
Targeting RB1 Loss in Cancers
by
 , ,
, ,
Paing Linn
1,2 ,
,
Susumu Kohno
1,
Jindan Sheng
1,
Nilakshi Kulathunga
1,
Hai Yu
1,
Zhiheng Zhang
1,
Dominic Voon
3,
Yoshihiro Watanabe
4 and
Chiaki Takahashi
1,* 1
Division of Oncology and Molecular Biology, Cancer Research Institute, Kanazawa University, Kanazawa 920-1192, Japan
2
Yangon General Hospital, Yangon, Myanmar
3
Institute of Frontier Sciences Initiative, Kanazawa University, Kanazawa 920-1192, Japan
4
iCREK, Kanazawa University Hospital, Kanazawa 920-8641, Japan
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(15), 3737; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153737
Submission received: 18 May 2021
/
Revised: 14 July 2021
/
Accepted: 21 July 2021
/
Published: 25 July 2021
(This article belongs to the Special Issue Multiple Roles of the RB Family of Tumor Suppressors and Implications for Cancer Therapy)
Abstract
:Simple Summary
Irreversible defects in RB1 tumor suppressor functions often predict poor outcomes in cancer patients. However, the RB1-defecient status can be a benefit as well for them, as it generates a variety of vulnerabilities induced through the upregulation of RB1 targets, relief from functional restrictions due to RB1 binding, presence of genes whose inactivation cause synthetic lethality with RB1 loss, or collateral synthetic lethality owing to simultaneous loss of neighboring genes.
Abstract
Retinoblastoma protein 1 (RB1) is encoded by a tumor suppressor gene that was discovered more than 30 years ago. Almost all mitogenic signals promote cell cycle progression by braking on the function of RB1 protein through mono- and subsequent hyper-phosphorylation mediated by cyclin-CDK complexes. The loss of RB1 function drives tumorigenesis in limited types of malignancies including retinoblastoma and small cell lung cancer. In a majority of human cancers, RB1 function is suppressed during tumor progression through various mechanisms. The latter gives rise to the acquisition of various phenotypes that confer malignant progression. The RB1-targeted molecules involved in such phenotypic changes are good quarries for cancer therapy. Indeed, a variety of novel therapies have been proposed to target RB1 loss. In particular, the inhibition of a number of mitotic kinases appeared to be synthetic lethal with RB1 deficiency. A recent study focusing on a neighboring gene that is often collaterally deleted together with RB1 revealed a pharmacologically targetable vulnerability in RB1-deficient cancers. Here we summarize current understanding on possible therapeutic approaches targeting functional or genomic aberration of RB1 in cancers.
1. Introduction
The discovery of RB1 gene had paved an avenue toward understanding the complicated functions of the human genome in preventing cells from carcinogenesis [1,2]. RB1 gene was thought to be mechanistically involved in the initiation of retinoblastoma as its inactivation following genetic mutation or deletion was prevalently identified in the pedigrees of hereditary cases [3]. This notion was later experimentally proven by the induction of retinoblastoma in mice following the ablation of multiple Rb family members [4]. Together with the presence of other cancer driving mutations in genes such as Trp53, RB1 loss can provoke carcinogenesis from various types of tissue. This suggests that RB1 may in fact be involved in the initiation and/or progression of previously unexpected varieties of cancer [5]. Of note, RB1 inactivation is detected in close to 90% of sporadic small cell lung cancers (SCLC), suggesting a contribution to their initiation, while its germ line mutation also often predisposes to SCLC [6]. Reportedly, RB1 inactivation occurs in 20~40% of osteosarcoma cases, yet it is thought to contribute to tumor progression based on pathological examination and linkage with unfavorable outcome [7,8]. RB1 reconstitution in RB1-deficient osteosarcoma-derived cells does not always completely attenuate their malignant phenotypes, presumably due to coexisting functional aberration in Trp53 tumor suppressor or other driver mutations [9,10]. These findings also indicate that RB1 may exert varied functions depending on cancer tissue type and timing during the course of tumor development.
Similar to osteosarcoma, RB1 is suggested to be inactivated during their progression in a majority of cancer types [7]. One of the best examples is prostate cancer. In primary/non-metastatic prostate cancers, RB1 deletion is identified with less than 10% prevalence, but this rises to higher than 30% in those attaining metastatic and/or castration-resistant phenotypes [11]. In addition to prostate cancer, many common types of cancers, including non-small cell lung cancer (NSCLC), breast cancer, bladder cancer, chronic myelogenous leukemia (CML), high-grade glioma, esophageal cancer, head and neck cancer and sarcoma, are thought to gain RB1 deficiency during their malignant progression [7].
The roles of RB1 in suppressing malignant progression of prostate cancer may not be solely explained by its primary function to constrain cell cycle progression. For example, RB1 controls the transcription of androgen receptor (AR) through the E2F family of transcription factors, hence its inactivation could enhance the refractoriness to endocrine therapy [12]. Moreover, the depletion of RB1 from RB1-proficient prostate cancer cells promoted gain of stem cell-like properties through increased expression of interleukin-6 (IL-6) and lysyl oxidase (LOX) [13]. Similarly, RB1 inactivation in breast cancers enhances stem cell-like behaviors and malignant progression through IL-6 and following activation of signal transducer and activator of transcription 3 (STAT3) activation [14,15,16,17,18]. RB1 loss in breast cancer cells activates fatty acid oxidation (FAO) and induces following Jun kinase (JNK) activation. Activated JNK then stimulates IL-6 production leading to gain of undifferentiated characters which are mediated by cell-autonomous STAT3 activation [14]. Later, the enhanced FAO following RB1 loss was explained by AMP-activated protein kinase (AMPK) activation and subsequent phosphorylation (downregulation) of acetyl-CoA carboxylase (ACC) [16]. Accordingly, the blockade of IL-6 by neutralizing antibody suppressed breast cancer progression is induced following RB1 inactivation [14]. Loss of Trp53 in mice generates well differentiated soft tissue sarcoma with low prevalence; additional loss of Rb1 converts this to undifferentiated pleomorphic type [17]. An increase in lineage plasticity has been observed in mouse Trp53-null osteosarcoma cells following Rb1 deletion [19]. Identification of molecules mediating RB1 functions in controlling undifferentiated characters of tumor cells may endow us new tools to target cancer stem cells [18]. Taken together, in addition to targeting aberrant cell cycle progression, functional suppression of the proteins aberrantly expressed as a result of RB1 inactivation may also be beneficial in cancer treatment.
This review aims to firstly shed light on such gene products and then focus on genes whose inactivation might exhibit synthetic lethality when RB1 is simultaneously inactivated. Finally, this review will discuss putative approaches to treat RB1-deficient cancers by targeting comparatively large-scale chromosomal aberration involving the RB1 loci.
2. Targeting Gene Products Upregulated Following RB1 Inactivation
The RB1/E2Fs/DPs complex primarily suppresses target gene transcription by recruiting histone deacetylase (HDAC). Loss of RB1 function liberates its most important counterpart, E2Fs, and these primarily transactivate target genes by recruiting histone acetylase (HAT). The target genes of the “transactivating” E2Fs (E2F1, E2F2 and E2F3a) contain a number of genes enrolled in cell cycle progression, DNA synthesis and replication, DNA damage repair and apoptosis [20]. In line with this, the deletion of E2f1 suppressed pituitary and thyroid tumorigenesis and prolonged the life span in Rb1-heterozygous mice [21]. Likewise, the loss of E2f2 or E2f3 in MMTV-Myc transgenic mice resulted in the cessation of mammary carcinogenesis [22]. E2F1 is frequently overexpressed in metastatic melanoma, and inhibition of E2F1 induced apoptosis and cellular senescence [23]. In specific types of cancer, higher expression of some of E2F family members correlates with advanced stage and poor prognosis [24]. Therefore, targeting E2Fs may yield certain benefits in cancer therapy. The E2F inhibitor HML006474 was designed based on a virtual screening of compounds that possibly inhibit DNA binding of the E2F4/DP2 complex. The E2F4-null cells were less sensitive to this compound compared to wild type, which supports the specificity of its action [25]. However, to date, no small molecules specifically targeting any of transactivating E2Fs have been described nor tested in clinical trials.
