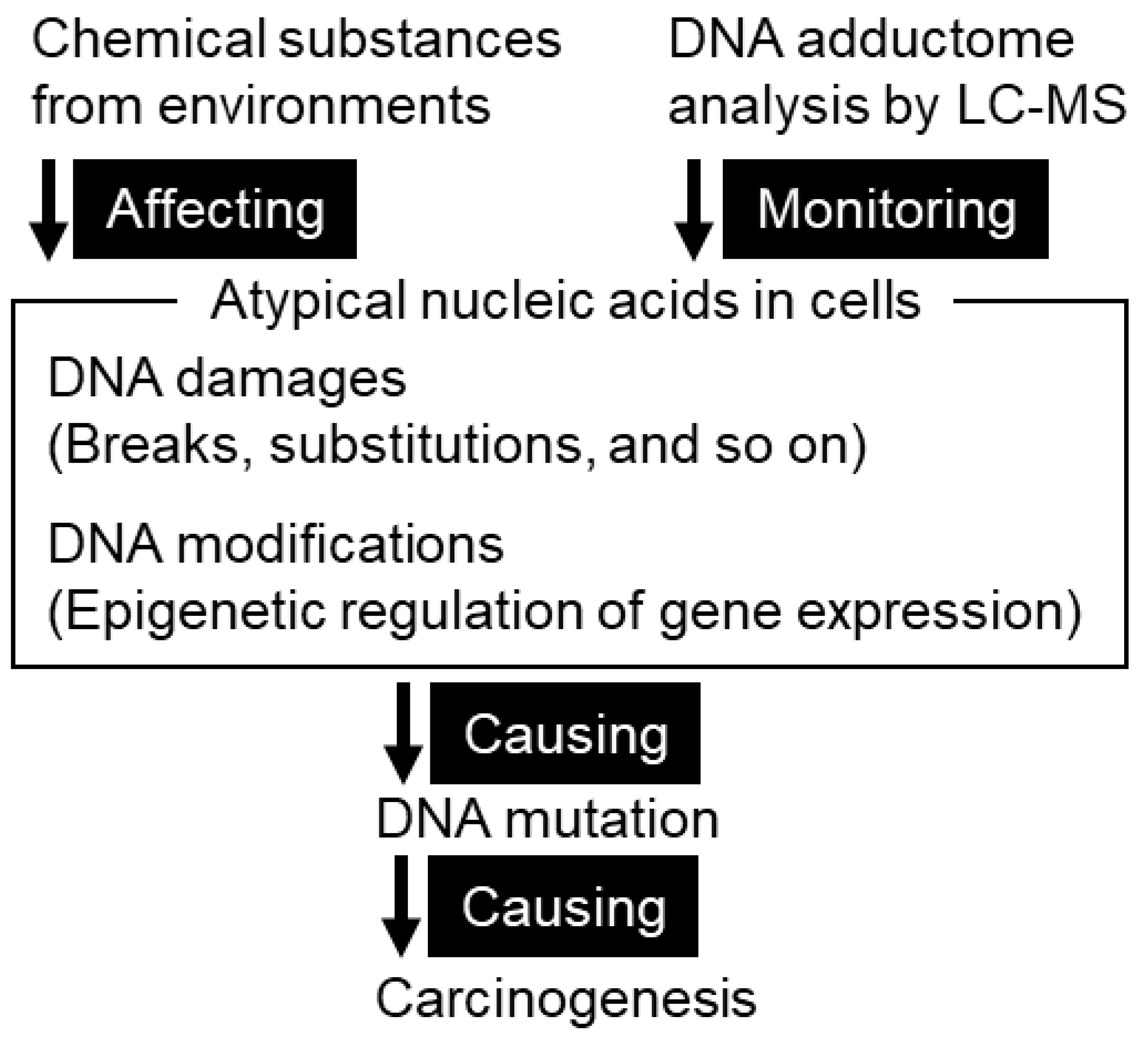
4. DNA Adducts as a Cause of Mutation
4.1. DNA Damage in Gastric Mucosae
4.1.1. Types of DNA Damage
4.1.2. Methodology for Identification of DNA Adducts
4.1.3. DNA Adductome
4.2. DNA Adduct Profile of Gastric Mucosae
4.2.1. Lipid Peroxidation-Induced DNA Adducts in Human Gastric Mucosa
4.2.2. Application of DNA Adductomics Technology to the Analysis of Cytosine Modifications in Gastric Cancer
4.2.3. Mass Spectrometric Profiling of DNA Adducts in the Human Stomach Associated with Damage from Environmental Factors
5. Field Cancerization and DNA Adductomics in the Stomach
5.1. Pathology of Gastric Cancer
5.1.1. Practice in Gastric Cancer Management: Detection, Diagnosis, and Therapy
5.1.2. Recent Molecular Characterization of Gastric Cancer
5.2. Field Cancerization
5.2.1. Original Concept of Field Cancerization
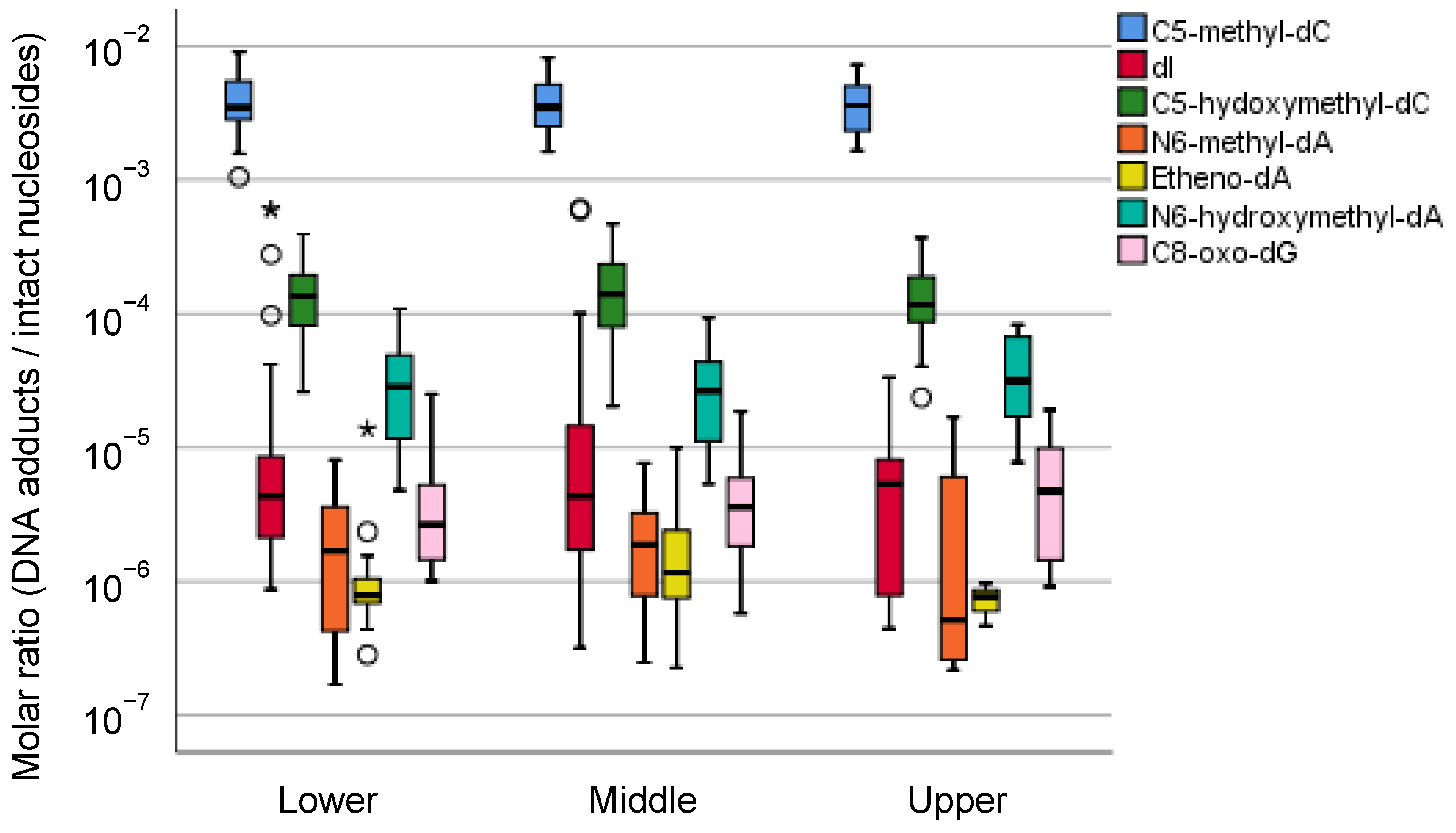
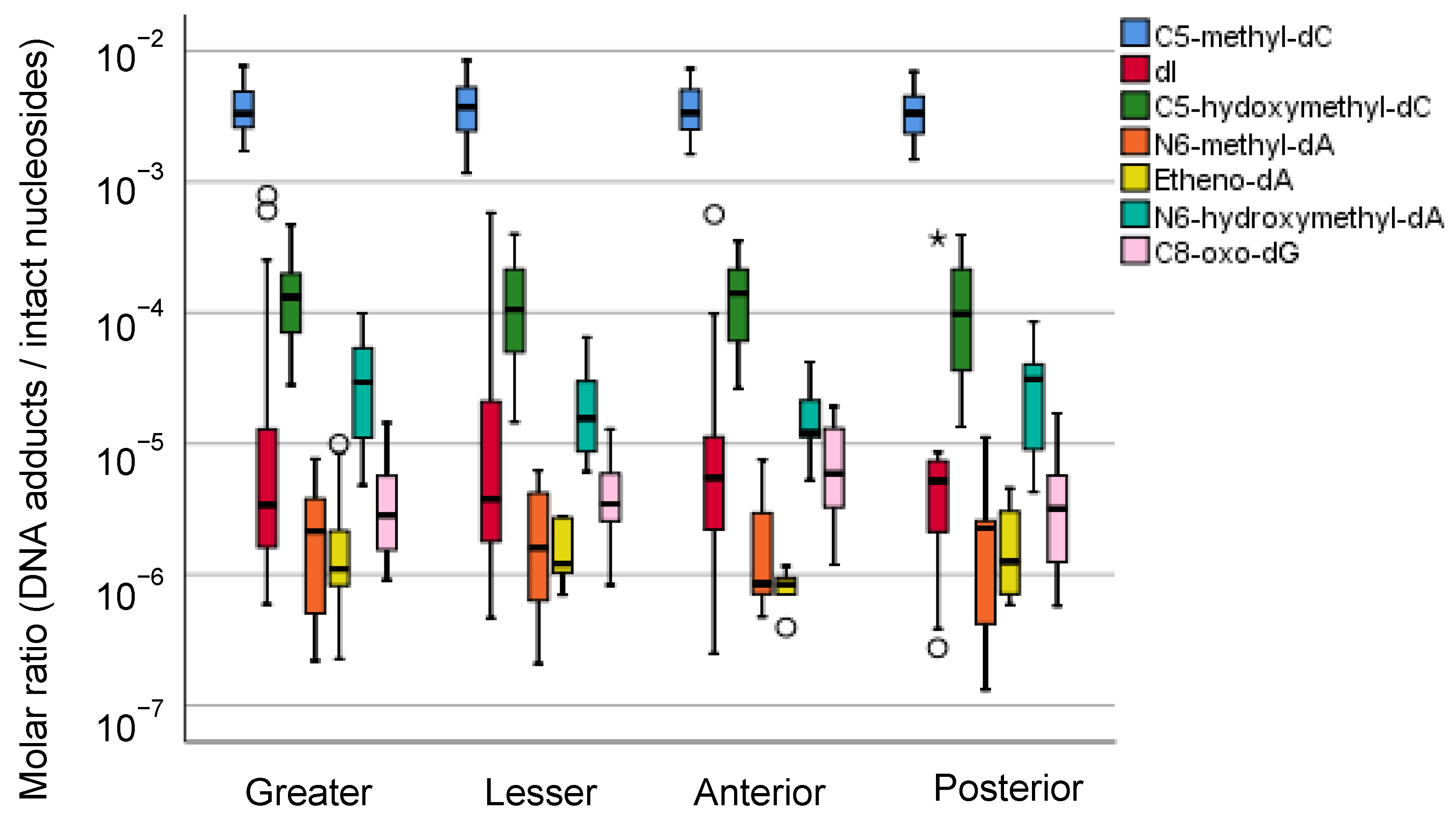
5.2.2. Field Cancerization of Gastric Mucosae and Preferential Cancer Location within the Stomach
5.2.3. Mass Spectrometric Profiling of DNA Adducts in the Local Part of the Human Stomach
6. Perspectives to Evaluate the Field Cancerization of Gastric Cancer
6.1. Mutational Signature Analysis Combined with Experimental Exposure and Perturbation of Genetic Backgrounds
6.2. Detection of DNA Damage by DNA Sequencing
6.3. Social and Scientific Significance of the Field Cancerization of Gastric Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Agency for Research on Cancer/World Health Organization. Global Cancer Observatory. 2021. Available online: https://gco.iarc.fr/ (accessed on 30 April 2021).
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Haenszel, W.; Kurihara, M. Studies of Japanese migrants. I. Mortality from cancer and other diseases among Japanese in the United States. J. Natl. Cancer Inst. 1968, 40, 43–68. [Google Scholar] [PubMed]
- Iwasaki, M.; Mameri, C.P.; Hamada, G.S.; Tsugane, S. Secular trends in cancer mortality among Japanese immigrants in the state of Sao Paulo, Brazil, 1979–2001. Eur. J. Cancer Prev. 2008, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hjerkind, K.V.; Qureshi, S.A.; Moller, B.; Weiderpass, E.; Deapen, D.; Kumar, B.; Ursin, G. Ethnic differences in the incidence of cancer in Norway. Int. J. Cancer 2017, 140, 1770–1780. [Google Scholar] [CrossRef]
- Sugimura, T.; Fujimura, S. Tumour production in glandular stomach of rat by N-methyl-N′-nitro-N-nitrosoguanidine. Nature 1967, 216, 943–944. [Google Scholar] [CrossRef]
- Matsukura, N.; Kawachi, T.; Sasajima, K.; Sano, T.; Sugimura, T.; Hirota, T. Induction of intestinal metaplasia in the stomachs of rats by N-methyl-N’-nitro-N-nitrosoguanidine. J. Natl. Cancer Inst. 1978, 61, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Fukase, K.; Kato, M.; Kikuchi, S.; Inoue, K.; Uemura, N.; Okamoto, S.; Terao, S.; Amagai, K.; Hayashi, S.; Asaka, M.; et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: An open-label, randomised controlled trial. Lancet 2008, 372, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Wynder, E.L.; Kmet, J.; Dungal, N.; Segi, M. An Epidemiological Investigation of Gastric Cancer. Cancer 1963, 16, 1461–1496. [Google Scholar] [CrossRef]
