Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer



Dipartimento di Scienze per la Qualità della Vita, Alma Mater Studiorum—Università di Bologna, Corso d’Augusto 237, 47921 Rimini, Italy
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(2), 304; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020304
Submission received: 16 December 2020
/
Revised: 9 January 2021
/
Accepted: 13 January 2021
/
Published: 15 January 2021
(This article belongs to the Special Issue Deregulation of Cell Death in Cancer)
Abstract
:Simple Summary
Anticancer therapeutic approaches based solely on apoptosis induction are often unsuccessful due to the activation of resistance mechanisms. The identification and characterization of compounds capable of triggering non-apoptotic, also called non-canonical cell death pathways, could represent an important strategy that may integrate or offer alternative approaches to the current anticancer therapies. In this review, we critically discuss the promotion of ferroptosis, necroptosis, and pyroptosis by natural compounds as a new anticancer strategy.
Abstract
Apoptosis has been considered the main mechanism induced by cancer chemotherapeutic drugs for a long time. This paradigm is currently evolving and changing, as increasing evidence pointed out that antitumor agents could trigger various non-canonical or non-apoptotic cell death types. A considerable number of antitumor drugs derive from natural sources, both in their naturally occurring form or as synthetic derivatives. Therefore, it is not surprising that several natural compounds have been explored for their ability to induce non-canonical cell death. The aim of this review is to highlight the potential antitumor effects of natural products as ferroptosis, necroptosis, or pyroptosis inducers. Natural products have proven to be promising non-canonical cell death inducers, capable of overcoming cancer cells resistance to apoptosis. However, as discussed in this review, they often lack a full characterization of their antitumor activity together with an in-depth investigation of their toxicological profile.
1. Introduction
Historically, cell death has been classified into two main categories: accidental [i.e., non-programmed cell death (PCD)] and PCD. Apoptosis and autophagy are both forms of PCD, while necrosis, instead, has been for a long time considered as a non-physiological process that occurs as a result of infection or injury [1]. However, in recent years accumulating evidence increasingly pointed out that various non-apoptotic forms of PCD, also called non-canonical, can be triggered independently of apoptosis or when the apoptotic process appears to be altered or inhibited [1,2,3]. Non-canonical cell deaths differ from the apoptotic process not only in morphological, but also in biochemical terms, and include various PCD pathways such as ferroptosis, necroptosis, and pyroptosis, which, on the contrary, can share the lytic nature with necrosis [1,4,5].
Nature is a never-ending source of preventive and curative agents, used since ancient times in traditional medicines to prevent and cure many human diseases [6]. Nature still continues to represent an inexhaustible source of pharmacologically active compounds, especially in the anticancer therapy field. Indeed, of the 185 new anticancer drugs discovered between 1981 and 2019, about 65% are natural or natural-based compounds [7]. Most of the discovered natural anticancer drugs originate from plants. There are about 250,000 plant species used for medicinal purposes, which played a crucial role for the treatment of different human diseases, according to the World Health Organization (WHO) [8]. Medicinal plants contain numerous compounds, known as primary and secondary metabolites [8]. By isolating bioactive compounds as drugs, developing bioactive compounds as semi-synthetic lead compounds, or using the whole or part of the plant, medicinal plants have been, and are still being used, as therapeutic antitumor agents [8]. The most effective drugs currently used in the oncological field are, among others, the vinca alkaloids vincristine and vinblastine, etoposide, paclitaxel, topotecan, and irinotecan, which all originate from terrestrial plants [9]. Interestingly, although until a few years ago apoptosis was the anticancer mechanism of action described for these compounds, it has been shown that some of them also induce non-canonical cell deaths [10,11]. Many other different natural compounds were thus explored and identified as promoters of non-canonical cell death.
The aim of this review is to highlight the antitumor effects of natural products as ferroptosis, necroptosis and pyroptosis inducers, and to critically analyze the limitations and challenges associated with the development of non-canonical cell death-based anticancer strategy. Although the activation of other kinds of non-apoptotic PCD, such as autophagy, anoikis, paraptosis, partanathos, netosis, or entosis could represent new promising mechanisms for the prevention or treatment of cancer, those pathways are not characterized yet. Moreover, the ability of natural compounds to trigger them is not substantial. For this reason, we focused our attention only on necroptosis, ferroptosis, and pyroptosis, for which an extensive set of information allows a comprehensive analysis. In particular, the most characterized compounds will be analyzed in detail, while the others will be included in the tables. Most of the natural products inducing non-canonical cell death have been studied in vitro. Only for some of them there are in vivo studies. Tables reporting in vitro studies are in the main text, while tables reporting in vivo studies as well as the effects of natural inducers of non-canonical cell death used in association are included in the Supplementary Materials.