As E2Fs target a wide variety of genes that could promote or suppress tumor development, it is much safer to consider strategies to target individual E2F target. As numerous review articles have already expounded on this point, this article picks up only a few. In particular, although cyclin E and cyclin dependent kinase 2 (CDK2) are representative targets of E2Fs during G1/S, and inhibitors to CDK2 are undergoing clinical trials [26], the efficacy of these agents and their dependency on the presence of intact RB1 remains a scantly addressed topic.
E2Fs target numerous genes involved in replication control, nucleotide synthesis, DNA damage response/repair, chromatin structure and mitotic progression. RB1 inactivation induces chromosomal instability (CIN), at least partially, by upregulating spindle assembly checkpoint protein mitotic arrest deficient 2 (MAD2). Overexpressed MAD2 may contribute to induced aneuploidy and stabilization of erroneous attachment of mitotic spindles [27,28]. Dysregulated expression of MAD2 is correlated to cancer progression and poor prognosis [29]. A small molecule, M2I-1, targeting MAD2-CDC20 interaction has been developed [30]. A report suggested that M2I-1 sensitizes cancer cells to anti-mitotic reagents [31] but, thus far, no report has tested its efficacy in RB1-deficient context. Many of mitotic kinases are upregulated following E2F dysregulation and inhibition of some of them has been indicated to be synthetically lethal with RB1 inactivation (see below).
Besides MAD2, numerous possibly druggable molecules have been indicated to be involved in the RB1 loss-induced CIN [32]. Aurora A and polo-like kinase 1 (PLK1) are upregulated following RB1 depletion, which may lead to aneuploidy or other mitotic abnormalities, further causing malignant progression [33]. Synthetic inhibitors to these kinases are now in clinical trials (Table 1). Mis-localization of CAPD3/condensin II has been proposed to mediate centromere dysfunction induced by RB1 loss [34]. However, synthetic inhibitors to this molecule are under development and it is not clear whether such inhibitors could exhibit anti-tumor activity.
In addition to mitotic catastrophe, replicative catastrophe induced by abnormally enhanced cell cycle progression induced by gain of function by E2Fs could provide a certain vulnerability especially upon chemotherapy or radiation therapy [35]. The observation that RB1 inactivation in thyroid calcitonin-producing (C) cells or fibroblastic cells induced DNA damage response followed by senescence supports this speculation [36].
As alluded to the introduction, mostly in an E2F-dependent manner, RB1 status affects the milieu surrounding tumor cells including chemokine/cytokine secretion, extracellular matrix, immune cells or remote organs to be metastasized. The molecules mediating such functions of RB1 could serve as good therapeutic targets [37,38]. C-C motif chemokine 2 (CCL2) appeared to be secreted from RB1-deficient breast cancer cells, thereby such tumor cells recruit immunosuppressive cells including myeloid-derived suppressive cells (MDSC), regulatory T cells (Treg) or macrophages, allowing malignant progression following RB1 inactivation. Moreover, blockade of CCL2 signaling in host mice by genetically deleting CCR2, encoding the receptor for CCL2, almost completely abrogated mammary carcinogenesis from MMTV-cre; Rb1flox/flox mice [14,38]. RB1 inactivation has been linked to upregulation of PD-L1 expression through direct interaction with NF-κB protein p65. Surprisingly, a particular phosphorylated form of RB1 inactivates p65 and therefore attenuates expression of PD-L1 and cIAPs. The peptide mimicking this phosphorylated form of RB1 improved the therapeutic efficacy in radiotherapy by suppressing radiation-induced PD-L1 expression [39]. These findings make clear contrast with the enhanced immunogenicity induced by locking RB1 in unphosphorylated form by the treatment with CDK4/6 inhibitor [40], and suggest that RB1 status might affect the efficacy of therapy by immune checkpoint inhibitors (ICIs).
A while ago, an unexpected twist arose from C. elegans studies that indicated lin-35, an RB1 orthologue, might be placed upstream of Ras signaling during vulval development [41,42]. In cultured cells, including embryonic fibroblasts and thyroid C cells, K-Ras and N-Ras are activated 5~10 times greater following RB1 loss, and this may promote or suppress tumor development in a highly context-dependent manner [36,43,44,45,46,47]. It has been estimated that mutation of K-Ras or N-Ras in the context of RB1 loss exerts a mitogenic effect that is 50~60 times greater than that of corresponding wild type Ras in the presence of RB1. Mechanistically, the RB1-Ras nexus has been explained by farnesylation and geranylgeranylation to anchor cytosolic Ras proteins to lipid bilayers, which are innervated by E2Fs and SREBPs [36,48], thus this axis is pharmacologically targetable. These findings led researchers to address RB1 functions in cholesterol and then lipid metabolism. ELOVL fatty acid elongase 6 (ELOVL6) and fatty acid desaturase 1 (SCD1) have been linked to RB1 via E2Fs and SREBPs, which explained the profound impact of RB1 loss on the lipidomic landscape [49]. The oleic acid, one of the immediate products of ELOVL6 and SCD1, appeared to promote mammary carcinogenesis by stabilizing c-Myc through GPR40 which is a receptor for oleic acid, providing a good rationale to develop antagonists to this receptor to prevent breast cancer associated with high fat diet in puberty [50].
Although the dependence on E2Fs is still unclear, human retinoblastoma cells highly express a proto-oncogene spleen tyrosine kinase (SYK) and depend on its expression for survival. Treatment of genetic retinoblastoma model with synthetic SYK inhibitor resulted in dramatic suppression of disease progression and prolongation of survival [51]. Furthermore, in retinoblastoma cells, ubiquitin-like, containing PFD and RING finger domains 1 (UHRF1) and helicase, lymphoid specific (HELLS) are overexpressed, which is linked to epigenetic activation of SYK [52,53].
3. Partners Other Than E2Fs Liberated upon RB1 Loss
More than 300 proteins have been indicated to directly bind to RB1 [48]. Varied mono-phosphorylation status of RB1 (possibly 14 variations) may increase the complexity of the relationship with specific binding partners [54]. As RB1 is typically not abundantly expressed in tissues, RB1 may carefully select partners depending on the cellular context. A number of chromatin modifiers, cyclins and signaling molecules, such as apoptosis signal-regulating kinase 1 (ASK1) equipped with LxCxE motif, bind to RB1 at the site also targeted by a number of oncogenic virus products carrying LxCxE motif including adenovirus E1A, simian virus 40 large T antigen and human papilloma virus E7 [55]. Such chromatin modifiers include attractive targets of cancer therapy. Notably, 5-azacytidine targets DNMT1 and has been used for the treatment of myelodysplastic syndrome [56]. Similarly, the utility of pan-HDAC inhibitors has long been evaluated in a variety of cancers [57]. The Suv39H1 inhibitor chaetocin exhibited significant therapeutic efficacy against acute myeloid leukemia when combined with other epigenetic drugs that include suberoylanilide hydroxamic acid (HDAC inhibitor) and JQ-1 (bromodomain inhibitor) [58]. However, how the loss of RB1 impacts the efficacies and sensitivities of these molecules is not well studied. One notable exception is TAI-1, which targets Hec-1 and exhibits synergy with multiple chemotherapeutic agents in the treatment of leukemia, breast and liver cancer. In these settings, the status of RB1 and Trp53 influenced the sensitivity to TAI-1 [59].
