
The Human Fetal and Adult Stem Cell Secretome Can Exert Cardioprotective Paracrine Effects against Cardiotoxicity and Oxidative Stress from Cancer Treatment

 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Collection of the hAFS- and the hMSC-Conditioned Medium
2.3. In Vitro Experimental Outline
2.4. Analysis of Intracellular Dox Fluorescence and Evaluation of Cell Apoptosis In Vitro
2.5. Analysis of mNVCM Mitochondrial Organization and Their In Vitro Transmembrane Potential
2.6. Biochemical Profiling on mNVCM and mNVFib
2.6.1. Assessment of Oxygen Consumption Rate (OCR)
2.6.2. Assay for Fo-F1 ATP Synthase Activity
2.6.3. Evaluation of Oxidative Phosphorylation Efficiency
2.6.4. Glucose Consumption Assay
2.6.5. Lactate Release Assay
2.7. Preclinical Model of Dox-Induced Cardiomyopathy
2.7.1. In Vivo Model
2.7.2. Gene Expression Analysis
2.7.3. Evaluation of Aerobic Metabolism Alterations, Lipid Peroxidation, and Antioxidant Enzymatic Defenses
2.8. Statistical Analyses
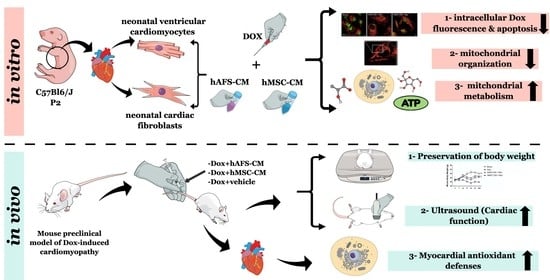
3. Results
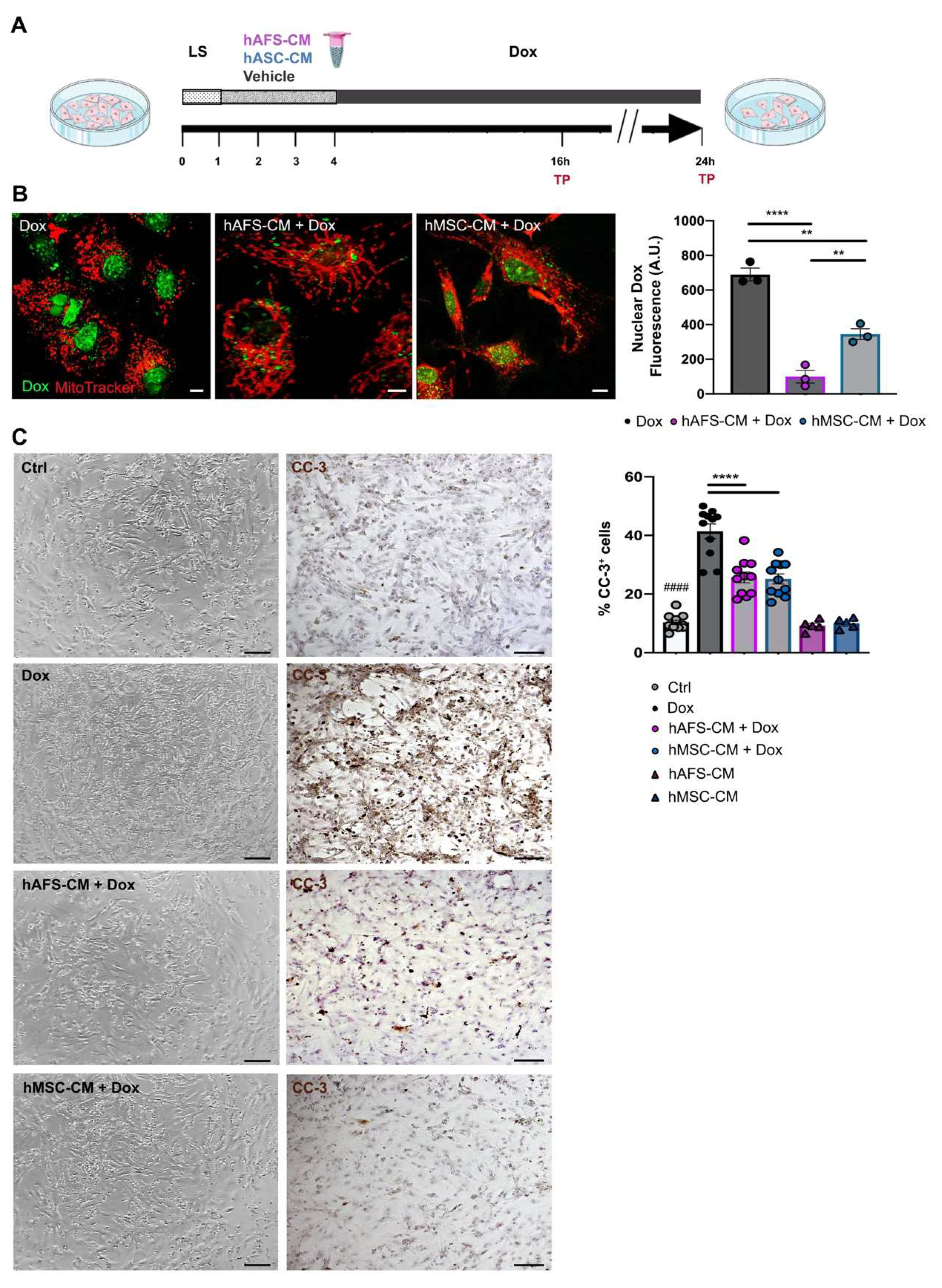
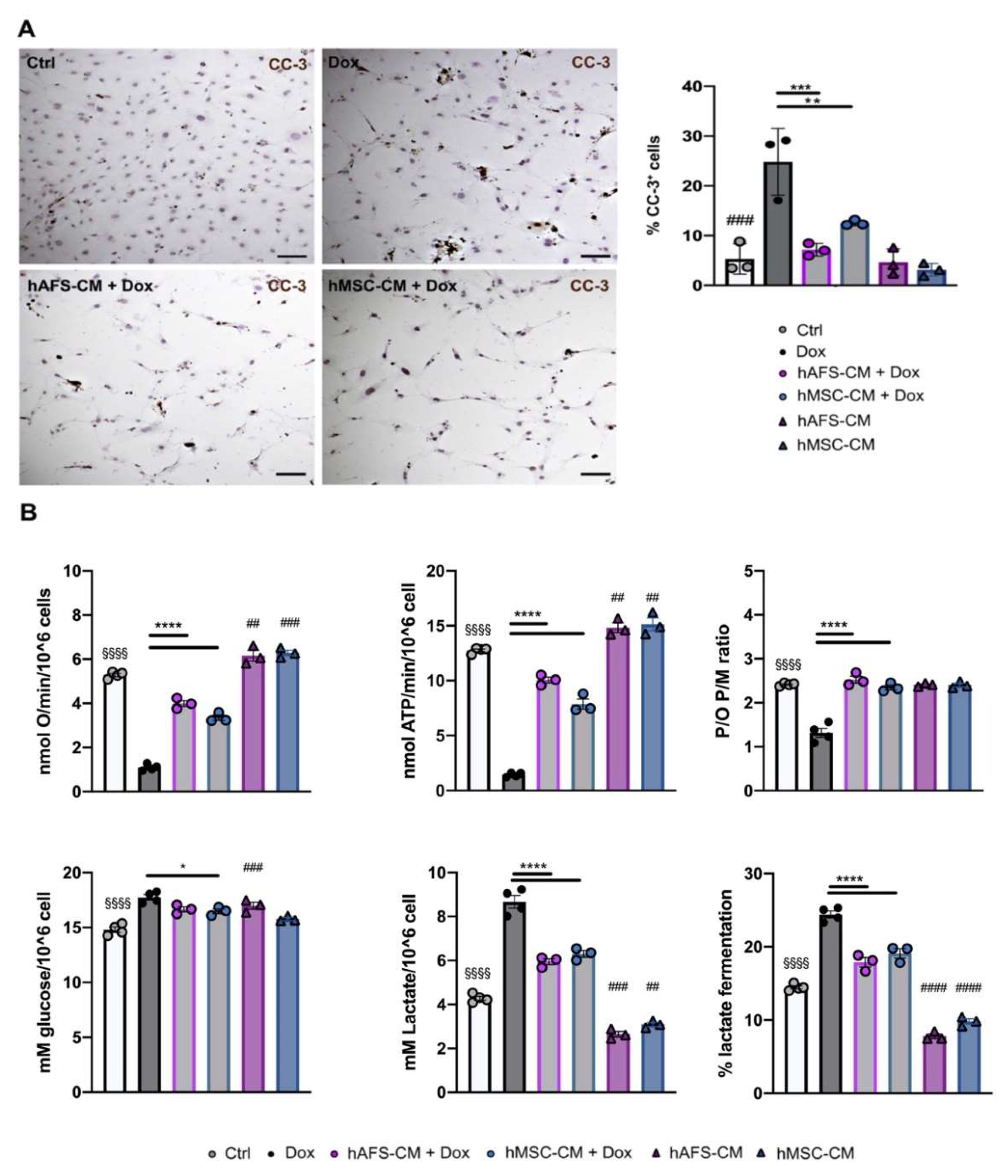
3.1. hAFS and hMSC Secretome Priming Preserves mNVCM from Unspecific Dox Uptake and from Dox-Induced Apoptosis
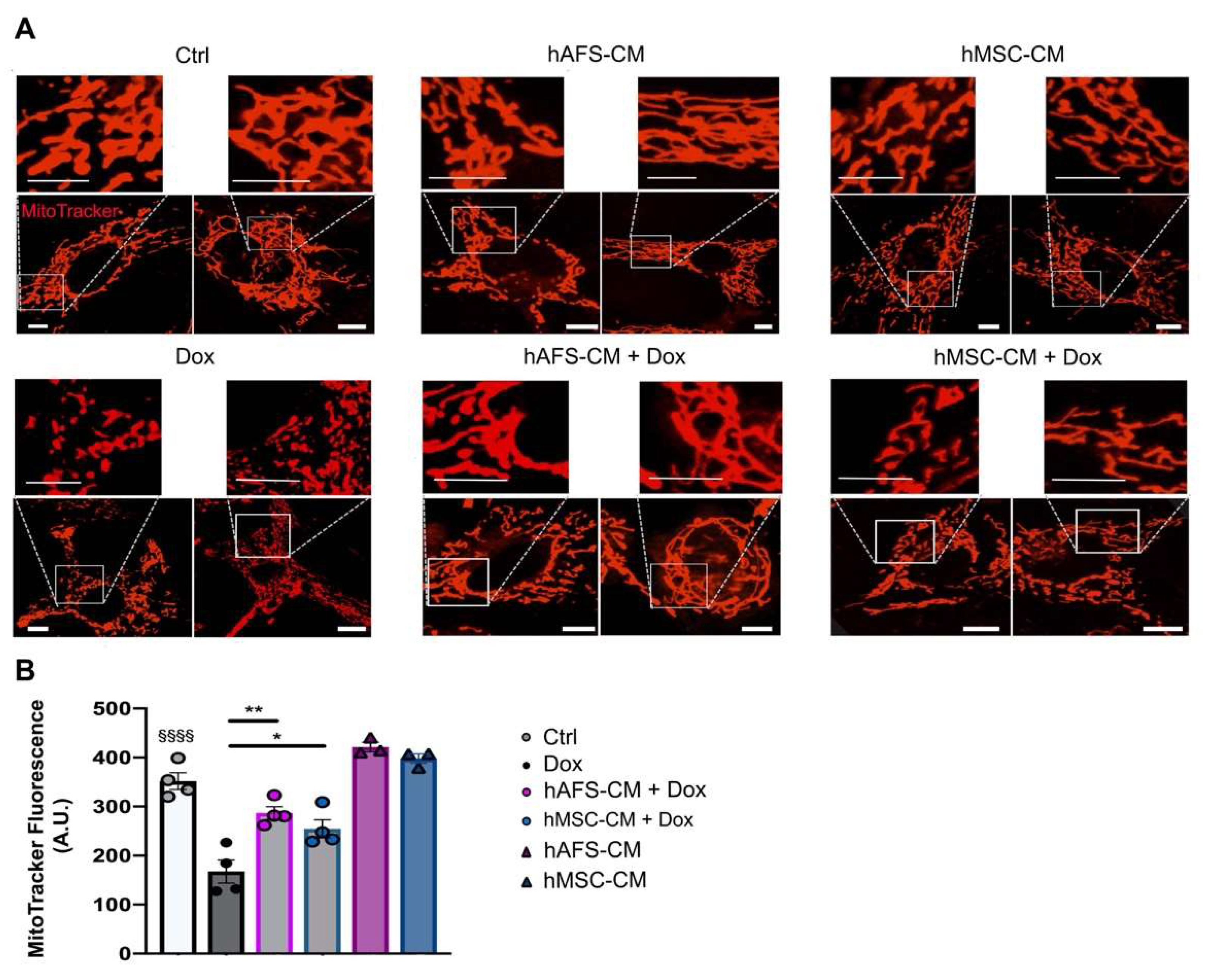
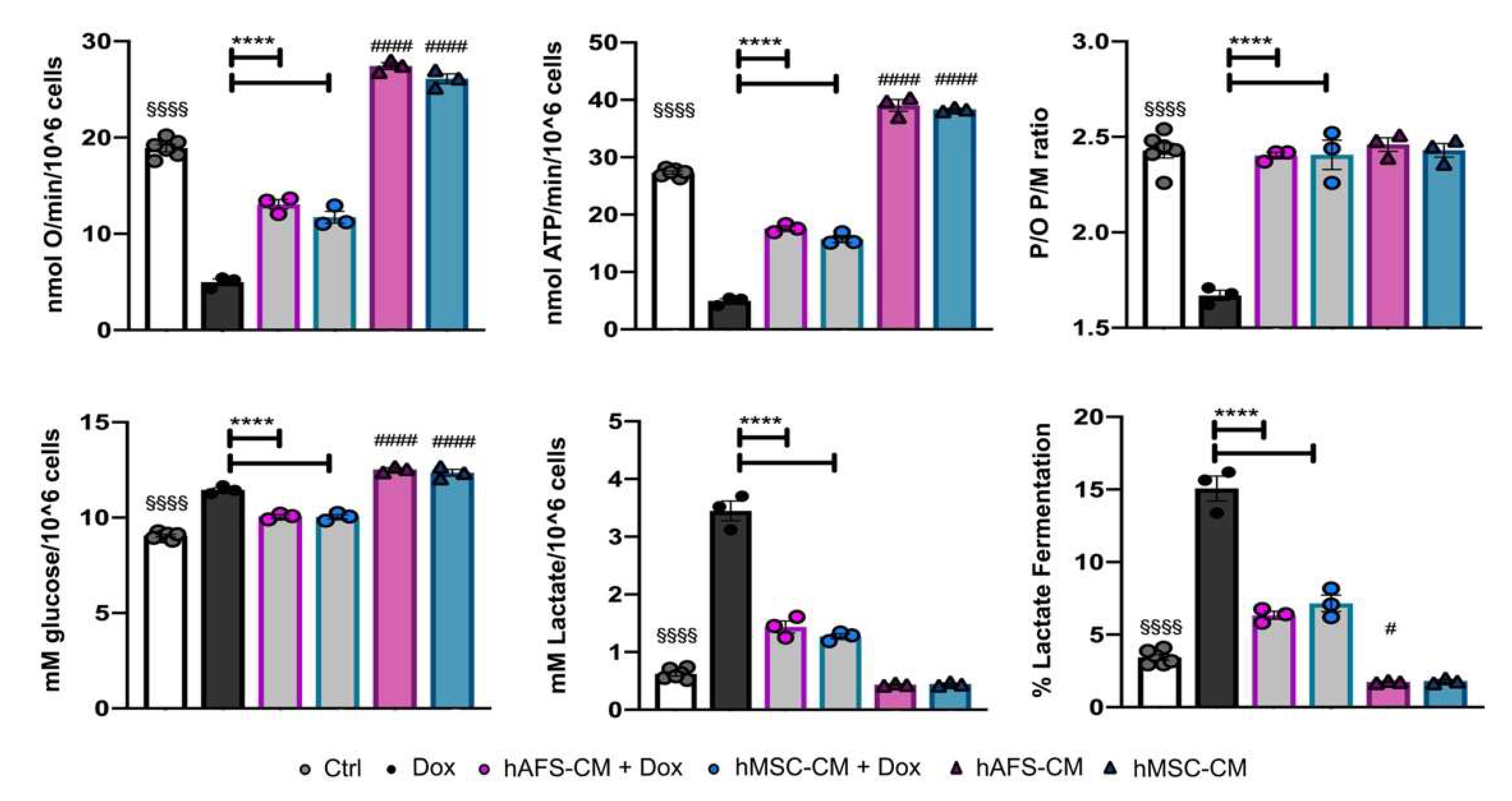
3.2. The Paracrine Potential of hAFS- and hMSC-CM Counteracts Dox-Induced Mitochondrial Dysfunction on mNVCM
3.3. Both hAFS- and hMSC-Secretome Preserve mNVFib from Dox-Induced Apotosis and Mitochondrial Impairment
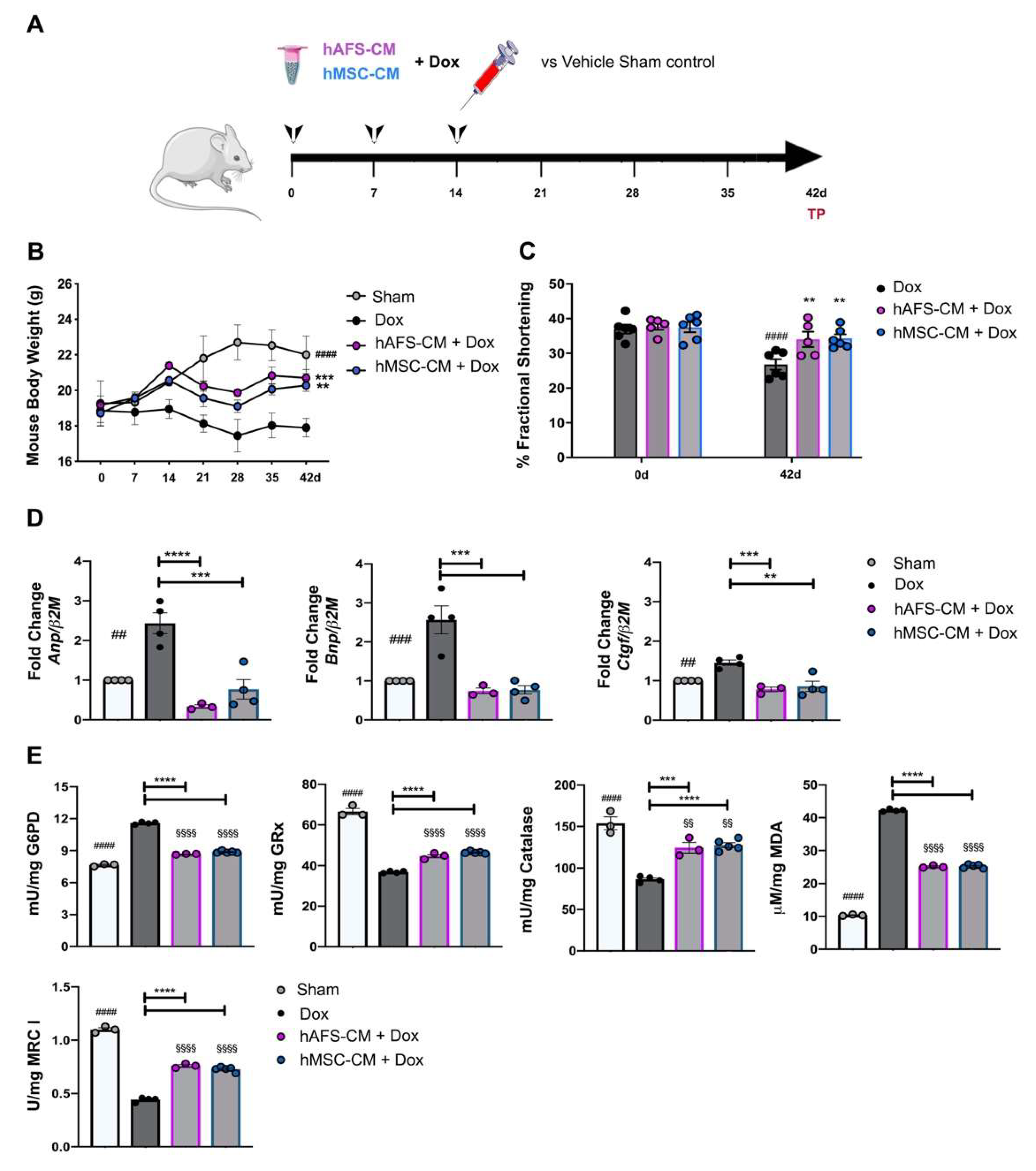
3.4. hAFS- and hMSC-CM Protect Against DOX Cardiotoxicity In Vivo