2. Ferroptosis
Ferroptosis, firstly discovered by Dixon et al. in 2012 [12], is a non-canonical cell death characterized by an iron-dependent accumulation of lipid reactive oxygen species (ROS), which leads to cell demise [13]. Ferroptosis differs from any other form of regulated cell death. Morphologically, it does not involve any typical apoptotic feature; it is not characterized by cytoplasmatic swelling or disruption of cell membrane, as in necrotic cell death; the formation of typical autophagic vacuoles is not observed [12]. Ferroptotic cells, instead, are morphologically characterized by a distinct shrinkage of mitochondria with enhanced membrane density and decrease/depletion of mitochondrial cristae [12].
Ferroptosis is caused by compounds able to antagonize glutathione peroxidase 4 (GPX4) in a direct way or through the inhibition of Xc− system. Xc− system is an amino acid antiporter responsible for intracellular transport of extracellular cystine by exchanging intracellular glutamate [14] (Figure 1). Once inside the cells, cystine is reduced to cysteine, an essential substrate for glutathione (GSH) synthesis [15]. Hence, the inhibition of Xc− system alters GSH biosynthesis, reducing the antioxidant activity of glutathione and selenium-dependent GPXs [16,17,18]. Among GPXs, GPX4 is the only one able to reduce hydrogen peroxides or organic hydroperoxides into water or corresponding alcohols by converting GSH into oxidized glutathione (GSSG) [19,20] (Figure 1). Then, the inhibition of GPX4, through direct or indirect mechanisms, leads to lipid ROS accumulation and activates the ferroptotic cell death cascade [12,21,22] (Figure 1).
Iron-dependent accumulation of lipid ROS can occur through non-enzymatic and/or enzymatic lipid peroxidation. Non-enzymatic lipid peroxidation, also called lipid autoxidation, consists in a free radical-driven chain reaction where ROS initiate the oxidation of polyunsaturated fatty acids (PUFAs). Within an autocatalytic process, autoxidation can be propagated leading to membrane destruction, and subsequent ferroptotic cell death [23]. Enzymatic lipid peroxidation is mostly driven by lipoxygenases (LOXs). LOXs, through their dioxygenase activity, catalyze oxygen insertion into PUFAs membrane, generating different lipid hydroperoxides (LOOH), which can start the autocatalytic process of lipid autoxidation mentioned above [22].
If the link between lipid metabolism and ferroptosis induction is well known, how lipid peroxidation leads to ferroptotic cell death is not clear yet. Two mechanisms have been hypothesized. The first hypothesis is that lipid hydroperoxides, produced by PUFAs peroxidation, generate reactive toxic products, i.e., 4-hydroxy-2-nonenal (4-HNE) or malondialdehyde (MDA), which consequently inactivate different survival proteins, leading to ferroptosis [24]. The second hypothesis is that extensive phospholipids peroxidation leads to structural and functional modifications of cellular membrane [23].
Natural Compounds as Ferroptosis Inducers
Several natural compounds, alone or in combination, have been found to induce ferroptosis in different in vitro (Table 1 and Table S1) and in vivo (Table S2) cancer models.
Amentoflavone is a flavonoid mainly found in Selaginella tamariscina (P. Beauv.) Spring and in other species of Selaginella, as well as in many other plant species [55]. Amentoflavone exhibits anticancer effects in several tumor cells by inducing apoptosis, autophagy and ferroptosis, and by inhibiting cell-cycle progression [27,56,57,58,59,60,61]. In U251 and U373 glioma cell lines and in a glioma xenograft model, but not in normal human astrocytes, it triggered ferroptotic cell death by reducing GSH and ferritin heavy chain (FTH) intracellular levels, thus leading to the accumulation of lipid ROS and malondialdehyde (MDA), a PUFAs oxidation product, and subsequent cell death [27] (Table 1 and Table S2). Hence, amentoflavone induces ferroptosis through the rupture of iron homeostasis by reducing the intracellular levels of FTH, which is involved in the intracellular iron storage [27]. Interestingly, both in vitro and in vivo, amentoflavone induced the degradation of FTH by activating autophagy via AMPK (AMP-activated protein kinase)/mTOR (mammalian target of rapamycin)/P70S6K (phosphoprotein 70 ribosomial protein S6 kinase) signaling pathway, suggesting the induction of autophagy-dependent ferroptosis [27]. Autophagy is known as a potent ferroptosis enhancer. Ferritinophagy, in particular, degrades the iron storage protein ferritin and increases the release of free iron, leading to ferroptosis induction [62,63,64].