SKP2 directly binds to RB1 [60], whereupon RB1 loss liberates SKP2 to promote the proteasomal degradation of p27KIP. SKP2 was initially identified as a cyclin A binding protein and later found to serve as a component of the SCFSKP2 E3 ubiquitin ligase complex targeting various substrates containing many of cell cycle regulators, including p27KIP. Additional loss of Skp2 loci appeared to increase the degree of apoptosis in Rb1-nullizygous mouse embryonic fibroblasts (MEFs) in an E2F-dependent manner. Similarly, Skp2 loss increased apoptotic cells in Rb1-deficient pituitary tumor developed in mice, also in an E2F-dependent manner [61]. Loss of SKP2 stabilizes p27KIP and simultaneously exposes cyclin A to p27KIP. Additionally, the loss of SKP2 lessens cyclin A activity to attenuate E2F1 function. Consequently, overactivated E2F1 behaves more proapoptotic rather than proliferative in the absence of RB1 [61].
Deficiency in terminal differentiation following RB1 inactivation would be another possible target in cancer treatment. Many of the tissue-specific transcription factors, including MYOD, CCAAT enhancer binding protein (C/EBP), glucocorticoid receptor (GR), GATA-binding factor 1 (GATA-1), PU-1, core-binding factor alpha 1 (CBFA-1), pancreas duodenum homeobox 1 (PDX1), runt-related transcription factor 2 (RUNX2) and nuclear factor of IL-6 expression (NF-IL6), directly or indirectly collaborate with RB1 to determine lineage specificity and to induce terminal differentiation [62]. As such, the loss of RB1 promotes undifferentiated behaviors of cancer cells, though the extent to which tissue-specific transcription factors also contribute to this phenotype is still unclear. Added to this, the functional recovery of tissue-specific transcription factors by pharmacological approaches may be difficult.
Yet how the loss of RB1 promotes undifferentiated behaviors of cancer cells might be at least partially explained by the pivotal roles of RB1 in controlling pluripotency factors including octamer-binding transcription factor 4 (OCT4) and SRY-box 2 (SOX2), by directly binding to the regulatory elements for these genes [63]. This study has been done in the context of inducible pluripotent stem cells (iPSs). Additionally, as abovementioned, IL-6 and LOX have been proposed to explain the effect of RB1 loss on the malignant progression of prostate and breast cancer [13,14]. Interference to these machineries may enable cancer therapy by inducing spontaneous differentiation.
4. Synthetic Lethality with RB1 Inactivation
It is commonly held that loss-of-function mutations or deletions of tumor suppressor genes are hard to pharmacologically target in cancer therapy. One possible approach to target cancers with an inactivated tumor suppressor is to find synthetic lethal targets in such tumor cells [64]. The discovery of synthetic lethal interaction between breast cancer susceptibility gene 1/2 (BRCA1/2) tumor suppressors and poly ADP-ribose polymerase (PARP) DNA repair protein provided an opportunity to use PARP inhibitors in clinical cancer therapy [65]. With the aim of identifying genes that are in synthetic lethal relationship with RB1, RNAi, shRNA, CRISPR/Cas9i or chemical libraries have been often employed on high-throughput screening bases. RB1 mutation contributes to the development of SCLC (see above). However, it has been difficult to develop methods to directly target RB1 mutation in SCLC due to the lack of ‘druggability’—intrinsic factors favoring the design of small chemical inhibitors. One notable success comes in a pharmacogenomic screening of RB1-mutant cancer cell lines that identified Aurora A, whose inhibition exhibited cell death selectively in RB1-mutant SCLC cells [66]. A CRISPR/Cas9-based screening identified Aurora B whose loss appeared to be synthetic lethal with RB1 loss in SCLC [67]. The latter study speculated that inhibition of Aurora B might affect mitotic fidelity in RB1-deficient cells to a lethal level. Another study using an RNAi library in RB1-knockout lung cancer cells identified Aurora A and reasoned its mechanism by E2F-mediated upregulation of microtubule destabilizer Stathmin/OP18. Aurora A inhibition activated Stathmin through reduction of its phosphorylation, inducing mitotic cell death in RB1-knockout lung cancer cells [68].
Aurora kinases maintain mitotic fidelity by driving centrosome maturation, spindle assembly, centrosome bi-orientation and cytokinesis as one member of mitotic kinases. Many mitotic kinases are aberrantly expressed in RB1-deficient cells in an E2F-dependent manner and polo-like kinase 1 (PLK1) is one of them [69]. This provides one of the mechanisms whereby RB1 controls mitotic fidelity. Additionally, RB1 inactivation is linked to chromosomal instability (CIN) at least partially through enhanced replication fork stalling [69]. Thereby, in specific contexts, RB1 inactivation induces DNA damage response associated with increased activity of ATM, ATR, checkpoint kinase 1 (CHK1) and CHK2 and increased staining for γ-H2AX at the level that subsequently triggers cellular senescence [36,48,70,71,72]. A study using a pharmacological screening revealed that the synthetic CDK4/6 inhibitor strongly antagonizes the toxicity induced by a PLK1 inhibitor and a CHK1 inhibitor in triple negative breast cancer (TNBC) cells [73]. Conversely, the loss of RB1 highly sensitized TNBC cells to these reagents. Actually, PLK1 and CHK1 were upregulated in RB1-deficient TNBCs. In the presence of PLK1 inhibitor, RB1-proficient cells arrest in G2/M, and thus escaped from cell death, but RB1-deficient cells maintain DNA replication and progress to higher aneuploidy and would hence undergo cell death when treated with PLK1 inhibitor. In the presence of CHK1 inhibitor, RB1-deficient cells exhibited several symptoms of S phase collapse that were absent in RB1-proficient cells [73]. These findings suggest that the unstable mitotic fidelity or dampen DNA damage response due to the inactivation of RB1, would expose tumor cells to novel vulnerability to agents that target these accumulated abnormalities to catastrophically trigger cell death, instead of resulting in CIN.
SCLC often harbor mutations in RB1 and Trp53, but rare mutation in BRCA1/2 nor other genes carrying homologous recombination functions. However, single usage of PARP inhibitors or combination therapy with another drug have been suggested to be effective in the treatment of SCLC [74]. Furthermore, in prostate cancers, simultaneous loss of BRCA2 and RB1 sensitizes tumor cells to PARP inhibitor [75]. In advanced prostate cancer cases, the co-deletion of RB1 and BRCA2 is frequently found. If even the lack of RB1 alone increases the sensitivity to PARP inhibitors is not clarified yet.
There are indeed many observations from basic and clinical studies indicating that loss of RB1 function tend to associate with favorable response to chemotherapy [76]. More efficient and prolonged response to chemotherapy was reported in RB1-deficient non-small cell lung cancers (NSCLC) compared to in their RB1-proficient counterparts. This led the authors of these studies to propose that RB1 deficiency has resulted in the bypassing of certain checkpoints [77]. Similarly, breast cancer cases with disrupted RB pathway tend to exhibit favorable response to neoadjuvant chemotherapy [78,79]. In TNBC, RB1 deficiency may better benefit from gamma irradiation and chemotherapy using doxorubicin and methotrexate rather than RB1-proficient [80]. Similarly, the loss of RB1 function together with K-Ras mutation predicts favorable response to platinum-based chemotherapy in a particular type of pancreatic neoplasia [81]. More recently, RB1 loss was identified as one of predictive markers of better response to a topoisomerase inhibitor irinotecan [82]. Therefore, whilst the loss of RB1 function is recognized as a hallmark of an overall poor prognosis for cancer patients, it may paradoxically provide clinically important vulnerabilities that could be targeted by genotoxic agents.