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xia, J.; Minamino, S.; Kuwabara, K.; Arai, S. Stem cell secretome as a new booster for regenerative medicine. Biosci. Trends 2019, 13, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Kucia, M.; Jadczyk, T.; Greco, N.J.; Wojakowski, W.; Tendera, M.; Ratajczak, J. Pivotal role of paracrine effects in stem cell therapies in regenerative medicine: Can we translate stem cell-secreted paracrine factors and microvesicles into better therapeutic strategies. Leukemia 2012, 26, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.K.; Rhee, J.W.; Wu, J.C. Adult stem cell therapy and heart failure, 2000 to 2016: A systematic review. JAMA Cardiol. 2016, 1, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Mirotsou, M.; Jayawardena, T.M.; Schmeckpeper, J.; Gnecchi, M.; Dzau, V.J. Paracrine mechanisms of stem cell reparative and regenerative actions in the heart. J. Mol. Cell Cardiol. 2011, 50, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnecchi, M.; He, H.; Noiseux, N.; Liang, O.D.; Zhang, L.; Morello, F.; Mu, H.; Melo, L.G.; Pratt, R.E.; Ingwall, J.S.; et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006, 20, 661–669. [Google Scholar] [CrossRef]
- Gnecchi, M.; Zhang, Z.; Ni, A.; Dzau, V.J. Paracrine Mechanisms in Adult Stem Cell Signaling and Therapy. Circ. Res. 2008, 103, 1204–1219. [Google Scholar] [CrossRef]
- Menasché, P. Cell therapy trials for heart regeneration—Lessons learned and future directions. Nat. Rev. Cardiol. 2018, 15, 659–671. [Google Scholar] [CrossRef]
- Hodgkinson, C.P.; Bareja, A.; Gomez, J.A.; Dzau, V.J. Emerging Concepts in Paracrine Mechanisms in Regenerative Cardiovascular Medicine and Biology. Circ. Res. 2016, 118, 95–107. [Google Scholar] [CrossRef] [Green Version]
