The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Background
2. The Physiological Role of Cellular Prion Protein (PrPC)
2.1. Structure, Biogenesis, and Intracellular Trafficking of PrPC
2.2. The Physiological and Beneficial Role of PrPC
2.3. PrPC Functions in Cell Survival and Stress Protection
3. The Multi-Faceted Role of PrPC in Cancer initiation and Progression
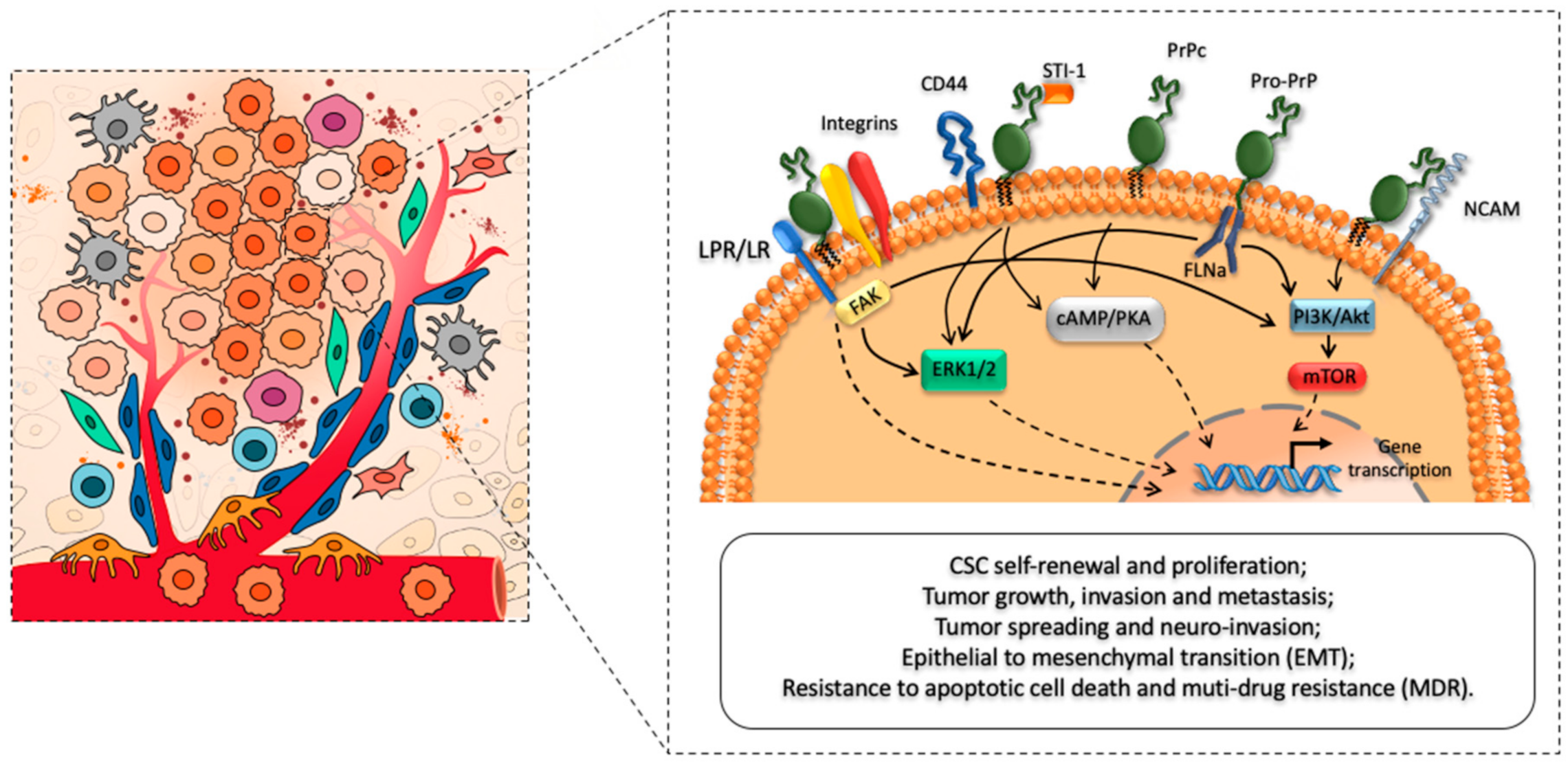
3.1. The Role of PrPC in CSC Stemness and Differentiation
3.2. The Role of PrPC in CSC Growth and Proliferation
3.3. The Role of PrPC in CSC Migration and Invasion
3.3.1. The Role of PrPC in Tumor Spreading and Neuro-Invasion
3.3.2. PrPC Spreading and PrPC-Mediated Epigenetic Effects
3.4. Role of PrPC in Multidrug Resistance (MDR)
4. PrPC as a Potential Biomarker in Human Cancers
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Ferrucci, M.; Gesi, M.; Di Poggio, A.B.; Giorgi, F.S.; Biagioni, F.; Paparelli, A. A hypothesis on prion disorders: Are infectious, inherited, and sporadic causes so distinct? Brain Res. Bull. 2006, 69, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Scrapie prions. Annu. Rev. Microbiol. 1989, 43, 345–374. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Scott, M.R.; Dearmond, S.J.; Cohen, F.E. Prion Protein Biology Review. Cell 1998, 93, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, R.K.; McKinley, M.P.; Bowman, K.A.; Braunfeld, M.B.; Barry, R.A.; Prusiner, S.B. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 2310–2314. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Epstein, F.H.; Prusiner, S.B. Prions and Neurodegenerative Diseases. N. Engl. J. Med. 1987, 317, 1571–1581. [Google Scholar] [CrossRef]
- Dearmond, S.J.; McKinley, M.P.; Barry, R.A.; Braunfeld, M.B.; McColloch, J.R.; Prusinert, S.B. Identification of prion amyloid filaments in scrapie-infected brain. Cell 1985, 41, 221–235. [Google Scholar] [CrossRef]
- Ma, J.; Lindquist, S. Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 2002, 298, 1785–1788. [Google Scholar] [CrossRef]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of prion protein aggregation in neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648–8669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schätzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Aguib, Y.; Schatzl, H.M. Autophagy, Prion Infection and their Mutual Interactions. Curr. Issues Mol. Biol. 2009, 12, 87–97. [Google Scholar] [PubMed] [Green Version]
- Speldewinde, S.H.; Doronina, V.A.; Grant, C.M. Autophagy protects against de novo formation of the [PSI+] prion in yeast. Mol. Biol. Cell 2015, 26, 4541–4551. [Google Scholar] [CrossRef]
- Prusiner, S.B. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schätzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.F.; Yu, K.H.; Jheng, C.P.; Chung, R.; Lee, C.I. Curcumin reduces amyloid fibrillation of prion protein and decreases reactive oxidative stress. Pathogens 2013, 2, 506–519. [Google Scholar] [CrossRef]
- Lee, J.H.; Moon, J.H.; Kim, S.W.; Jeong, J.K.; Nazim, U.M.D.; Lee, Y.J.; Seol, J.W.; Park, S.Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef] [Green Version]
- Comincini, S.; Facoetti, A.; Del Vecchio, I.; Peoc’h, K.; Laplanche, J.L.; Magrassi, L.; Ceroni, M.; Ferretti, L.; Nano, R. Differential expression of the prion-like protein doppel gene (PRND) in astrocytomas: A new molecular marker potentially involved in tumor progression. Anticancer Res. 2004, 24, 1507–1517. [Google Scholar]
- Comincini, S.; Ferrara, V.; Arias, A.; Malovini, A.; Azzalin, A.; Ferretti, L.; Benericetti, E.; Cardarelli, M.; Gerosa, M.; Passarin, M.G.; et al. Diagnostic value of PRND gene expression profiles in astrocytomas: Relationship to tumor grades of malignancy. Oncol. Rep. 2007, 17, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, L.; Ryan, Y.; Hilton, D.A.; Lyons-Rimmer, J.; Dave, F.; Maze, E.A.; Adams, C.L.; Rigby-Jones, R.; Ammoun, S.; Hanemann, C.O. Cellular prion protein (PrP C) in the development of Merlin-deficient tumours. Oncogene 2017, 36, 6132–6142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res. Ther. 2017, 8, 76. [Google Scholar] [CrossRef]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2015, 34, 3305–3314. [Google Scholar] [CrossRef]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Pan, Y.; Shi, Y.; Guo, C.; Jin, X.; Sun, L.; Liu, N.; Qiao, T.; Fan, D. Overexpression and significance of prion protein in gastric cancer and multidrug-resistant gastric carcinoma cell line SGC7901/ADR. Int. J. Cancer 2005, 113, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Wang, W.; Wu, Q.; Lu, Y.; Su, T.; Gu, N.; Li, K.; Wang, J.; Du, R.; Zhao, X.; et al. MGr1-Antigen/37 kDa laminin receptor precursor promotes cellular prion protein induced multi-drug-resistance of gastric cancer. Oncotarget 2017, 8, 71630–71641. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Du, J.P.; Li, S.J.; Zhai, L.P.; Yang, X.Y.; Wang, Z.H.; Wu, Z.T.; Han, Y. Octarepeat peptides of prion are essential for multidrug resistance in gastric cancer cells. J. Dig. Dis. 2012, 13, 143–152. [Google Scholar] [CrossRef]
- de Lacerda, T.C.S.; Costa-Silva, B.; Giudice, F.S.; Dias, M.V.S.; de Oliveira, G.P.; Teixeira, B.L.; dos Santos, T.G.; Martins, V.R. Prion protein binding to HOP modulates the migration and invasion of colorectal cancer cells. Clin. Exp. Metastasis 2016, 33, 441–451. [Google Scholar] [CrossRef]
- Lee, J.H.; Yun, C.W.; Lee, S.H. Cellular prion protein enhances drug resistance of colorectal cancer cells via regulation of a survival signal pathway. Biomol. Ther. 2018, 26, 313–321. [Google Scholar] [CrossRef]
- Roucou, X.; Giannopoulos, P.N.; Zhang, Y.; Jodoin, J.; Goodyer, C.G.; LeBlanc, A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005, 12, 783–795. [Google Scholar] [CrossRef] [Green Version]
- Meslin, F.; Hamaï, A.; Gao, P.; Jalil, A.; Cahuzac, N.; Chouaib, S.; Mehrpour, M. Silencing of prion protein sensitizes breast adriamycin-resistant carcinoma cells to TRAIL-mediated cell death. Cancer Res. 2007, 67, 10910–10919. [Google Scholar] [CrossRef] [Green Version]
- Gil, M.; Kim, Y.K.; Kim, K.E.; Kim, W.; Park, C.S.; Lee, K.J. Cellular prion protein regulates invasion and migration of breast cancer cells through MMP-9 activity. Biochem. Biophys. Res. Commun. 2016, 470, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Dagdanova, A.; Hescheler, J.; Wartenberg, M. Redox-regulation of intrinsic prion expression in multicellular prostate tumor spheroids. Free Radic. Biol. Med. 1999, 27, 1276–1283. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Yin, S.; Xu, J.; Petrolla, A.A.; Singh, N.; Tartakoff, A.; Abbott, D.W.; Xin, W.; et al. Binding of pro-prion to filamin A disrupts cytoskeleton and correlates with poor prognosis in pancreatic cancer. J. Clin. Investig. 2009, 119, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, S.; Huang, D.; Cui, M.; Hu, H.; Zhang, L.; Wang, W.; Parameswaran, N.; Jackson, M.; Osborne, B.; et al. Cellular Prion Protein Mediates Pancreatic Cancer Cell Survival and Invasion through Association with and Enhanced Signaling of Notch1. Am. J. Pathol. 2016, 186, 2945–2956. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, M.; Giambelluca, M.A.; Scavuzzo, M.C.; Di Franco, G.; Guadagni, S.; Palmeri, M.; Furbetta, N.; Gianardi, D.; Funel, N.; Pollina, L.E.; et al. The occurrence of prion protein in surgically resected pancreatic adenocarcinoma. Pancreatology 2020, 20, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lin, C.H.; Shih, N.C.; Liu, H.L.; Wang, W.C.; Lin, K.Y.; Liu, Z.Y.; Tseng, Y.J.; Chang, H.K.; Lin, Y.C.; et al. Cellular prion protein transcriptionally regulated by NFIL3 enhances lung cancer cell lamellipodium formation and migration through JNK signaling. Oncogene 2020, 39, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Shi, Q.; Zhang, N.S.; Xiao, K.; Chen, L.N.; Yang, X.D.; Ji, J.F.; Dong, X.P. Expression of prion protein is closely associated with pathological and clinical progression and abnormalities of p53 in head and neck squamous cell carcinomas. Oncol. Rep. 2016, 35, 817–824. [Google Scholar] [CrossRef]
- Sollazzo, V.; Galasso, M.; Volinia, S.; Carinci, F. Prion proteins (PRNP and PRND) are over-expressed in osteosarcoma. J. Orthop. Res. 2012, 30, 1004–1012. [Google Scholar] [CrossRef]