The acceleration of cell cycle progression following RB1 inactivation may provide further exploitable vulnerabilities in cancer therapy. Cell division cycle 25 proteins (CDC25s) are dual phosphatases, of which CDC25A and CDC25B are prominent in the frequency of their overexpression in cancers. Such a status in TNBC predicts poor prognosis. One of their most important substrates is cyclin dependent kinase 1 (CDK1), which cooperates with cyclin B to promote mitotic processes. Liu et al. screened a kinome/phosphatase inhibitor library for compounds cytotoxic to RB1, PTEN and Trp53-deficient TNBC cells, and identified a CDC25 inhibitor [83]. WEE1 is a nuclear kinase that phosphorylates CDK1 at the same site (Tyr15) as CDC25s [84]. The authors demonstrated that a WEE1 inhibitor synergizes with a CDC25 inhibitor. How inhibitors of proteins with such opposing functions synergize is explained by their distinct roles during cell cycle progression [85]. WEE1 phosphorylates CDK1 during S phase at Thr14 and Tyr15 enabling a separate phosphorylation at Thr161 by CDK-activating kinase (CAK) during G2 phase [86]. This additional phosphorylation at Thr161 allows CDC25s to fully activate CDK1 during M phase by executing dephosphorylation at Thr14 and Tyr15 [87]. In short, WEE1 and CDC25s cooperate to promote the M phase progression in a sequential manner [88]. Other researchers demonstrated that the treatment of TNBC cells with a CDC25 inhibitor collaterally activates a resistant mechanism that is supported by AKT activity [89]. Thus, an PI3K inhibitor would synergize with a CDC25 inhibitor. Although suppression of CDK1 activity was thought to be a key mechanism of the action of these inhibitors, CDK1 antagonist did not fully recapitulate it. This result indicates that there may be CDK1-indepdent functions of CDC25 and WEE1 in driving RB1-deficient cancers.
Prior to these reports, a study using Drosophila melanogaster system uncovered that inactivation of rbf (the fly orthologue of RB1) and gig (the fly orthologue of tuberous sclerosis complex 2 (TSC2)) synergistically induces cell death in imaginal disc cells placed under stress conditions [90]. TSC2 together with TSC1 inhibits the function of a complex containing TORC1 and thereby suppresses tumor growth. Germline mutation in either of these genes causes an autosomal dominant tumor-prone syndrome. The phenotypes observed in rbf-gig double deficient fly cells appeared to depend on E2F transcription factors and the TOR (target of rapamycin) pathway. The synthetic lethal interaction between RB1 and TSC2 was also a case in multiple human prostate cancer cell lines. Inhibition of TSC2 in RB1-deficient cancer cells increased oxidative stress in a superoxide dismutase 2 (SOD2)-dependent manner, causing apoptosis [90]. The authors reasoned that this phenomenon was caused by the suppression of de novo lipid synthesis [90]. A follow-up study done by the same group indicated that dtsc1 (the fly orthologue of TSC1) also exhibited synthetic lethality when inactivated together with rbf, which is associated with deregulated G1/S control, increased DNA damage and energy stress [91]. These findings suggest that the mTOR pathway may provide a vulnerable point in the treatment of RB1-deficient cancers in human.
An ‘ORFeome’ RNAi screen in Caenorhabditis elegans identified zfp-2, which encodes a zinc finger transcription factor, as a gene in a synthetic lethal relationship with lin-35 (the C. elegans orthologue of RB1). These genes appeared to cooperate in somatic gonadal development [92].
A study made use of multiple publicly available large-scale shRNA and siRNA screening data bases to explore genes in the synthetic lethal relationship with RB1 in TNBCs and newly identified NUP88 and NUP214 nuclear pore components associated with the MAD2 spindle check protein. It further explored a kinase and bromodomain containing transcription factor called transcription initiation factor TFIID subunit 1 (TAF1) and rediscovered SKP2 and SKP1 [93]. As above mentioned, MAD2 is overexpressed in RB1-deficient cells and critically contributes to CIN [69]. TAF1 has been demonstrated to directly bind to RB1 to carry out basal transcription [94].
The products of most of genes so far identified due to their synthetic lethal relationship with RB1 are pharmacologically targetable (Table 1). Although loss of RB1 function is generally associated with poor prognosis, it simultaneously gives rise to pharmacologically tractable vulnerabilities leading tumor cells to carry lethal levels of DNA damage, CIN, oxidative stress or E2F hyperactivation, causing cell death hopefully at a therapeutically satisfactory degree. As RB1 function is lost in a wide variety of cancer types at varied stages, synthetic lethality screening in RB1-deficient cells needs to be extended to more varies of cancers.
5. Targeting Loss of a Gene Neighboring RB1
The frequency that RB1 gene loci undergo homozygous deletion in cancer is the fourth highest among tumor suppressor genes following INK4A, PTEN and SMAD4 [95]. Genetic deletion of SMAD4 that occurs in almost 30% of pancreatic ductal adenocarcinoma (PDAC) often associates simultaneous deletion of malic enzyme 2 (ME2) gene, as these genes locate within 200 kb distance in the same chromosome. ME2 and its isozyme ME3 share oxidative decarboxylase function to convert malate to pyruvate and to regenerate NADPH. Depletion of ME3 in ME2-deficient pancreatic cancer cells induced lethal effects [96]. A similar collateral vulnerability has been found in the glycolytic gene enolase 1 (ENO1) and ENO2. ENO1 locates in the 1p36 locus which is frequently deleted in glioblastoma [97]. These studies demonstrated that targeting genes involved in genomic collapse in the chromosomal region surrounding tumor suppressor genes or frequently collapsed genome regions may generate therapeutic benefits through collateral lethality.
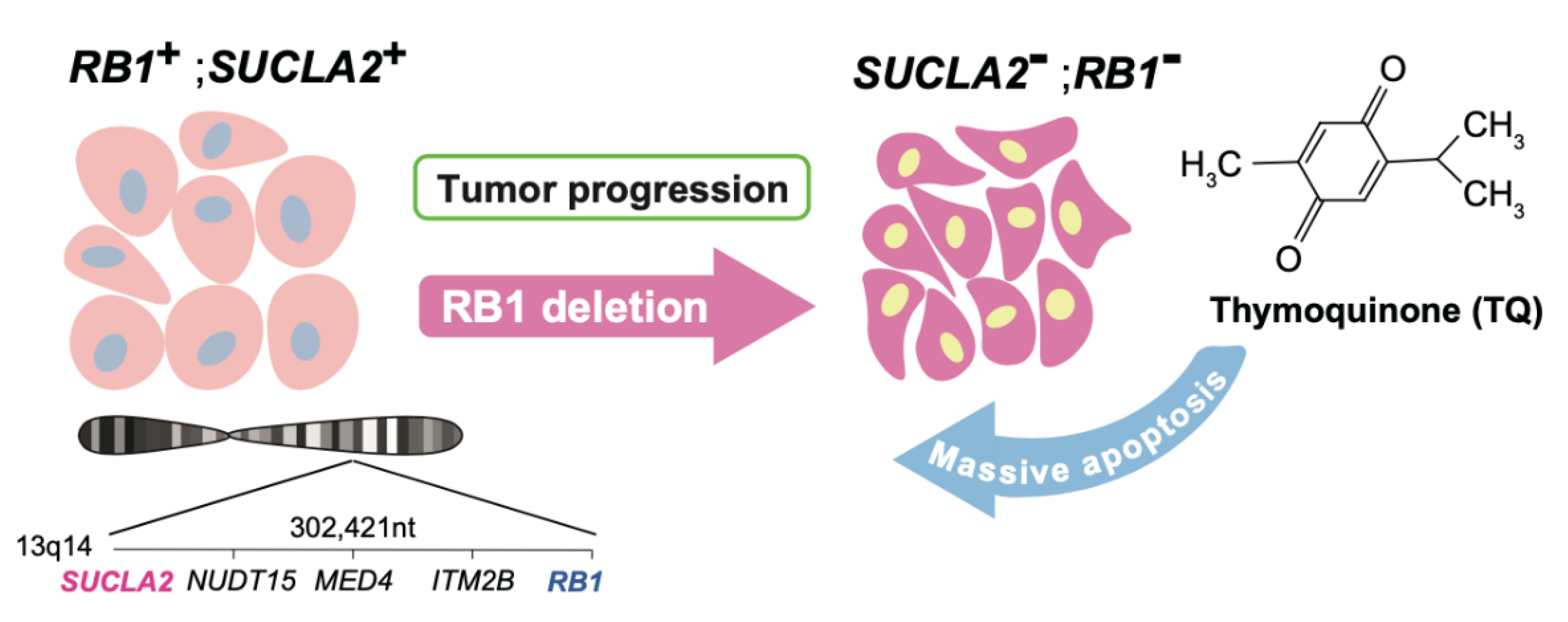
Kohno et al. attempted a survey of metabolic genes located near to tumor suppressor genes that are frequently deleted in cancers. They then examined the frequency of simultaneous loss of such genes upon tumor suppressor gene loss in individual cancer type. This effort discovered succinyl lyase A2 (SUCLA2) gene that locates around 302 kb away from RB1 tumor suppressor gene in human chromosome 13q14.2. The germ line mutation or deletion in SUCLA2 gene is causative of mitochondrial encephalomyopathy and methylmalonic aciduria [98]. There is a case report of a patient who had undergone a genomic deletion involving both SUCLA2 and RB1 gene. The patient developed encephalomyopathy and retinoblastoma simultaneously [99]. The simultaneous loss of RB1 and SUCLA2 was detected in various cancer types with varied prevalence. Surprisingly, particularly in prostate cancers, SUCLA2 gene deletion was found to take place upon RB1 loss with near to 100% prevalence [100].