- Sid-Otmane, C.; Perrault, L.P.; Ly, H.Q. Mesenchymal stem cell mediates cardiac repair through autocrine, paracrine and endocrine axes. J. Transl. Med. 2020, 18, 336. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, W.; Li, L.; Peng, Y.; Chen, P.; Huang, H.; Guo, Y.; Xia, X.; Wang, Y.; Wang, H.; et al. The Relative Contribution of Paracine Effect versus Direct Differentiation on Adipose-Derived Stem Cell Transplantation Mediated Cardiac Repair. PLoS ONE 2013, 8, e59020. [Google Scholar] [CrossRef] [PubMed]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Kenigsberg, B.; Wellstein, A.; Barac, A. Left Ventricular Dysfunction in Cancer Treatment: Is it Relevant? JACC Heart Fail. 2018, 6, 87–95. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [Green Version]
- Raschi, E.; Diemberger, I.; Cosmi, B.; De Ponti, F. ESC position paper on cardiovascular toxicity of cancer treatments: Challenges and expectations. Intern. Emerg. Med. 2018, 13, 1–9. [Google Scholar] [CrossRef]
- Anker, M.S.; Hadzibegovic, S.; Lena, A.; Belenkov, Y.; Bergler-Klein, J.; de Boer, R.A.; Farmakis, D.; von Haehling, S.; Iakobishvili, Z.; Maack, C.; et al. Recent advances in cardio-oncology: A report from the ‘Heart Failure Association 2019 and World Congress on Acute Heart Failure 2019’. ESC Heart Fail. 2019, 6, 1140–1148. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines. Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Varricchi, G.; Ameri, P.; Cadeddu, C.; Ghigo, A.; Madonna, R.; Marone, G.; Mercurio, V.; Monte, I.; Novo, G.; Parrella, P.; et al. Antineoplastic drug-induced cardiotoxicity: A redox perspective. Front. Physiol. 2018, 9, 167. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Jairaj Naik, T.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Sala, V.; De Santis, M.C.; Cimino, J.; Cappello, P.; Pianca, N.; Di Bona, A.; Margaria, J.P.; Martini, M.; Lazzarini, E.; et al. Phosphoinositide 3-Kinase Gamma Inhibition Protects from Anthracycline Cardiotoxicity and Reduces Tumor Growth. Circulation 2018, 138, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial determinants of doxorubicin- induced cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, E.; Balbi, C.; Altieri, P.; Pfeffer, U.; Gambini, E.; Canepa, M.; Varesio, L.; Bosco, M.C.; Coviello, D.; Pompilio, G.; et al. The human amniotic fluid stem cell secretome effectively counteracts doxorubicin-induced cardiotoxicity. Sci. Rep. 2016, 6, 29994. [Google Scholar] [CrossRef] [PubMed]
- De Coppi, P.; Bartsch, G.; Siddiqui, M.M.; Xu, T.; Santos, C.C.; Perin, L.; Mostoslavsky, G.; Serre, A.C.; Snyder, E.Y.; Yoo, J.J.; et al. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 2007, 25, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Sicco, C.L.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal stem cell-derived extracellular vesicles as mediators of anti-inflammatory effects: Endorsement of macrophage polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Radisic, M.; Euloth, M.; Yang, L.; Langer, R.; Freed, L.E.; Vunjak-Novakovic, G. High-density seeding of myocyte cells for cardiac tissue engineering. Biotechnol. Bioeng. 2003, 82, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Dufour, C.; Cesaro, S.; Bottega, R.; Faleschini, M.; Cuccarolo, P.; Corsolini, F.; Usai, C.; Columbaro, M.; Cipolli, M.; et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci. Rep. 2016, 6, 25441. [Google Scholar] [CrossRef] [Green Version]
- Ravera, S.; Aluigi, M.G.; Calzia, D.; Ramoino, P.; Morelli, A.; Panfoli, I. Evidence for ectopic aerobic ATP production on C6 glioma cell plasma membrane. Cell. Mol. Neurobiol. 2011, 31, 313–321. [Google Scholar] [CrossRef]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta Bioenerg. 2005, 1706, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016, 7, 70715–70737. [Google Scholar] [CrossRef] [Green Version]