- Li, C.; Yu, S.; Nakamura, F.; Pentikäinen, O.T.; Singh, N.; Yin, S.; Xin, W.; Sy, M.S. Pro-prion binds filamin A, facilitating its interaction with integrin β1, and contributes to melanomagenesis. J. Biol. Chem. 2010, 285, 30328–30339. [Google Scholar] [CrossRef] [Green Version]
- Ke, J.; Wu, G.; Zhang, J.; Li, H.; Gao, S.; Shao, M.; Gao, Z.; Sy, M.S.; Cao, Y.; Yang, X.; et al. Melanoma migration is promoted by prion protein via Akt-hsp27 signaling axis. Biochem. Biophys. Res. Commun. 2020, 523, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Antonacopoulou, A.G.; Palli, M.; Marousi, S.; Dimitrakopoulos, F.I.; Kyriakopoulou, U.; Tsamandas, A.C.; Scopa, C.D.; Papavassiliou, A.G.; Kalofonos, H.P. Prion protein expression and the M129V polymorphism of the PRNP gene in patients with colorectal cancer. Mol. Carcinog. 2010, 49, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Déry, M.A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; LeBlanc, A.C. Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Ma, J.; Zhang, W.; Gong, C.; He, J.; Wang, Y.; Yu, G.; Yuan, C.; Wang, X.; Sun, Y.; et al. The role of prion protein expression in predicting gastric cancer prognosis. J. Cancer 2016, 7, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Le Corre, D.; Ghazi, A.; Balogoun, R.; Pilati, C.; Aparicio, T.; Martin-Lannerée, S.; Marisa, L.; Djouadi, F.; Poindessous, V.; Crozet, C.; et al. The cellular prion protein controls the mesenchymal-like molecular subtype and predicts disease outcome in colorectal cancer. EBioMedicine 2019, 46, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsaro, A.; Bajetto, A.; Thellung, S.; Begani, G.; Villa, V.; Nizzari, M.; Pattarozzi, A.; Solari, A.; Gatti, M.; Pagano, A.; et al. Cellular prion protein controls stem cell-like properties of human glioblastoma tumor-initiating cells. Oncotarget 2016, 7, 38638–38657. [Google Scholar] [CrossRef]
- Du, L.; Rao, G.; Wang, H.; Li, B.; Tian, W.; Cui, J.; He, L.; Laffin, B.; Tian, X.; Hao, C.; et al. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res. 2013, 73, 2682–2694. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Pan, Y.; Zhang, D.; Guo, C.; Shi, Y.; Wang, J.; Chen, Y.; Wang, X.; Liu, J.; Guo, X.; et al. Cellular prion protein promotes proliferation and G1/S transition of human gastric cancer cells SGC7901 and AGS. FASEB J. 2007, 21, 2247–2256. [Google Scholar] [CrossRef]
- McEwan, J.F.; Windsor, M.L.; Cullis-Hill, S.D. Antibodies to prion protein inhibit human colon cancer cell growth. Tumor Biol. 2009, 30, 141–147. [Google Scholar] [CrossRef]
- Yun, C.W.; Yun, S.; Lee, J.H.; Han, Y.S.; Yoon, Y.M.; An, D.; Lee, S.H. Silencing prion protein in ht29 human colorectal cancer cells enhances anticancer response to fucoidan. Anticancer Res. 2016, 36, 4449–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, D.; Liu, Y.; Mao, Y.; Gao, L.; Zhang, H.; Luan, S.; Huang, F.; Li, Q. TMZ-induced PrPc/par-4 interaction promotes the survival of human glioma cells. Int. J. Cancer 2012, 130, 309–318. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.; Jimenez, C.R.; Carvalho, B.; Belien, J.A.M.; Delis-Van Diemen, P.M.; Mongera, S.; Piersma, S.R.; Vikas, M.; Navani, S.; Pontén, F.; et al. Cell surface proteomics identifies glucose transporter type 1 and prion protein as candidate biomarkers for colorectal adenoma-to-carcinoma progression. Gut 2012, 61, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Shang, Y.; Liu, C.; Li, J.; Hu, H.; Liang, C.; Han, Y.; Zhang, W.; Liang, J.; Wu, K. Overexpression of PrPc, combined with MGr1-Ag/37LRP, is predictive of poor prognosis in gastric cancer. Int. J. Cancer 2014, 135, 2329–2337. [Google Scholar] [CrossRef]
- Wang, J.H.; Du, J.P.; Zhang, Y.H.; Zhao, X.J.; Fan, R.Y.; Wang, Z.H.; Wu, Z.T.; Han, Y. Dynamic changes and surveillance function of prion protein expression in gastric cancer drug resistance. World J. Gastroenterol. 2011, 17, 3986–3993. [Google Scholar] [CrossRef]
- Meslin, F.; Conforti, R.; Mazouni, C.; Morel, N.; Tomasic, G.; Drusch, F.; Yacoub, M.; Sabourin, J.C.; Grassi, J.; Delaloge, S.; et al. Efficacy of adjuvant chemotherapy according to Prion protein expression in patients with estrogen receptor-negative breast cancer. Ann. Oncol. 2007, 18, 1793–1798. [Google Scholar] [CrossRef]