- Ohnami, S.; Sato, Y.; Yoshimura, K.; Ohnami, S.; Sakamoto, H.; Aoki, K.; Ueno, H.; Ikeda, M.; Morizane, C.; Shimada, K.; et al. His595Tyr polymorphism in the methionine synthase reductase (MTRR) gene is associated with pancreatic cancer risk. Gastroenterology 2008, 135, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Saeki, N.; Saito, A.; Choi, I.J.; Matsuo, K.; Ohnami, S.; Totsuka, H.; Chiku, S.; Kuchiba, A.; Lee, Y.S.; Yoon, K.A.; et al. A functional single nucleotide polymorphism in mucin 1, at chromosome 1q22, determines susceptibility to diffuse-type gastric cancer. Gastroenterology 2011, 140, 892–902. [Google Scholar] [CrossRef]
- Tanikawa, C.; Kamatani, Y.; Toyoshima, O.; Sakamoto, H.; Ito, H.; Takahashi, A.; Momozawa, Y.; Hirata, M.; Fuse, N.; Takai-Igarashi, T.; et al. Genome-wide association study identifies gastric cancer susceptibility loci at 12q24.11-12 and 20q11.21. Cancer Sci. 2018, 109, 4015–4024. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Yoshimura, K.; Saeki, N.; Katai, H.; Shimoda, T.; Matsuno, Y.; Saito, D.; Sugimura, H.; Tanioka, F.; Shunji, K.; et al. Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nat. Genet. 2008, 40, 730–740. [Google Scholar] [CrossRef]
- Haenszel, W.; Kurihara, M.; Segi, M.; Lee, R.K. Stomach cancer among Japanese in Hawaii. J. Natl. Cancer Inst. 1972, 49, 969–988. [Google Scholar]
- Iwasaki, M.; Mameri, C.P.; Hamada, G.S.; Tsugane, S. Cancer mortality among Japanese immigrants and their descendants in the state of Sao Paulo, Brazil, 1999–2001. Jpn. J. Clin. Oncol. 2004, 34, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, T.; El-Omar, E.M.; Xiao, F.; Shirai, N.; Takashima, M.; Sugimura, H. Interleukin 1beta polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology 2002, 123, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Akabane, M.; Inami, T.; Matsushima, S.; Ishibashi, T.; Ichinowatari, Y.; Miyajima, Y.; Watanabe, S. Urinary salt excretion and stomach cancer mortality among four Japanese populations. Cancer Causes Control. 1991, 2, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Merliss, R.R. Talc-treated rice and Japanese stomach cancer. Science 1971, 173, 1141–1142. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.J. Carcinogenesis of gastrointestinal cancer. Front. Gastrointest. Res. 1979, 4, 1–16. [Google Scholar] [CrossRef]
- Wynder, E.L.; Reddy, B.S.; McCoy, G.D.; Weisburger, J.H.; Williams, G.M. Diet and cancer of the gastrointestinal tract. Adv. Intern. Med. 1977, 22, 397–419. [Google Scholar] [PubMed]
- Lijinsky, W.; Reuber, M.D.; Blackwell, B.N. Carcinogenicity of nitrosotrialkylureas in Fischer 344 rats. J. Natl. Cancer Inst. 1980, 65, 451–453. [Google Scholar]
- Sugimura, T.; Wakabayashi, K. Gastric carcinogenesis: Diet as a causative factor. Med. Oncol. Tumor Pharmacother. 1990, 7, 87–92. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Tornaletti, S.; Pfeifer, G.P. Complete and tissue-independent methylation of CpG sites in the p53 gene: Implications for mutations in human cancers. Oncogene 1995, 10, 1493–1499. [Google Scholar]