- Ravera, S.; Cossu, V.; Tappino, B.; Nicchia, E.; Dufour, C.; Cavani, S.; Sciutto, A.; Bolognesi, C.; Columbaro, M.; Degan, P.; et al. Concentration-dependent metabolic effects of metformin in healthy and Fanconi anemia lymphoblast cells. J. Cell. Physiol. 2018, 233, 1736–1751. [Google Scholar] [CrossRef]
- Cappelli, E.; Degan, P.; Bruno, S.; Pierri, F.; Miano, M.; Raggi, F.; Farruggia, P.; Mecucci, C.; Crescenzi, B.; Naim, V.; et al. The passage from bone marrow niche to bloodstream triggers the metabolic impairment in Fanconi Anemia mononuclear cells. Redox Biol. 2020, 36, 101618. [Google Scholar] [CrossRef]
- Cossu, V.; Marini, C.; Piccioli, P.; Rocchi, A.; Bruno, S.; Orengo, A.M.; Emionite, L.; Bauckneht, M.; Grillo, F.; Capitanio, S.; et al. Obligatory role of endoplasmic reticulum in brain FDG uptake. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1184–1196. [Google Scholar] [CrossRef]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [Green Version]
- Asnani, A.; Moslehi, J.J.; Adhikari, B.B.; Baik, A.H.; Beyer, A.M.; de Boer, R.A.; Ghigo, A.; Grumbach, I.M.; Jain, S.; Zhu, H. Preclinical Models of Cancer Therapy–Associated Cardiovascular Toxicity: A Scientific Statement From the American Heart Association. Circ. Res. 2021, 129, e21–e34. [Google Scholar] [CrossRef] [PubMed]
- Berthonneche, C.; Peter, B.; Schüpfer, F.; Hayoz, P.; Kutalik, Z.; Abriel, H.; Pedrazzini, T.; Beckmann, J.S.; Bergmann, S.; Maurer, F. Cardiovascular response to beta-adrenergic blockade or activation in 23 inbred mouse strains. PLoS ONE 2009, 4, e6610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, M.; Wallach, C.; Ehmke, H.; Schwoerer, A.P. Genetic background dominates the susceptibility to ventricular arrhythmias in a murine model of β-adrenergic stimulation. Sci. Rep. 2018, 8, 2312. [Google Scholar] [CrossRef]