- Oesch, B.; Westaway, D.; Wälchli, M.; McKinley, M.P.; Kent, S.B.H.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Makrinou, E.; Collinge, J.; Antoniou, M. Genomic characterization of the human prion protein (PrP) gene locus. Mamm. Genome 2002, 13, 696–703. [Google Scholar] [CrossRef]
- Acevedo-Morantes, C.Y.; Wille, H. The structure of human prions: From biology to structural models—Considerations and pitfalls. Viruses 2014, 6, 3875–3892. [Google Scholar] [CrossRef]
- Rane, N.S.; Yonkovich, J.L.; Hegde, R.S. Protection from cytosolic prion protein toxicity by modulation of protein translocation. EMBO J. 2004, 23, 4550–4559. [Google Scholar] [CrossRef] [Green Version]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Wiseman, F.K.; Cancellotti, E.; Piccardo, P.; Iremonger, K.; Boyle, A.; Brown, D.; Ironside, J.W.; Manson, J.C.; Diack, A.B. The Glycosylation Status of PrP C Is a Key Factor in Determining Transmissible Spongiform Encephalopathy Transmission between Species. J. Virol. 2015, 89, 4738–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.W.; Wang, L.Q.; Huang, J.J.; Pan, K.; Chen, J.; Liang, Y. Glycosylation Significantly Inhibits the Aggregation of Human Prion Protein and Decreases Its Cytotoxicity. Sci. Rep. 2018, 8, 12603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xin, W.; Sy, M.S. Binding of pro-prion to filamin A: By design or an unfortunate blunder. Oncogene 2010, 29, 5329–5345. [Google Scholar] [CrossRef] [Green Version]
- Lewis, V.; Hooper, N.M. The role of lipid rafts in prion protein biology. Front. Biosci. 2011, 16, 151–168. [Google Scholar] [CrossRef] [Green Version]
- Hooper, N.M.; Taylor, D.R.; Watt, N.T. Mechanism of the metal-mediated endocytosis of the prion protein. Biochem. Soc. Trans. 2008, 36, 1272–1276. [Google Scholar] [CrossRef]
- Peters, P.J.; Mironov, A.; Peretz, D.; Van Donselaar, E.; Leclerc, E.; Erpel, S.; DeArmond, S.J.; Burton, D.R.; Williamson, R.A.; Vey, M.; et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell Biol. 2003, 162, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.R.; Hooper, N.M. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007, 402, 17–23. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, C.; Booth, S.A.; Beniac, D.R.; Coulthart, M.B.; Booth, T.F.; McNicol, A. Cellular prion protein is released on exosomes from activated platelets. Blood 2006, 107, 3907–3911. [Google Scholar] [CrossRef] [PubMed]
- Vella, L.J.; Sharples, R.A.; Lawson, V.A.; Masters, C.L.; Cappai, R.; Hill, A.F. Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 2007, 211, 582–590. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef] [Green Version]
- Hajj, G.N.M.; Arantes, C.P.; Dias, M.V.S.; Roffé, M.; Costa-Silva, B.; Lopes, M.H.; Porto-Carreiro, I.; Rabachini, T.; Lima, F.R.; Beraldo, F.H.; et al. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci. 2013, 70, 3211–3227. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M.; Maria Merino, A. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Ryskalin, L.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. mTOR Modulates Intercellular Signals for Enlargement and Infiltration in Glioblastoma Multiforme. Cancers (Basel) 2020, 12, 2486. [Google Scholar] [CrossRef]
- Büeler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; Dearmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Büeler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Sailer, A.; Büeler, H.; Fischer, M.; Aguzzi, A.; Weissmann, C. No propagation of prions in mice devoid of PrP. Cell 1994, 77, 967–968. [Google Scholar] [CrossRef]
- Richt, J.A.; Kasinathan, P.; Hamir, A.N.; Castilla, J.; Sathiyaseelan, T.; Vargas, F.; Sathiyaseelan, J.; Wu, H.; Matsushita, H.; Koster, J.; et al. Production of cattle lacking prion protein. Nat. Biotechnol. 2007, 25, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, J.; Xu, Y.; Zhu, C.; Yu, H.; Liu, S.; Hongying, S.; Chen, J.; Xu, X.; Wu, Y.; et al. Generation of goats lacking prion protein. Mol. Reprod. Dev. 2009, 76, 3. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Limanaqi, F.; Familiari, P.; Frati, A.; Fornai, F. Prion protein in glioblastoma multiforme. Int. J. Mol. Sci. 2019, 20, 5107. [Google Scholar] [CrossRef] [Green Version]