- Shiao, Y.H.; Palli, D.; Buzard, G.S.; Caporaso, N.E.; Amorosi, A.; Saieva, C.; Fraumeni, J.F., Jr.; Anderson, L.M.; Rice, J.M. Implications of p53 mutation spectrum for cancer etiology in gastric cancers of various histologic types from a high-risk area of central Italy. Carcinogenesis 1998, 19, 2145–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, T.; Shibata, T.; Okamoto, Y.; Yamazaki, J.; Kawamura, T.; Horiguchi, N.; Okubo, M.; Nakano, N.; Ishizuka, T.; Nagasaka, M.; et al. Mutation spectrum of TP53 gene predicts clinicopathological features and survival of gastric cancer. Oncotarget 2016, 7, 42252–42260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M.; Risques, R.A.; Toyota, M.; Capella, G.; Moreno, V.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001, 61, 4689–4692. [Google Scholar] [PubMed]
- Klungland, A.; Laake, K.; Hoff, E.; Seeberg, E. Spectrum of mutations induced by methyl and ethyl methanesulfonate at the hprt locus of normal and tag expressing Chinese hamster fibroblasts. Carcinogenesis 1995, 16, 1281–1285. [Google Scholar] [CrossRef]
- Richardson, K.K.; Richardson, F.C.; Crosby, R.M.; Swenberg, J.A.; Skopek, T.R. DNA base changes and alkylation following in vivo exposure of Escherichia coli to N-methyl-N-nitrosourea or N-ethyl-N-nitrosourea. Proc. Natl. Acad. Sci. USA 1987, 84, 344–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, N.; Bertrand, D.; Hillmer, A.M.; Zang, Z.J.; Yao, F.; Jacques, P.E.; Teo, A.S.; Cutcutache, I.; Zhang, Z.; Lee, W.H.; et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Siu, H.C.; Leung, S.Y.; Stratton, M.R. A mutational signature in gastric cancer suggests therapeutic strategies. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative Molecular Analysis of Gastrointestinal Adenocarcinomas. Cancer Cell 2018, 33, 721–735.e28. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.S.; Kim, K.M.; Ting, J.C.; Yu, K.; Fu, J.; Liu, S.; Cristescu, R.; Nebozhyn, M.; Gong, L.; Yue, Y.G.; et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Ramnarayanan, K.; Zhu, F.; Srivastava, S.; Xu, C.; Tan, A.L.K.; Lee, M.; Tay, S.; Das, K.; Xing, M.; et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell 2018, 33, 137–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology 2014, 147, 407–417. [Google Scholar] [CrossRef]
- Suzuki, A.; Katoh, H.; Komura, D.; Kakiuchi, M.; Tagashira, A.; Yamamoto, S.; Tatsuno, K.; Ueda, H.; Nagae, G.; Fukuda, S.; et al. Defined lifestyle and germline factors predispose Asian populations to gastric cancer. Sci. Adv. 2020, 6, eaav9778. [Google Scholar] [CrossRef]
- Phillips, D.H.; Arlt, V.M. The 32P-postlabeling assay for DNA adducts. Nat. Protoc. 2007, 2, 2772–2781. [Google Scholar] [CrossRef]
- Kanaly, R.A.; Hanaoka, T.; Sugimura, H.; Toda, H.; Matsui, S.; Matsuda, T. Development of the adductome approach to detect DNA damage in humans. Antioxid. Redox Signal. 2006, 8, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.H.; Kageyama, S.; Matsuda, S.; Kanemoto, K.; Sasada, Y.; Oka, M.; Shinmura, K.; Mori, H.; Kawai, K.; Kasai, H.; et al. Detection of lipid peroxidation-induced DNA adducts caused by 4-oxo-2(E)-nonenal and 4-oxo-2(E)-hexenal in human autopsy tissues. Chem. Res. Toxicol. 2010, 23, 1442–1448. [Google Scholar] [CrossRef]
- Ishino, K.; Kato, T.; Kato, M.; Shibata, T.; Watanabe, M.; Wakabayashi, K.; Nakagama, H.; Totsuka, Y. Comprehensive DNA adduct analysis reveals pulmonary inflammatory response contributes to genotoxic action of magnetite nanoparticles. Int. J. Mol. Sci. 2015, 16, 3474–3492. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Villalta, P.W.; Turesky, R.J. Data-Independent Mass Spectrometry Approach for Screening and Identification of DNA Adducts. Anal. Chem. 2017, 89, 11728–11736. [Google Scholar] [CrossRef]