- Shah, A.P.; Siedlecka, U.; Gandhi, A.; Navaratnarajah, M.; Abou Al-Saud, S.; Yacoub, M.H.; Terracciano, C.M. Genetic background affects function and intracellular calcium regulation of mouse hearts. Cardiovasc. Res. 2010, 87, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Budde, T.; Haney, J.; Bien, S.; Schwebe, M.; Riad, A.; Tschöpe, C.; Staudt, A.; Jedlitschky, G.; Felix, S.B.; Kroemer, H.K.; et al. Acute exposure to doxorubicin results in increased cardiac P-glycoprotein expression. J. Pharm. Sci. 2011, 100, 3951–3958. [Google Scholar] [CrossRef] [PubMed]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II poisons: From early studies to new perspectives. Int. J. Mol. Sci. 2018, 19, 3480. [Google Scholar] [CrossRef] [Green Version]
- Sala, V.; Della Sala, A.; Hirsch, E.; Ghigo, A. Signaling Pathways Underlying Anthracycline Cardiotoxicity. Antioxid. Redox Signal. 2020, 32, 1098–1114. [Google Scholar] [CrossRef]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Toan, S.; Mui, D.; Zhou, H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol. 2020, 231, e13590. [Google Scholar]
- Murabito, A.; Hirsch, E.; Ghigo, A. Mechanisms of Anthracycline-Induced Cardiotoxicity: Is Mitochondrial Dysfunction the Answer? Front. Cardiovasc. Med. 2020, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaseelan, R.; Poizat, C.; Wu, H.Y.; Kedes, L. Molecular mechanisms of doxorubicin-induced cardiomyopathy. Selective suppression of Reiske iron-sulfur protein, ADP/ATP translocase, and phosphofructokinase genes is associated with ATP depletion in rat cardiomyocytes. J. Biol. Chem. 1997, 272, 5828–5832. [Google Scholar] [CrossRef] [Green Version]