- Prestori, F.; Rossi, P.; Bearzatto, B.; Lainé, J.; Necchi, D.; Diwakar, S.; Schiffmann, S.N.; Axelrad, H.; D’Angelo, E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J. Neurosci. 2008, 28, 7091–7103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senatore, A.; Colleoni, S.; Verderio, C.; Restelli, E.; Morini, R.; Condliffe, S.B.; Bertani, I.; Mantovani, S.; Canovi, M.; Micotti, E.; et al. Mutant PrP Suppresses Glutamatergic Neurotransmission in Cerebellar Granule Neurons by Impairing Membrane Delivery of VGCC α 2δ-1 Subunit. Neuron 2012, 74, 300–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roffé, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, M.J.; Cheng, C.Z.; Zhou, B.; Zimonjic, D.B.; Mani, S.A.; Kaba, M.; Gifford, A.; Reinhardt, F.; Popescu, N.C.; Guo, W.; et al. Enrichment of a population of mammary gland cells that form mammospheres and have in vivo repopulating activity. Cancer Res. 2007, 67, 8131–8138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, A.; Pericuesta, E.; Ramírez, M.; Gutierrez-Adan, M. Prion Protein Expression Regulates Embryonic Stem Cell Pluripotency and Differentiation. PLoS ONE 2011, 6, e18422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Steele, A.D.; Lindquist, S.; Lodish, H.F. Prion protein is expressed on long-term repopulating hematopoietic stem cells and its important for their self-renewal. Proc. Natl. Acad. Sci. USA 2006, 103, 2184–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, T.G.; Silva, I.R.; Costa-Silva, B.; Lepique, A.P.; Martins, V.R.; Lopes, M.H. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells 2011, 29, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Mangé, A.; Dong, L.; Lehmann, S.; Schachner, M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 2003, 22, 227–233. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Gauczynski, S.; Peyrin, J.M.; Haïk, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.P.; Dormont, D.; Lasmézas, C.I.; et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001, 20, 5863–5875. [Google Scholar] [CrossRef] [Green Version]
- Linden, R.; Cordeiro, Y.; Lima, L.M.T.R. Allosteric function and dysfunction of the prion protein. Cell. Mol. Life Sci. 2012, 69, 1105–1124. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Martins, V.R.; Beraldo, F.H.; Hajj, G.N.; Lopes, M.H.; Lee, K.S.; Prado, M.A.; Linden, R. Prion protein: Orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 2010, 12, 63–86. [Google Scholar]
- Chiarini, L.B.; Freitas, A.R.O.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Linden, R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002, 21, 3317–3326. [Google Scholar] [CrossRef]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.M.; Muras, A.G.; Mancini, G.L.; Castro, R.M.P.S.; Ribeiro, K.C.B.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graner, E.; Mercadante, A.F.; Zanata, S.M.; Forlenza, O.V.; Cabral, A.L.B.; Veiga, S.S.; Juliano, M.A.; Roesler, R.; Walz, R.; Minetti, A.; et al. Cellular prion protein binds laminin and mediates neuritogenesis. Mol. Brain Res. 2000, 76, 85–92. [Google Scholar] [CrossRef]
- Rushworth, J.V.; Griffiths, H.H.; Watt, N.T.; Hooper, N.M. Prion protein-mediated toxicity of amyloid-β oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 2013, 288, 8935–8951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.; Kellermann, O.; et al. Neuritogenesis: The prion protein controls β1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar] [CrossRef]
- Llorens, F.; Carulla, P.; Villa, A.; Torres, J.M.; Fortes, P.; Ferrer, I.; del Río, J.A. PrPC regulates epidermal growth factor receptor function and cell shape dynamics in Neuro2a cells. J. Neurochem. 2013, 127, 124–138. [Google Scholar] [CrossRef]
- Kuwahara, C.; Takeuchi, A.M.; Nishimura, T.; Haraguchi, K.; Kubosaki, A.; Matsumoto, Y.; Saeki, K.; Matsumoto, Y.; Yokoyama, T.; Itohara, S.; et al. Prions prevent neuronal cell-line death. Nature 1999, 400, 225–226. [Google Scholar] [CrossRef]
- Roucou, X.; Guo, Q.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A.C. Cytosolic Prion Protein Is Not Toxic and Protects against Bax-mediated Cell Death in Human Primary Neurons. J. Biol. Chem. 2003, 278, 40877–40881. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef]