- Carra, A.; Guidolin, V.; Dator, R.P.; Upadhyaya, P.; Kassie, F.; Villalta, P.W.; Balbo, S. Targeted High Resolution LC/MS(3) Adductomics Method for the Characterization of Endogenous DNA Damage. Front. Chem. 2019, 7, 658. [Google Scholar] [CrossRef]
- Tretyakova, N.; Goggin, M.; Sangaraju, D.; Janis, G. Quantitation of DNA adducts by stable isotope dilution mass spectrometry. Chem. Res. Toxicol. 2012, 25, 2007–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Turesky, R.J.; Tarifa, A.; DeCaprio, A.P.; Cooke, M.S.; Walmsley, S.J.; Villalta, P.W. Development of a DNA Adductome Mass Spectral Database. Chem. Res. Toxicol. 2020, 33, 852–854. [Google Scholar] [CrossRef]
- Hemminki, K. Nucleic acid adducts of chemical carcinogens and mutagens. Arch. Toxicol. 1983, 52, 249–285. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Tao, H.; Goto, M.; Yamada, H.; Suzuki, M.; Wu, Y.; Xiao, N.; He, Q.; Guo, W.; Cai, Z.; et al. Lipid peroxidation-induced DNA adducts in human gastric mucosa. Carcinogenesis 2013, 34, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Shinmura, K.; Matsushima, Y.; Ishino, K.; Yamada, H.; Totsuka, Y.; Matsuda, T.; Nakagama, H.; Sugimura, H. Human DNA glycosylase enzyme TDG repairs thymine mispaired with exocyclic etheno-DNA adducts. Free Radic. Biol. Med. 2014, 76, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavli, B.; Sundheim, O.; Akbari, M.; Otterlei, M.; Nilsen, H.; Skorpen, F.; Aas, P.A.; Hagen, L.; Krokan, H.E.; Slupphaug, G. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem. 2002, 277, 39926–39936. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, A.E.; O’Brien, P.J. Kinetic mechanism for the flipping and excision of 1,N(6)-ethenoadenine by human alkyladenine DNA glycosylase. Biochemistry 2009, 48, 11357–11369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Kurabe, N.; Matsushima, Y.; Suzuki, M.; Kahyo, T.; Ohnishi, I.; Tanioka, F.; Tajima, S.; Goto, M.; Yamada, H.; et al. Robust quantitative assessments of cytosine modifications and changes in the expressions of related enzymes in gastric cancer. Gastric Cancer 2015, 18, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, I.; Iwashita, Y.; Matsushita, Y.; Ohtsuka, S.; Yamashita, T.; Inaba, K.; Fukazawa, A.; Ochiai, H.; Matsumoto, K.; Kurono, N.; et al. Mass spectrometric profiling of DNA adducts in the human stomach associated with damage fromenvironmental factors. Genes Environ. 2021, 43, 1–13. [Google Scholar] [CrossRef]
- Xiong, J.; Ye, T.T.; Ma, C.J.; Cheng, Q.Y.; Yuan, B.F.; Feng, Y.Q. N 6-Hydroxymethyladenine: A hydroxylation derivative of N6-methyladenine in genomic DNA of mammals. Nucleic Acids Res. 2019, 47, 1268–1277. [Google Scholar] [CrossRef] [Green Version]
- Lauren, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- WHO. Digestive System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Guo, R.J.; Arai, H.; Kitayama, Y.; Igarashi, H.; Hemmi, H.; Arai, T.; Hanai, H.; Sugimura, H. Microsatellite instability of papillary subtype of human gastric adenocarcinoma and hMLH1 promoter hypermethylation in the surrounding mucosa. Pathol. Int. 2001, 51, 240–247. [Google Scholar] [CrossRef]