- Plecitá-Hlavatá, L.; Lessard, M.; Šantorová, J.; Bewersdorf, J.; Ježek, P. Mitochondrial oxidative phosphorylation and energetic status are reflected by morphology of mitochondrial network in INS-1E and HEP-G2 cells viewed by 4Pi microscopy. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Ferree, A.; Shirihai, O. Mitochondrial dynamics: The intersection of form and function. Adv. Exp. Med. Biol. 2012, 748, 13–40. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER–mitochondria contact sites: Role in metabolic diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef] [Green Version]
- Flis, V.V.; Daum, G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 2013, 5, a013235. [Google Scholar] [CrossRef]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Singla, D.K.; Ahmed, A.; Singla, R.; Yan, B. Embryonic stem cells improve cardiac function in doxorubicin-induced cardiomyopathy mediated through multiple mechanisms. Cell Transplant. 2012, 21, 1919–1930. [Google Scholar] [CrossRef] [Green Version]
- Merino, H.; Singla, D.K. Notch-1 mediated cardiac protection following embryonic and induced pluripotent stem cell transplantation in doxorubicin-induced heart failure. PLoS ONE 2014, 9, e101024. [Google Scholar] [CrossRef]
- Singla, D.K. Akt-mTOR Pathway Inhibits Apoptosis and Fibrosis in Doxorubicin-Induced Cardiotoxicity Following Embryonic Stem Cell Transplantation. Cell Transplant. 2015, 24, 1031–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, D.K.; Abdelli, L.S. Embryonic Stem Cells and Released Factors Stimulate c-kit(+)/FLK-1(+) Progenitor Cells and Promote Neovascularization in Doxorubicin-Induced Cardiomyopathy. Cell Transplant. 2015, 24, 1043–1052. [Google Scholar] [CrossRef]
- Dargani, Z.T.; Singla, D.K. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H460–H471. [Google Scholar] [CrossRef] [PubMed]