- Rachidi, W.; Vilette, D.; Guiraud, P.; Arlotto, M.; Riondel, J.; Laude, H.; Lehmann, S.; Favier, A. Expression of prion protein increases cellular copper binding and antioxidant enzyme activities but not copper delivery. J. Biol. Chem. 2003, 278, 9064–9072. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Panzera, A.; Rogawski, D.; Greene, L.E.; Eisenberg, E. Cellular prion protein (PrPc) protects neuronal cells from the effect of huntingtin aggregation. J. Cell Sci. 2007, 120, 2663–2671. [Google Scholar] [CrossRef] [Green Version]
- Edenhofer, F.; Rieger, R.; Famulok, M.; Wendler, W.; Weiss, S.; Winnacker, E.L. Prion protein PrPc interacts with molecular chaperones of the Hsp60 family. J. Virol. 1996, 70, 4724–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Lindquist, S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 14955–14960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambold, A.S.; Miesbauer, M.; Rapaport, D.; Bartke, T.; Baier, M.; Winklhofer, K.F.; Tatzelt, J. Association of Bcl-2 with misfolded prion protein is linked to the toxic potential of cytosolic PrP. Mol. Biol. Cell 2006, 17, 3356–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Guo, M.; Shen, A.; Mei, F.; Peng, X.; Gong, R.; Guo, D.; Wu, J.; Tien, P.; Xiao, G. Bovine PrPC directly interacts with αB-crystalline. FEBS Lett. 2005, 579, 5419–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U.; Hayer-Hartl, M. Protein folding. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camberg, J.L.; Doyle, S.M.; Johnston, D.M.; Wickner, S. Molecular Chaperones. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 456–460. [Google Scholar]
- Satoh, J.; Onoue, H.; Arima, K.; Yamamura, T. The 14-3-3 Protein Forms a Molecular Complex with Heat Shock Protein Hsp60 and Cellular Prion Protein. J. Neuropathol. Exp. Neurol. 2005, 64, 858–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahabadi, H.M.; Taghibiglou, C. Cellular prion protein (Prpc): Putative interacting partners and consequences of the interaction. Int. J. Mol. Sci. 2020, 21, 7058. [Google Scholar] [CrossRef]
- Shyu, W.C.; Harn, H.J.; Saeki, K.; Kubosaki, A.; Matsumoto, Y.; Onodera, T.; Chen, C.J.; Hsu, Y.D.; Chiang, Y.H. Molecular modulation of expression of prion protein by heat shock. Mol. Neurobiol. 2002, 26, 1–12. [Google Scholar] [CrossRef]
- Mangé, A.; Béranger, F.; Peoc’h, K.; Onodera, T.; Frobert, Y.; Lehmann, S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell 2004, 96, 125–132. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Gillott, A.; Thomas, D.A.; Perera, W.S.S.; Hooper, N.M. Reactive oxygen species-mediated β-clevage of the prion protein in the cellular response to oxidative stress. J. Biol. Chem. 2005, 280, 35914–35921. [Google Scholar] [CrossRef] [Green Version]
- Haigh, C.L.; McGlade, A.R.; Collins, S.J. MEK1 transduces the prion protein N2 fragment antioxidant effects. Cell. Mol. Life Sci. 2015, 72, 1613–1629. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lannerée, S.; Hirsch, T.Z.; Hernandez-Rapp, J.; Halliez, S.; Vilotte, J.L.; Launay, J.M.; Mouillet-Richard, S. PrP C from stem cells to cancer. Front. Cell Dev. Biol. 2014, 2, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adorno-Cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-Hubert, E.; Sinyuk, M.; et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015, 75, 924–929. [Google Scholar] [CrossRef] [Green Version]
- Go, G.; Yun, C.W.; Yoon, Y.M.; Lim, J.H.; Lee, J.H.; Lee, S.H. Role of PrPC in cancer stem cell characteristics and drug resistance in colon cancer cells. Anticancer Res. 2020, 40, 5611–5620. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhao, L.; Liang, J.; Liu, J.; Shi, Y.; Liu, N.; Zhang, G.; Jin, H.; Gao, J.; Xie, H.; et al. Cellular prion protein promotes invasion and metastasis of gastric cancer. FASEB J. 2006, 20, 1886–1888. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kakeya, T.; Yamazaki, T.; Takekida, K.; Nakamura, N.; Matsuda, H.; Takatori, K.; Tanimura, A.; Tanamoto, K.I.; Sawada, J.I. G1-dependent prion protein expression in human glioblastoma cell line T98G. Biol. Pharm. Bull. 2002, 25, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Erlich, R.B.; Kahn, S.A.; Lima, F.R.S.; Muras, A.G.; Martins, R.A.P.; Linden, R.; Chiarini, L.B.; Martins, V.R.; Neto, V.M. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 2007, 55, 1690–1698. [Google Scholar] [CrossRef]