SUCLA2 contributes to a complex of enzymes that converts succinyl-CoA to succinate in the TCA cycle and generates GTP from GDP. Indeed, depletion of SUCLA2 in prostate cancer cells resulted in lower mitochondrial/respiratory activity and enhanced glycolysis. These metabolic phenotypes continuously sustained as long as SUCLA2 was downregulated and thus were likely to be targeted aiming for treatment. Kohno et al. sought the ways to make use of this finding. First, they attempted to additionally inhibit SUCLG2 isozyme in the presence of RB1-SUCLA2 deletion, as it might completely disrupt the conversion from succinyl-CoA to succinate. However, it did not kill such cells with satisfactory efficacy [100].
Kohno et al. next screened a number of chemical libraries to find compounds that selectively kill RB1-SUCLA2-deficeint prostate cancer cells [100]. This attempt hit several compounds including 2-isopropyl-5-methylbenzo-1,4-quinone (thymoquinone), a natural compound which is known as one of major bioactive ingredients of Nigella Sativa [101] (Figure 1). Although its anticancer and antioxidative functions were reported, the molecular target has been unidentified. In vivo treatment of immunodeficient mice xenografted with RB1-SUCLA2-deficeint prostate cancer cells with thymoquinone resulted in a significantly selective suppression of tumorous growth via apoptosis [100]. These findings indicated that the loss of a gene neighboring RB1 can be pharmacologically targeted in cancer therapy. Homozygous deletion of RB1 is estimated to occur in more than 30% of advanced prostate cancer cases [11]. Heterozygous deletion of RB1 is also frequent in advanced prostate cancers, which is naturally associated with heterozygous loss of SUCLA2. Thymoquinone appeared to be effective to SUCLA2-heterozyous prostate cancers as well [100]. Interestingly, the Aurora A inhibitor that targets RB1 loss in a synthetic lethal manner synergized with thymoquinone in prostate cancer cells doubly deficient of RB1 and SUCLA2 [100]. Furthermore, more varieties of cancers (bladder, ovarian, uterine, liver, cervical, esophageal, breast cancers, sarcoma, diffuse large B-cell lymphoma, etc.) are estimated to carry RB1-SUCLA2 deletion with up to 5% prevalence [100]. The development of drugs with stronger thymoquinone-like activity and identification of target molecule are awaited. Additionally, the discovery of biomarkers that help quick diagnosis of SUCLA2 deficiency in tumor tissue would be forthcoming [100].
In addition to SUCLA2, there are some more genes neighboring RB1 locating even closer to RB1. Lee et al. demonstrated that among such genes, loss of integral membrane protein 2B (ITM2B), chromosomal condensation 1-llike (CHC1L) and purinergic receptor P2Y G protein-coupled protein 5 (P2RY5) function may contribute to enhance tumor promotion in RB1-deficient bladder cancer [102]. In a sharp contrast, loss of SUCLA2 would endow a certain disadvantage from the views of metabolism and pharmacological vulnerability. Probably, this explains why RB1-SUCLA2 co-deletion is not quite prevalent in cancers other than prostate cancer [100].
Dyson’s group carried out shRNA screenings to identify genes assisting RB1-induced cellular senescence and discovered large tumor suppressor 2 (LATS2) [103]. Interestingly, LATS2 locus are frequently deleted simultaneously with RB1 locus. LATS2 is primarily a component of Hippo pathway, but appeared to phosphorylate dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), which then phosphorylates LIN52 a subunit of DREAM (DP-1, RB1, E2Fs and MuvB) complex. Therefore, loss of LATS2 impairs functions of DREAM complex, cooperating with RB1 loss to promote tumor development [104]. In this scenario, in sharp contrast to SUCLA2, loss of LATS2 generates certain growth advantages to RB1-deficient cancer. It is of interest to address if pharmacological enhancement of LATS2 function in RB1-deficient cancer is feasible. As described above, BRCA2 gene is also often co-deleted with RB1 [74]. These tumors are good therapeutic targets of PARP inhibitors.
6. Conclusions
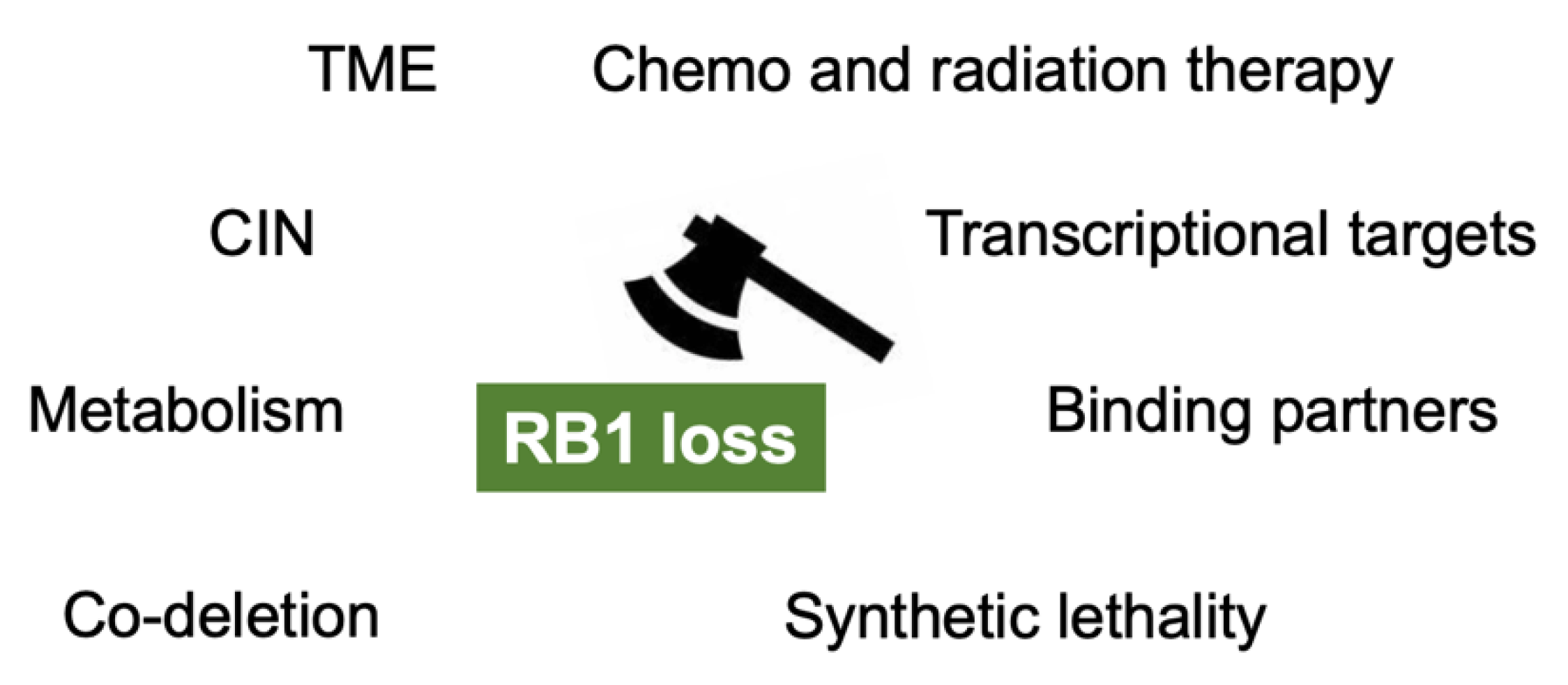
Inactivation of RB1 functions and the RB pathway is a major hallmark of cancer. Irreversible dysfunction of RB1 often predicts poor prognosis in many types of cancer. However, the RB1-defecient status in cancers may turn out to be a disguised blessing for cancer patients as it generates a variety of therapeutic benefits through the upregulation of RB1 targets, the presence of genes which are in a synthetic lethal relationship with RB1, and collateral synthetic lethality due to SUCLA2 loss (Figure 2).