- Song, J.P.; Kitayama, Y.; Igarashi, H.; Guo, R.J.; Wang, Y.J.; Kobayashi, T.; Konno, H.; Kataoka, H.; Tanaka, M.; Sugimura, H. Centromere numerical abnormality in the papillary, papillotubular type of early gastric cancer, a further characterization of a subset of gastric cancer. Int. J. Oncol. 2002, 21, 1205–1211. [Google Scholar] [CrossRef]
- Chan, K.; Brown, I.S.; Kyle, T.; Lauwers, G.Y.; Kumarasinghe, M.P. Chief cell-predominant gastric polyps: A series of 12 cases with literature review. Histopathology 2016, 68, 825–833. [Google Scholar] [CrossRef]
- Ushiku, T.; Shinozaki, A.; Shibahara, J.; Iwasaki, Y.; Tateishi, Y.; Funata, N.; Fukayama, M. SALL4 represents fetal gut differentiation of gastric cancer, and is diagnostically useful in distinguishing hepatoid gastric carcinoma from hepatocellular carcinoma. Am. J. Surg. Pathol. 2010, 34, 533–540. [Google Scholar] [CrossRef]
- Japanese Gastric Cancer Association. Japanese classification of gastric carcinoma: 3rd English edition. Gastric Cancer 2011, 14, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Oota, K.; Sobin, L.H. Histological Typing of Gastric and Oesophageal Tumours, 1st ed.; World Health Organization: Geneva, Switzerland, 1977; Volume 18. [Google Scholar]
- Japanese Gastric Cancer Association. Japanese gastric cancer treatment guidelines 2018 (5th edition). Gastric Cancer 2020, 24, 1–21. [Google Scholar]
- Tan, P.; Yeoh, K.G. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology 2015, 149, 1153–1162. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Xing, R.; Zhou, Y.; Yu, J.; Yu, Y.; Nie, Y.; Luo, W.; Yang, C.; Xiong, T.; Wu, W.K.K.; Li, Z.; et al. Whole-genome sequencing reveals novel tandem-duplication hotspots and a prognostic mutational signature in gastric cancer. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Gao, X.; Peng, X.; Chen, M.J.M.; Li, Z.; Wei, B.; Wen, X.; Wei, B.; Dong, Y.; Bu, Z.; et al. Multi-omics characterization of molecular features of gastric cancer correlated with response to neoadjuvant chemotherapy. Sci. Adv. 2020, 6, eaay4211. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dang, M.; Harada, K.; Han, G.; Wang, F.; Pizzi, M.P.; Zhao, M.; Tatlonghari, G.; Zhang, S.; Hao, D.; et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat. Med. 2021, 27, 141–151. [Google Scholar] [CrossRef]
- Song, L.; Song, M.; Camargo, M.C.; Van Duine, J.; Williams, S.; Chung, Y.; Kim, K.M.; Lissowska, J.; Sivins, A.; Gao, W.; et al. Identification of anti-Epstein-Barr virus (EBV) antibody signature in EBV-associated gastric carcinoma. Gastric. Cancer 2021, 24, 858–867. [Google Scholar] [CrossRef]
- Okabe, A.; Huang, K.K.; Matsusaka, K.; Fukuyo, M.; Xing, M.; Ong, X.; Hoshii, T.; Usui, G.; Seki, M.; Mano, Y.; et al. Cross-species chromatin interactions drive transcriptional rewiring in Epstein-Barr virus-positive gastric adenocarcinoma. Nat. Genet. 2020, 52, 919–930. [Google Scholar] [CrossRef]
- Re, V.; Brisotto, G.; Repetto, O.; De Zorzi, M.; Caggiari, L.; Zanussi, S.; Alessandrini, L.; Canzonieri, V.; Miolo, G.; Puglisi, F.; et al. Overview of Epstein-Barr-Virus-Associated Gastric Cancer Correlated with Prognostic Classification and Development of Therapeutic Options. Int. J. Mol. Sci. 2020, 21, 9400. [Google Scholar] [CrossRef]