- Azzalin, A.; Sbalchiero, E.; Barbieri, G.; Palumbo, S.; Muzzini, C.; Comincini, S. The doppel (Dpl) protein influences in vitro migration capability in astrocytoma-derived cells. Cell. Oncol. 2008, 30, 491–501. [Google Scholar] [CrossRef]
- Kim, Y.C.; Won, S.Y.; Jeong, B.H. Identification of Prion Disease-Related Somatic Mutations in the Prion Protein Gene (PRNP) in Cancer Patients. Cells 2020, 9, 1480. [Google Scholar] [CrossRef]
- Yang, L.; Gao, Z.; Hu, L.; Wu, G.; Yang, X.; Zhang, L.; Zhu, Y.; Wong, B.S.; Xin, W.; Sy, M.S.; et al. Glycosylphos-phatidylinositol anchor modification machinery deficiency is responsible for the formation of pro-prion protein (PrP) in BxPC-3 protein and increases cancer cell motility. J. Biol. Chem. 2016, 291, 3905–3917. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Tao, L.; Xu, J.; Li, Q.; Yu, J.; Jin, Y.; Chen, Q.; Xu, Z.; Zou, Q.; Liu, X. CD44/Cellular prion protein interact in multidrug resistant breast cancer cells and correlate with responses to neoadjuvant chemotherapy in breast cancer patients. Mol. Carcinog. 2014, 53, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yun, C.W.; Han, Y.S.; Kim, S.M.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.J.; Lee, S.H. Melatonin and 5-fluorouracil co-suppress colon cancer stem cells by regulating cellular prion protein-Oct4 axis. J. Pineal Res. 2018, 65, e12519. [Google Scholar] [CrossRef] [PubMed]
- Chieng, C.K.-L.; Say, Y.-H. Cellular prion protein contributes to LS 174T colon cancer cell carcinogenesis by increasing invasiveness and resistance against doxorubicin-induced apoptosis. Tumor Biol. 2015, 36, 8107–8120. [Google Scholar] [CrossRef]
- Li, Q.Q.; Cao, X.X.; Xu, J.D.; Chen, Q.; Wang, W.J.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. The role of P-glycoprotein/cellular prion protein interaction in multidrug-resistant breast cancer cells treated with paclitaxel. Cell. Mol. Life Sci. 2009, 66, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Esch, D.; Vahokoski, J.; Groves, M.R.; Pogenberg, V.; Cojocaru, V.; Vom Bruch, H.; Han, D.; Drexler, H.C.A.; Araúzo-Bravo, M.J.; Ng, C.K.L.; et al. A unique Oct4 interface is crucial for reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 295–301. [Google Scholar] [CrossRef]
- Li, Q.Q.; Sun, Y.P.; Ruan, C.P.; Xu, X.Y.; Ge, J.H.; He, J.; De Xu, Z.; Wang, Q.; Gao, W.C. Cellular prion protein promotes glucose uptake through the Fyn-HIF-2α-Glut1 pathway to support colorectal cancer cell survival. Cancer Sci. 2011, 102, 400–406. [Google Scholar] [CrossRef]
- Tao, Z.; Li, T.; Ma, H.; Yang, Y.; Zhang, C.; Hai, L.; Liu, P.; Yuan, F.; Li, J.; Yi, L.; et al. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 2018, 9, 1063. [Google Scholar] [CrossRef]
- Wang, Q.; Qian, J.; Wang, F.; Ma, Z. Cellular prion protein accelerates colorectal cancer metastasis via the Fyn-SP1-SATB1 axis. Oncol. Rep. 2012, 28, 2029–2034. [Google Scholar] [CrossRef] [Green Version]
- Mangé, A.; Milhavet, O.; Umlauf, D.; Harris, D.; Lehmann, S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002, 514, 159–162. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Muras, A.G.; Hajj, G.N.M.; Ribeiro, K.B.; Nomizo, R.; Nonogaki, S.; Chammas, R.; Martins, V.R. Prion protein ablation increases cellular aggregation and embolization contributing to mechanisms of metastasis. Int. J. Cancer 2009, 125, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.E.; Dai, H.; Tahir, S.A.; Li, R.; Timme, T.; Ittmann, M.; Frolov, A.; Wheeler, T.M.; Rowley, D.; Thompson, T.C. Stromal antiapoptotic paracrine loop in perineural invasion of prostatic carcinoma. Cancer Res. 2006, 66, 5159–5164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skancke, M.; Arnott, S.M.; Amdur, R.L.; Siegel, R.S.; Obias, V.J.; Umapathi, B.A. Lymphovascular invasion and perineural invasion negatively impact overall survival for stage II adenocarcinoma of the colon. Dis. Colon Rectum 2019, 62, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Han, Y.S.; Yoon, Y.M.; Yun, C.W.; Yun, S.P.; Kim, S.M.; Kwon, H.Y.; Jeong, D.; Baek, M.J.; Lee, H.J.; et al. Role of HSPA1L as a cellular prion protein stabilizer in tumor progression via HIF-1α/GP78 axis. Oncogene 2017, 36, 6555–6567. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.N.; Olcott, E.; Jeffrey, R.B. Extrapancreatic perineural invasion in pancreatic adenocarcinoma. Abdom. Radiol. 2018, 43, 323–331. [Google Scholar] [CrossRef]