- Baglioni, S.; Cantini, G.; Poli, G.; Francalanci, M.; Squecco, R.; Franco, A.; Borgogni, E.; Frontera, S.; Nesi, G.; Liotta, F.; et al. Functional differences in visceral and subcutaneous fat pads originate from differences in the adipose stem cell. PLoS ONE 2012, 7, e36596. [Google Scholar] [CrossRef]
- Siegel, G.; Kluba, T.; Hermanutz-Klein, U.; Bieback, K.; Northoff, H.; Schäfer, R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksu, A.E.; Rubin, J.P.; Dudas, J.R.; Marra, K.G. Role of gender and anatomical region on induction of osteogenic differentiation of human adipose-derived stem cells. Ann. Plast. Surg. 2008, 60, 306–322. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.Y.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, F.; Bruno, S.; Costa, A.; Li, M.; Russo, M.; Cimino, J.; Altieri, P.; Ruggeri, C.; Gorgun, C.; De Biasio, P.; et al. The Human Fetal and Adult Stem Cell Secretome Can Exert Cardioprotective Paracrine Effects against Cardiotoxicity and Oxidative Stress from Cancer Treatment. Cancers 2021, 13, 3729. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153729
Villa F, Bruno S, Costa A, Li M, Russo M, Cimino J, Altieri P, Ruggeri C, Gorgun C, De Biasio P, et al. The Human Fetal and Adult Stem Cell Secretome Can Exert Cardioprotective Paracrine Effects against Cardiotoxicity and Oxidative Stress from Cancer Treatment. Cancers. 2021; 13(15):3729. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153729
Chicago/Turabian StyleVilla, Federico, Silvia Bruno, Ambra Costa, Mingchuan Li, Michele Russo, James Cimino, Paola Altieri, Clarissa Ruggeri, Cansu Gorgun, Pierangela De Biasio, and et al. 2021. "The Human Fetal and Adult Stem Cell Secretome Can Exert Cardioprotective Paracrine Effects against Cardiotoxicity and Oxidative Stress from Cancer Treatment" Cancers 13, no. 15: 3729. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153729