- Chakraborty, P.; Ghatak, S.; Chenkual, S.; Pachuau, L.; Zohmingthanga, J.; Bawihtlung, Z.; Khenglawt, L.; Pautu, J.L.; Maitra, A.; Chhakchhuak, L.; et al. Panel of significant risk factors predicts early stage gastric cancer and indication of poor prognostic association with pathogens and microsatellite stability. Genes Environ. 2021, 43, 1–15. [Google Scholar] [CrossRef]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Lochhead, P.; Chan, A.T.; Nishihara, R.; Fuchs, C.S.; Beck, A.H.; Giovannucci, E.; Ogino, S. Etiologic field effect: Reappraisal of the field effect concept in cancer predisposition and progression. Mod. Pathol. 2015, 28, 14–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar] [PubMed]
- Ushijima, T. Epigenetic field for cancerization. J. Biochem. Mol. Biol. 2007, 40, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, S.A.; Greaves, L.C.; Gutierrez-Gonzalez, L.; Rodriguez-Justo, M.; Deheragoda, M.; Leedham, S.J.; Taylor, R.W.; Lee, C.Y.; Preston, S.L.; Lovell, M.; et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology 2008, 134, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology 2009, 136, 542–550. [Google Scholar] [CrossRef]
- Pereira, A.; Moreira, F.; Vinasco-Sandoval, T.; Cunha, A.; Vidal, A.; Ribeiro-Dos-Santos, A.M.; Pinto, P.; Magalhaes, L.; Assumpcao, M.; Demachki, S.; et al. miRNome Reveals New Insights Into the Molecular Biology of Field Cancerization in Gastric Cancer. Front. Genet. 2019, 10, 592. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, N.; Ogawa, S. Clonal expansion in non-cancer tissues. Nat. Rev. Cancer 2021, 21, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef]
- Kim, K.; Cho, Y.; Sohn, J.H.; Kim, D.H.; Do, I.G.; Lee, H.J.; Do, S.I.; Ahn, S.; Lee, H.W.; Chae, S.W. Clinicopathologic characteristics of early gastric cancer according to specific intragastric location. BMC Gastroenterol. 2019, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Fang, C.; Shi, J.; Sun, Q.; Wu, H.; Gold, J.S.; Weber, H.C.; Guan, W.; Zhang, Y.; Yu, C.; et al. Differences in Clinicopathology of Early Gastric Carcinoma between Proximal and Distal Location in 438 Chinese Patients. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef]
- Kang, D.H.; Choi, C.W.; Kim, H.W.; Park, S.B.; Kim, S.J.; Nam, H.S.; Ryu, D.G. Location characteristics of early gastric cancer treated with endoscopic submucosal dissection. Surg. Endosc. 2017, 31, 4673–4679. [Google Scholar] [CrossRef]
- Bakhti, S.Z.; Latifi-Navid, S.; Zahri, S.; Yazdanbod, A. Inverse association of Helicobacter pylori cagPAI genotypes with risk of cardia and non-cardia gastric adenocarcinoma. Cancer Med. 2019, 8, 4928–4937. [Google Scholar] [CrossRef]
- Yao, Q.; Qi, X.; Xie, S.H. Sex difference in the incidence of cardia and non-cardia gastric cancer in the United States, 1992–2014. BMC Gastroenterol. 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Ghidini, M.; Donida, B.M.; Totaro, L.; Ratti, M.; Pizzo, C.; Benzoni, I.; Lomiento, D.; Aldighieri, F.; Toppo, L.; Ranieri, V.; et al. Prognostic factors associated with survival in a large cohort of gastric cancer patients resected over a decade at a single Italian center: The Cremona experience. Clin. Transl. Oncol. 2020, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Butt, J.; Varga, M.G.; Wang, T.; Tsugane, S.; Shimazu, T.; Zheng, W.; Abnet, C.C.; Yoo, K.Y.; Park, S.K.; Kim, J.; et al. Smoking, Helicobacter Pylori Serology, and Gastric Cancer Risk in Prospective Studies from China, Japan, and Korea. Cancer Prev. Res. 2019, 12, 667–674. [Google Scholar] [CrossRef] [Green Version]