Author Contributions
P.L., S.K., J.S. and C.T. wrote and D.V. edited the manuscript. All other authors provided information necessary for preparing this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Japan Agency for Medical Research and Development, 21cm0106182h0006.
Acknowledgments
This study was supported by Funding Program for Next Generation World-Leading Researchers (LS049) from cabinet office of Japan, Grant-in-Aid for Scientific Research (17H03576, 17K19586, 19K22555 and 20H03509 to CT, and 17K14992 and 20K07612 to SK) from MEXT, Project for Cancer Research and Therapeutic (P-CREATE) (19cm0106164h0001, 20cm0106164h0001 and 21cm0106182h0006) from Japan Agency for Medical Research and Development (AMED), and TaNeDS (C1010568) and a collaborative research fund (C1010818) from Daiichi-Sankyo Co. Ltd.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.L.; Polski, A.; Cavenee, W.K.; Dryja, T.P.; Murphree, A.L.; Gallie, B.L. The RB1 Story: Characterization and Cloning of the First Tumor Suppressor Gene. Genes 2019, 10, 879. [Google Scholar] [CrossRef] [Green Version]
- Macpherson, D. Insights from mouse models into human retinoblastoma. Cell Div. 2008, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Wikenheiser-Brokamp, K.A. Retinoblastoma family proteins: Insights gained through genetic manipulation of mice. Cell. Mol. Life Sci. 2006, 63, 767–780. [Google Scholar] [CrossRef]
- Wikenheiser-Brokamp, K.A. Retinoblastoma regulatory pathway in lung cancer. Curr. Mol. Med. 2006, 6, 783–793. [Google Scholar] [CrossRef]
- Viatour, P.; Sage, J. Newly identified aspects of tumor suppression by RB. Dis. Model. Mech. 2011, 4, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Feugeas, O.; Guriec, N.; Babin-Boilletot, A.; Marcellin, L.; Simon, P.; Babin, S.; Thyss, A.; Hofman, P.; Terrier, P.; Kalifa, C.; et al. Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J. Clin. Oncol. 1996, 14, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Yee, J.K.; Shew, J.Y.; Chen, P.L.; Bookstein, R.; Friedmann, T.; Lee, E.Y.; Lee, W.H. Suppression of the neoplastic phenotype by replacement of the RB gene in human cancer cells. Science 1988, 242, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Ookawa, K.; Tsuchida, S.; Adachi, J.; Yokota, J. Differentiation induced by RB expression and apoptosis induced by p53 expression in an osteosarcoma cell line. Oncogene 1997, 14, 1389–1396. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Yeow, W.S.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.E.; Witkiewicz, A.K.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salah, M.; Nishimoto, Y.; Kohno, S.; Kondoh, A.; Kitajima, S.; Muranaka, H.; Nishiuchi, T.; Ibrahim, A.; Yoshida, A.; Takahashi, C. An in vitro system to characterize prostate cancer progression identified signaling required for self-renewal. Mol. Carcinog. 2016, 55, 1974–1989. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB-IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef]
- Yoshida, A.; Kitajima, S.; Li, F.; Chaoyang, C.; Takegami, Y.; Kohno, S.; Wan, Y.; Hayashi, N.; Muranaka, H.; Nishimoto, Y.; et al. MicroRNA-140 mediates RB tumor suppressor function to control stem cell-like activity through interleukin-6. Oncotarget 2017, 8, 13872–13885. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Kitajima, S.; Kohno, S.; Yoshida, A.; Tange, S.; Sasaki, S.; Okada, N.; Nishimoto, Y.; Muranaka, H.; Nagatani, N.; et al. Retinoblastoma Inactivation Induces a Protumoral Microenvironment via Enhanced CCL2 Secretion. Cancer Res. 2019, 79, 3903–3915. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Kohno, S.; Kondoh, A.; Sasaki, N.; Nishimoto, Y.; Li, F.; Mohammed, M.S.A.; Muranaka, H.; Nagatani, N.; Suzuki, M.; et al. Undifferentiated State Induced by Rb-p53 Double Inactivation in Mouse Thyroid Neuroendocrine Cells and Embryonic Fibroblasts. Stem Cells 2015, 33, 1657–1669. [Google Scholar] [CrossRef]
- Kohno, S.; Kitajima, S.; Sasaki, N.; Takahashi, C. Retinoblastoma tumor suppressor functions shared by stem cell and cancer cell strategies. World J. Stem Cells 2016, 8, 170–184. [Google Scholar] [CrossRef]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [Green Version]
- Bracken, A.P.; Ciro, M.; Cocito, A.; Helin, K. E2F target genes: Unraveling the biology. Trends Biochem. Sci. 2004, 29, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, L.; Bronson, R.; Williams, B.O.; Dyson, N.J.; Harlow, E.; Jacks, T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(+/−)mice. Nat. Genet. 1998, 18, 360–364. [Google Scholar] [CrossRef]
- Fujiwara, K.; Yuwanita, I.; Hollern, D.P.; Andrechek, E.R. Prediction and genetic demonstration of a role for activator E2Fs in Myc-induced tumors. Cancer Res. 2011, 71, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Rouaud, F.; Hamouda-Tekaya, N.; Cerezo, M.; Abbe, P.; Zangari, J.; Hofman, V.; Ohanna, M.; Mograbi, B.; El-Hachem, N.; Benfodda, Z.; et al. E2F1 inhibition mediates cell death of metastatic melanoma. Cell Death Dis. 2018, 9, 527. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Lu, F.; Shao, Y. The E2F family as potential biomarkers and therapeutic targets in colon cancer. PeerJ 2020, 8, e8562. [Google Scholar] [CrossRef]
- Ma, Y.; Kurtyka, C.A.; Boyapalle, S.; Sung, S.S.; Lawrence, H.; Guida, W.; Cress, W.D. A small-molecule E2F inhibitor blocks growth in a melanoma culture model. Cancer Res. 2008, 68, 6292–6299. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2019, 62, 4233–4251. [Google Scholar] [CrossRef]
- Sotillo, R.; Hernando, E.; Diaz-Rodriguez, E.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Schvartzman, J.M.; Duijf, P.H.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Perez, I.S. MAD2 in the Spotlight as a Cancer Therapy Regulator. Int. J. Cancer Oncol. 2016, 3, 1–8. [Google Scholar] [CrossRef]
- Kastl, J.; Braun, J.; Prestel, A.; Moller, H.M.; Huhn, T.; Mayer, T.U. Mad2 Inhibitor-1 (M2I-1): A Small Molecule Protein-Protein Interaction Inhibitor Targeting the Mitotic Spindle Assembly Checkpoint. ACS Chem. Biol. 2015, 10, 1661–1666. [Google Scholar] [CrossRef]
- Li, J.; Dang, N.; Martinez-Lopez, N.; Jowsey, P.A.; Huang, D.; Lightowlers, R.N.; Gao, F.; Huang, J.Y. M2I-1 disrupts the in vivo interaction between CDC20 and MAD2 and increases the sensitivities of cancer cell lines to anti-mitotic drugs via MCL-1s. Cell Div. 2019, 14, 5. [Google Scholar] [CrossRef]
- Sage, J.; Straight, A.F. RB’s original CIN? Genes Dev. 2010, 24, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Amato, A.; Lentini, L.; Schillaci, T.; Iovino, F.; Di Leonardo, A. RNAi mediated acute depletion of retinoblastoma protein (pRb) promotes aneuploidy in human primary cells via micronuclei formation. BMC Cell Biol. 2009, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