- Piro, J.R.; Harris, B.T.; Nishina, K.; Soto, C.; Morales, R.; Rees, J.R.; Supattapone, S. Prion Protein Glycosylation Is Not Required for Strain-Specific Neurotropism. J. Virol. 2009, 83, 5321–5328. [Google Scholar] [CrossRef] [Green Version]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted cellular prion protein binds doxorubicin and correlates with anthracycline resistance in breast cancer. JCI Insight 2019, 4, e124092. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Tsai, C.N.; Lin, C.Y.; Chen, H.W.; Lee, Y.S.; Chao, A.; Wang, T.H.; Wang, H.S.; Lai, C.H. Secreted Stress-Induced Phosphoprotein 1 Activates the ALK2-SMAD Signaling Pathways and Promotes Cell Proliferation of Ovarian Cancer Cells. Cell Rep. 2012, 2, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.H.; Chao, A.; Tsai, C.L.; Chang, C.L.; Chen, S.H.; Lee, Y.S.; Chen, J.K.; Lin, Y.J.; Chang, P.Y.; Wang, C.J.; et al. Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol. Cell. Proteomics 2010, 9, 1873–1884. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; You, H.; Qi, J.; Yang, C.; Ren, Y.; Cheng, H. Autocrine and paracrine STIP1 signaling promote osteolytic bone metastasis in renal cell carcinoma. Oncotarget 2017, 8, 17012–17026. [Google Scholar] [CrossRef] [Green Version]
- Sahi, C.; Knox, J.J.; Clemons, M.; Joshua, A.M.; Broom, R. Renal cell carcinoma bone metastases: Clinical advances. Ther. Adv. Med. Oncol. 2010, 2, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; You, H.; Liu, F.; An, H.; Shi, Y.; Yu, Q.; Fan, D. Differentially expressed gene profiles between multidrug resistant gastric adenocarcinoma cells and their parental cells. Cancer Lett. 2002, 185, 211–218. [Google Scholar] [CrossRef]
- Barbieri, G.; Palumbo, S.; Gabrusiewicz, K.; Azzalin, A.; Marchesi, N.; Spedito, A.; Biggiogera, M.; Sbalchiero, E.; Mazzini, G.; Miracco, C.; et al. Silencing of cellular prion protein (PrPc) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy 2011, 7, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, C.J.; Kawamata, F.; Liu, C.; Ham, S.; Győrffy, B.; Munn, A.L.; Wei, M.Q.; Möller, A.; Whitehall, V.; Wiegmans, A.P. EGFR and Prion protein promote signaling via FOXO3a-KLF5 resulting in clinical resistance to platinum agents in colorectal cancer. Mol. Oncol. 2019, 13, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, Y.; Kakeya, T.; Sakai, A.; Takatori, K.; Nakamura, N.; Matsuda, H.; Yamazaki, T.; Tanamoto, K.I.; Sawada, J.I. Propagation of a protease-resistant form of prion protein in long-term cultured human glioblastoma cell line T98G. J. Gen. Virol. 2004, 85, 3449–3457. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
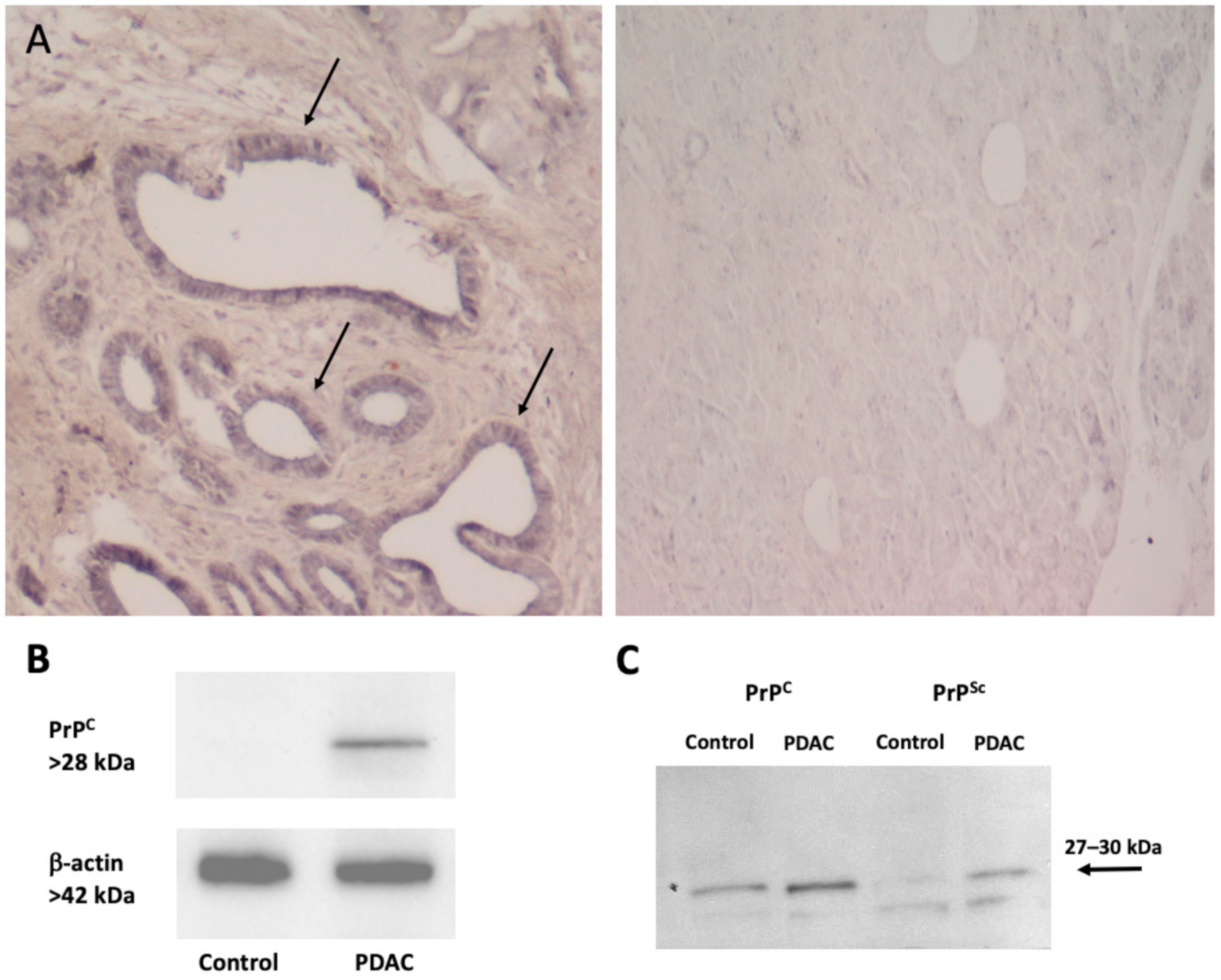
| Protein | Control | Tumor | Ratio T/C |
|---|---|---|---|
| PrPC | 32.80 ± 3.5 | 44.34 ± 5.4 | 1.35 ± 0.1 |
| PrPSc | 22.57 ± 4.1 | 50.36 ± 6.8 * | 2.26 ± 0.1 |
| Ratio PrPSc/PrPC | 0.68 ± 0.1 | 1.13 ± 0.1 * | 1.70 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Giambelluca, M.A.; Morelli, L.; Frati, A.; Fornai, F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers 2021, 13, 170. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020170
Ryskalin L, Biagioni F, Busceti CL, Giambelluca MA, Morelli L, Frati A, Fornai F. The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer. Cancers. 2021; 13(2):170. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020170
Chicago/Turabian StyleRyskalin, Larisa, Francesca Biagioni, Carla L. Busceti, Maria A. Giambelluca, Luca Morelli, Alessandro Frati, and Francesco Fornai. 2021. "The Role of Cellular Prion Protein in Promoting Stemness and Differentiation in Cancer" Cancers 13, no. 2: 170. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020170