- Pereira, S.A.; Antunes, A.M.M. Special Issue “Adductomics: Elucidating the Environmental Causes of Disease”. High-Throughput 2019, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Vidal, A.F.; Ribeiro-Dos-Santos, A.M.; Vinasco-Sandoval, T.; Magalhaes, L.; Pinto, P.; Anaissi, A.K.M.; Demachki, S.; de Assumpcao, P.P.; Dos Santos, S.E.B.; Ribeiro-Dos-Santos, A. The comprehensive expression analysis of circular RNAs in gastric cancer and its association with field cancerization. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, N.; Ushijima, T. Epigenetic impact of infection on carcinogenesis: Mechanisms and applications. Genome Med. 2016, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, A.; Tsukamoto, T.; Takamura-Enya, T.; Watanabe, N.; Kaminishi, M.; Sugimura, T.; Tatematsu, M.; Ushijima, T. Frequent hypomethylation in multiple promoter CpG islands is associated with global hypomethylation, but not with frequent promoter hypermethylation. Cancer Sci. 2004, 95, 58–64. [Google Scholar] [CrossRef]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, L.; Pandiri, A.R.; Li, Y.R.; Droop, A.; Hewinson, J.; Quail, M.A.; Iyer, V.; Shepherd, R.; Herbert, R.A.; Campbell, P.J.; et al. The mutational signature profile of known and suspected human carcinogens in mice. Nat. Genet. 2020, 52, 1189–1197. [Google Scholar] [CrossRef]
- Zhivagui, M.; Ng, A.W.T.; Ardin, M.; Churchwell, M.I.; Pandey, M.; Renard, C.; Villar, S.; Cahais, V.; Robitaille, A.; Bouaoun, L.; et al. Experimental and pan-cancer genome analyses reveal widespread contribution of acrylamide exposure to carcinogenesis in humans. Genome Res. 2019, 29, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Totsuka, Y.; Lin, Y.; He, Y.; Ishino, K.; Sato, H.; Kato, M.; Nagai, M.; Elzawahry, A.; Totoki, Y.; Nakamura, H.; et al. DNA Adductome Analysis Identifies N-Nitrosopiperidine Involved in the Etiology of Esophageal Cancer in Cixian, China. Chem. Res. Toxicol. 2019, 32, 1515–1527. [Google Scholar] [CrossRef] [Green Version]
- Zavadil, J.; Rozen, S.G. Experimental Delineation of Mutational Signatures is an Essential Tool in Cancer Epidemiology and Prevention. Chem. Res. Toxicol. 2019, 32, 2153–2155. [Google Scholar] [CrossRef]
- Totsuka, Y.; Watanabe, M.; Lin, Y. New horizons of DNA adductome for exploring environmental causes of cancer. Cancer Sci. 2021, 112, 7. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.; Cooke, S.L.; Weiss, J.; Bailly, A.P.; Alexandrov, L.B.; Marshall, J.; Raine, K.; Maddison, M.; Anderson, E.; Stratton, M.R.; et al. C. elegans whole-genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency. Genome Res. 2014, 24, 1624–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, B.; Volkova, N.V.; Hong, Y.; Schofield, P.; Campbell, P.J.; Gerstung, M.; Gartner, A. Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 2018, 28, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkova, N.V.; Meier, B.; Gonzalez-Huici, V.; Bertolini, S.; Gonzalez, S.; Vohringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Zou, X.; Koh, G.; Nanda, A.; Degasperi, A.; Urgo, K.; Roumeliotis, T.; Agu, C.; Side, L.; Brice, G.; Perez-Aloso, V.; et al. Dissecting mutational mechanisms underpinning signatures caused.by replication errors and endogenous DNA damage. BiorXiv 2020. [Google Scholar] [CrossRef]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015, 29, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Li, W.; Adebali, O.; Yang, Y.; Oztas, O.; Selby, C.P.; Sancar, A. Genome-wide mapping of nucleotide excision repair with XR-seq. Nat. Protoc. 