- Shamma, A.; Takegami, Y.; Miki, T.; Kitajima, S.; Noda, M.; Obara, T.; Okamoto, T.; Takahashi, C. Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 2009, 15, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Takahashi, C. Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci. 2017, 108, 1726–1731. [Google Scholar] [CrossRef]
- Kitajima, S.; Li, F.; Takahashi, C. Tumor Milieu Controlled by RB Tumor Suppressor. Int. J. Mol. Sci. 2020, 21, 2450. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e26. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Lu, X.; Horvitz, H.R. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 1998, 95, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Ceol, C.J.; Horvitz, H.R. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell 2001, 7, 461–473. [Google Scholar] [CrossRef]
- Lee, K.Y.; Ladha, M.H.; McMahon, C.; Ewen, M.E. The retinoblastoma protein is linked to the activation of Ras. Mol. Cell Biol. 1999, 19, 7724–7732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, C.; Bronson, R.T.; Socolovsky, M.; Contreras, B.; Lee, K.Y.; Jacks, T.; Noda, M.; Kucherlapati, R.; Ewen, M.E. Rb and N-ras function together to control differentiation in the mouse. Mol. Cell Biol. 2003, 23, 5256–5268. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Contreras, B.; Bronson, R.T.; Loda, M.; Ewen, M.E. Genetic interaction between Rb and K-ras in the control of differentiation and tumor suppression. Mol. Cell Biol. 2004, 24, 10406–10415. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Contreras, B.; Iwanaga, T.; Takegami, Y.; Bakker, A.; Bronson, R.T.; Noda, M.; Loda, M.; Hunt, J.L.; Ewen, M.E. Nras loss induces metastatic conversion of Rb1-deficient neuroendocrine thyroid tumor. Nat. Genet. 2006, 38, 118–123. [Google Scholar] [CrossRef]
- Takahashi, C.; Ewen, M.E. Genetic interaction between Rb and N-ras: Differentiation control and metastasis. Cancer Res. 2006, 66, 9345–9348. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, C.; Sasaki, N.; Kitajima, S. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci. 2012, 103, 1182–1188. [Google Scholar] [CrossRef]
- Muranaka, H.; Hayashi, A.; Minami, K.; Kitajima, S.; Kohno, S.; Nishimoto, Y.; Nagatani, N.; Suzuki, M.; Kulathunga, L.A.N.; Sasaki, N.; et al. A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis 2017, 6, e350. [Google Scholar] [CrossRef]
- Kulathunga, N.; Kohno, S.; Linn, P.; Nishimoto, Y.; Horike, S.I.; Zaraiskii, M.I.; Kumar, S.; Muranaka, H.; Takahashi, C. Peripubertal high-fat diet promotes c-Myc stabilization in mammary gland epithelium. Cancer Sci. 2020, 111, 2336–2348. [Google Scholar] [CrossRef]
- Zhang, J.; Benavente, C.A.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J.; et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef]
- Zocchi, L.; Mehta, A.; Wu, S.C.; Wu, J.; Gu, Y.; Wang, J.; Suh, S.; Spitale, R.C.; Benavente, C.A. Chromatin remodeling protein HELLS is critical for retinoblastoma tumor initiation and progression. Oncogenesis 2020, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Benavente, C.A.; Finkelstein, D.; Johnson, D.A.; Marine, J.C.; Ashery-Padan, R.; Dyer, M.A. Chromatin remodelers HELLS and UHRF1 mediate the epigenetic deregulation of genes that drive retinoblastoma tumor progression. Oncotarget 2014, 5, 9594–9608. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaprio, J.A. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology 2009, 384, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.S.; Chen, J.Y.; Tsai, H.J.; Chen, T.Y.; Hung, W.C. The SUV39H1 inhibitor chaetocin induces differentiation and shows synergistic cytotoxicity with other epigenetic drugs in acute myeloid leukemia cells. Blood Cancer J. 2015, 5, e313. [Google Scholar] [CrossRef]
- Huang, L.Y.; Lee, Y.S.; Huang, J.J.; Chang, C.C.; Chang, J.M.; Chuang, S.H.; Kao, K.J.; Tsai, Y.J.; Tsai, P.Y.; Liu, C.W.; et al. Characterization of the biological activity of a potent small molecule Hec1 inhibitor TAI-1. J. Exp. Clin. Cancer Res. 2014, 33, 6. [Google Scholar] [CrossRef] [Green Version]
- Ji, P.; Jiang, H.; Rekhtman, K.; Bloom, J.; Ichetovkin, M.; Pagano, M.; Zhu, L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 2004, 16, 47–58. [Google Scholar] [CrossRef]
- Lu, Z.; Bauzon, F.; Fu, H.; Cui, J.; Zhao, H.; Nakayama, K.; Nakayama, K.I.; Zhu, L. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat. Commun. 2014, 5, 3463. [Google Scholar] [CrossRef] [Green Version]
- Skapek, S.X.; Pan, Y.R.; Lee, E.Y. Regulation of cell lineage specification by the retinoblastoma tumor suppressor. Oncogene 2006, 25, 5268–5276. [Google Scholar] [CrossRef] [Green Version]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells Lacking the RB1 Tumor Suppressor Gene Are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 5105. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.L.; Dyson, N.J. RB: Mitotic implications of a tumour suppressor. Nat. Rev. Cancer 2012, 12, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Shamma, A.; Suzuki, M.; Hayashi, N.; Kobayashi, M.; Sasaki, N.; Nishiuchi, T.; Doki, Y.; Okamoto, T.; Kohno, S.; Muranaka, H.; et al. ATM mediates pRB function to control DNMT1 protein stability and DNA methylation. Mol. Cell Biol. 2013, 33, 3113–3124. [Google Scholar] [CrossRef] [Green Version]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.E.; Roes, M.V.; Passos, D.T.; DeWeerd, M.C.; Chaikovsky, A.C.; Sage, J.; Howlett, C.J.; Dick, F.A. RB1 Deletion in Retinoblastoma Protein Pathway-Disrupted Cells Results in DNA Damage and Cancer Progression. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farago, A.F.; Yeap, B.Y.; Stanzione, M.; Hung, Y.P.; Heist, R.S.; Marcoux, J.P.; Zhong, J.; Rangachari, D.; Barbie, D.A.; Phat, S.; et al. Combination Olaparib and Temozolomide in Relapsed Small-Cell Lung Cancer. Cancer Discov. 2019, 9, 1372–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, G.; Armenia, J.; Mazzu, Y.Z.; Nandakumar, S.; Stopsack, K.H.; Atiq, M.O.; Komura, K.; Jehane, L.; Hirani, R.; Chadalavada, K.; et al. Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Zagorski, W.A.; Knudsen, E.S.; Reed, M.F. Retinoblastoma deficiency increases chemosensitivity in lung cancer. Cancer Res. 2007, 67, 8264–8273. [Google Scholar] [CrossRef] [Green Version]
- Trere, D.; Brighenti, E.; Donati, G.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Montanaro, L.; Derenzini, M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann. Oncol. 2009, 20, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Ertel, A.; McFalls, J.; Valsecchi, M.E.; Schwartz, G.; Knudsen, E.S. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2012, 18, 5110–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, T.J.; Liu, J.C.; Vizeacoumar, F.; Sun, T.; Maclean, N.; Egan, S.E.; Schimmer, A.D.; Datti, A.; Zacksenhaus, E. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PLoS ONE 2013, 8, e78641. [Google Scholar] [CrossRef] [Green Version]
- Hijioka, S.; Hosoda, W.; Matsuo, K.; Ueno, M.; Furukawa, M.; Yoshitomi, H.; Kobayashi, N.; Ikeda, M.; Ito, T.; Nakamori, S.; et al. Rb Loss and KRAS Mutation Are Predictors of the Response to Platinum-Based Chemotherapy in Pancreatic Neuroendocrine Neoplasm with Grade 3: A Japanese Multicenter Pancreatic NEN-G3 Study. Clin. Cancer Res. 2017, 23, 4625–4632. [Google Scholar] [CrossRef] [Green Version]
- Coussy, F.; El-Botty, R.; Chateau-Joubert, S.; Dahmani, A.; Montaudon, E.; Leboucher, S.; Morisset, L.; Painsec, P.; Sourd, L.; Huguet, L.; et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z.; et al. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent Activating Kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef] [PubMed]
- Lew, D.J.; Kornbluth, S. Regulatory roles of cyclin dependent kinase phosphorylation in cell cycle control. Curr. Opin. Cell Biol. 1996, 8, 795–804. [Google Scholar] [CrossRef]
- Zacksenhaus, E.; Liu, J.C.; Granieri, L.; Vorobieva, I.; Wang, D.Y.; Ghanbari-Azarnier, R.; Li, H.; Ali, A.; Chung, P.E.D.; Ju, Y.; et al. CDC25 as a common therapeutic target for triple-negative breast cancer—The challenges ahead. Mol. Cell Oncol. 2018, 5, e1481814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gordon, G.M.; Du, C.H.; Xu, J.; Du, W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell 2010, 17, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Gordon, G.M.; Zhang, T.; Zhao, J.; Du, W. Deregulated G1-S control and energy stress contribute to the synthetic-lethal interactions between inactivation of RB and TSC1 or TSC2. J. Cell Sci. 2013, 126, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceron, J.; Rual, J.F.; Chandra, A.; Dupuy, D.; Vidal, M.; van den Heuvel, S. Large-scale RNAi screens identify novel genes that interact with the C. elegans retinoblastoma pathway as well as splicing-related components with synMuv B activity. BMC Dev. Biol. 2007, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Brough, R.; Gulati, A.; Haider, S.; Kumar, R.; Campbell, J.; Knudsen, E.; Pettitt, S.J.; Ryan, C.J.; Lord, C.J. Identification of highly penetrant Rb-related synthetic lethal interactions in triple negative breast cancer. Oncogene 2018, 37, 5701–5718. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Siegert, J.L.; Ruppert, S.; Robbins, P.D. Rb interacts with TAF(II)250/TFIID through multiple domains. Oncogene 1997, 15, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrozzo, R.; Dionisi-Vici, C.; Steuerwald, U.; Lucioli, S.; Deodato, F.; Di Giandomenico, S.; Bertini, E.; Franke, B.; Kluijtmans, L.A.; Meschini, M.C.; et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain 2007, 130, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matilainen, S.; Isohanni, P.; Euro, L.; Lonnqvist, T.; Pihko, H.; Kivela, T.; Knuutila, S.; Suomalainen, A. Mitochondrial encephalomyopathy and retinoblastoma explained by compound heterozygosity of SUCLA2 point mutation and 13q14 deletion. Eur. J. Hum. Genet. 2015, 23, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Linn, P.; Nagatani, N.; Watanabe, Y.; Kumar, S.; Soga, T.; Takahashi, C. Pharmacologically targetable vulnerability in prostate cancer carrying RB1-SUCLA2 deletion. Oncogene 2020, 39, 5690–5707. [Google Scholar] [CrossRef]
- Khader, M.; Eckl, P.M. Thymoquinone: An emerging natural drug with a wide range of medical applications. Iran. J. Basic Med. Sci. 2014, 17, 950–957. [Google Scholar]
- Lee, S.; Jeong, J.; Majewski, T.; Scherer, S.E.; Kim, M.S.; Tuziak, T.; Tang, K.S.; Baggerly, K.; Grossman, H.B.; Zhou, J.H.; et al. Forerunner genes contiguous to RB1 contribute to the development of in situ neoplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 13732–13737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschop, K.; Conery, A.R.; Litovchick, L.; Decaprio, J.A.; Settleman, J.; Harlow, E.; Dyson, N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev. 2011, 25, 814–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschop, K.; Dyson, N. Identifying players in the functional network around pRB. Cell Cycle 2011, 10, 3814–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Thymoquinone targets SUCLA2 loss that collaterally takes place with RB1 homozygous or heterozygous deletion.
Figure 1.
Thymoquinone targets SUCLA2 loss that collaterally takes place with RB1 homozygous or heterozygous deletion.

Figure 2.
The mechanisms which enable us to target RB1 loss in cancers. TME: tumor microenvironment; CIN: chromosomal instability.
Figure 2.
The mechanisms which enable us to target RB1 loss in cancers. TME: tumor microenvironment; CIN: chromosomal instability.


{kind=link}
{kind=link}
Table 1.
Genes whose inactivation exerts synthetic lethality in the presence of RB1 deficiency, and representative inhibitors to the gene products and phase of clinical trials. ND: no data. The information was obtained by searching online accessible catalogues provided by Selleck, Santa Cruz and Abcam.
Table 1.
Genes whose inactivation exerts synthetic lethality in the presence of RB1 deficiency, and representative inhibitors to the gene products and phase of clinical trials. ND: no data. The information was obtained by searching online accessible catalogues provided by Selleck, Santa Cruz and Abcam.
| Gene | Inhibitor | Phase of Clinical Trial |
|---|---|---|
| Aurora A, B | Alisertib | III |
| Tozasertib | II | |
| Barasertib | I | |
| MNL8054 | I | |
| Danusertib | II | |
| AT9283 | II | |
| KW-2449 | I | |
| SNS-341 | I | |
| ENMD-2076 | II | |
| BI-847325 | I | |
| PLK1 | Volasertib | III |
| Rigosertib | III | |
| BI2536 | II | |
| CHK1 | AZD7762 | I |
| MK-8779 | II | |
| PF-477736 | I | |
| CDC25A, B | K-252a | ND |
| NSC663284 | ND | |
| NSC95397 | ND | |
| WEE1 | Adabosertib | II |
| TSC1, 2 | ND | ND |
| SKP2 | SKPin C1 | ND |
| TAF1 | CeMMEC-1, 13 | ND |
| BAY299 | ND | |
| NUP88 | ND | ND |
| NUP214 | ND | ND |
| PARP | Olaparib | FDA-approved |
| Veliparib | III | |
| Rucapatib | III | |
| Iniparib | III | |
| SYK | Fostamatinib | III |
| R406 | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Linn, P.; Kohno, S.; Sheng, J.; Kulathunga, N.; Yu, H.; Zhang, Z.; Voon, D.; Watanabe, Y.; Takahashi, C. Targeting RB1 Loss in Cancers. Cancers 2021, 13, 3737. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153737
AMA Style
Linn P, Kohno S, Sheng J, Kulathunga N, Yu H, Zhang Z, Voon D, Watanabe Y, Takahashi C. Targeting RB1 Loss in Cancers. Cancers. 2021; 13(15):3737. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153737
Chicago/Turabian StyleLinn, Paing, Susumu Kohno, Jindan Sheng, Nilakshi Kulathunga, Hai Yu, Zhiheng Zhang, Dominic Voon, Yoshihiro Watanabe, and Chiaki Takahashi. 2021. "Targeting RB1 Loss in Cancers" Cancers 13, no. 15: 3737. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153737
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.