2019, 14, 248–282. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hu, J.; Adebali, O.; Adar, S.; Yang, Y.; Chiou, Y.Y.; Sancar, A. Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene. Proc. Natl. Acad. Sci. USA 2017, 114, 6752–6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, C.M.; Selby, C.P.; Yang, Y.; Hsu, D.S.; Sancar, A. Genome-wide single-nucleotide resolution of oxaliplatin-DNA adduct repair in drug-sensitive and -resistant colorectal cancer cell lines. J. Biol. Chem. 2020, 295, 7584–7594. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hu, J.; Selby, C.P.; Li, W.; Yimit, A.; Jiang, Y.; Sancar, A. Single-nucleotide resolution analysis of nucleotide excision repair of ribosomal DNA in humans and mice. J. Biol. Chem. 2019, 294, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Yimit, A.; Adebali, O.; Sancar, A.; Jiang, Y. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, T.A.; Spittle, K.E.; Turner, S.W.; Korlach, J. Direct detection and sequencing of damaged DNA bases. Genome Integr. 2011, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, D.; Liu, Q.; Wang, K.; Egli, D. Detection of base analogs incorporated during DNA replication by nanopore sequencing. Nucleic Acids Res. 2020, 48, e88. [Google Scholar] [CrossRef] [PubMed]
- Nookaew, I.; Jenjaroenpun, P.; Du, H.; Wang, P.; Wu, J.; Wongsurawat, T.; Moon, S.H.; Huang, E.; Wang, Y.; Boysen, G. Detection and Discrimination of DNA Adducts Differing in Size, Regiochemistry, and Functional Group by Nanopore Sequencing. Chem. Res. Toxicol. 2020, 33, 2944–2952. [Google Scholar] [CrossRef] [PubMed]
- Sasazuki, S.; Sasaki, S.; Tsugane, S.; Japan Public Health Center Study, G. Cigarette smoking, alcohol consumption and subsequent gastric cancer risk by subsite and histologic type. Int. J. Cancer 2002, 101, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Fahey, M.T.; Sasaki, S.; Baba, S. Alcohol consumption and all-cause and cancer mortality among middle-aged Japanese men: Seven-year follow-up of the JPHC study Cohort, I. Japan Public Health Center. Am. J. Epidemiol. 1999, 150, 1201–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwashita, Y.; Ohnishi, I.; Matsushita, Y.; Ohtsuka, S.; Yamashita, T.; Inaba, K.; Fukazawa, A.; Ochiai, H.; Matsumoto, K.; Kurono, N.; et al. Geospatial Assessments of DNA Adducts in the Human Stomach: A Model of Field Cancerization. Cancers 2021, 13, 3728. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153728
Iwashita Y, Ohnishi I, Matsushita Y, Ohtsuka S, Yamashita T, Inaba K, Fukazawa A, Ochiai H, Matsumoto K, Kurono N, et al. Geospatial Assessments of DNA Adducts in the Human Stomach: A Model of Field Cancerization. Cancers. 2021; 13(15):3728. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153728
Chicago/Turabian StyleIwashita, Yuji, Ippei Ohnishi, Yuto Matsushita, Shunsuke Ohtsuka, Takashi Yamashita, Keisuke Inaba, Atsuko Fukazawa, Hideto Ochiai, Keigo Matsumoto, Nobuhito Kurono, and et al. 2021. "Geospatial Assessments of DNA Adducts in the Human Stomach: A Model of Field Cancerization" Cancers 13, no. 15: 3728. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153728