Two other natural compounds that trigger autophagy-dependent ferroptosis are dihydroartemisinin (DHA) and typhaneoside. DHA is a semi-synthetic derivative of artemisinin, a sesquiterpene lactone derived from Artemisia annua L. currently used as antimalarial agent, which promotes ferroptosis in glioma cells [40] and ferroptosis together with apoptosis in acute myeloid leukemia (AML) cancer cells [39] (Table 1). Typhaneoside, a flavonoid found in the extract of Pollen Typhae, triggered apoptotic and ferroptotic cell death in AML cancer cells [52] (Table 1). In particular, in AML cancer cells, as for amentoflavone, both DHA and typhaneoside induced autophagy-dependent ferroptosis [39,52] by raising the degradation of ferritin through ferritinophagy; moreover, autophagy inhibition mitigated ferroptosis induction by the two natural compounds [39,52] (Table 1). In another experimental setting, DHA did not trigger ferroptosis itself, but it sensitized resistant cancer cells to ferroptosis. In particular, in vitro [mouse embryonic fibroblasts (MEFs) and human osteosarcoma HT1080 cells] and in vivo (GPX4 iKO H292-xenografted female athymic nude-Foxn 1nu/Foxn1+ mice), DHA perturbed iron homeostasis leading to an increase in intracellular iron levels, which concurred to the restoration of RSL3′s and erastin’s ability to induce ferroptosis (Table 1 and Table S2) [65].
Artesunate is another semi-synthetic derivative of artemisinin. It induces ferroptosis in pancreatic [34,36], ovarian [35], head and neck cancer (HNC) [33], T-cell leukemia/lymphoma (ATLL) [32], and in Burkitt’s lymphoma [31] through the modulation of different molecular targets (Table 1). One of these targets is the endoplasmic reticulum (ER). ER stress is a condition of oxidative stress and perturbations in the ER folding machinery provoked by the accumulation of unfolded/misfolded proteins. ER stress activates a signaling process, called unfolded protein response (UPR), in order to lessen ER stress and to restore ER homeostasis [66,67]. In DAUDI and CA-46 lymphoma cells, artesunate triggered ferroptosis and ER stress through the activation of ATF4 (activating transcription factor-4)-CHOP (C/EBP [CCAAT-enhancer-binding protein] homologous protein)-CHAC1 (glutathione-specific γ-glutamylcyclotransferase 1) pathway and PERK [protein kinase RNA (PKR)-like ER kinase] branch of UPR [31] (Table 1). As proposed by the authors [31], the upregulation of CHAC1 possessing a GSH degradation activity [68,69] probably contributes to artesunate-induced ferroptosis [31]. Besides, it is well known that ATF4 could be upregulated by the depletion of amino acids [70], such as that of intracellular cysteine caused by ferroptosis inducers through the system Xc− inhibition. Hence, artesunate might induce ER stress in Burkitt’s lymphoma cells by altering the system Xc−, even if it has to be confirmed. ER stress is also involved in artesunate-induced ferroptosis in KRas mutant pancreatic cancer cells (PaTU8988 and AsPC-1) and in AsPC-1 xenografted BALB/c nude mice [34] (Table 1 and Table S2). Indeed, knockdown of glucose-regulated protein 78 (GRP78), which is considered the master regulator of the UPR signaling process [71], and the inhibition of the three UPR transducers [PERK, IRE1 (inositol requiring protein-1) and ATF6 (activating transcription factor-6] [72], enhanced artesunate-induced ferroptosis in vitro and in vivo [34] (Table 1 and Table S2). Of note, artesunate triggered ferroptosis in a most efficient way in pancreatic cancer cells carrying mutationally-active KRas mutations (i.e., AsPC-1) rather than in pancreatic cancer cells expressing wild type KRas (i.e., COLO-357 and BxPC-3) [36]. This outcome is not odd since KRas mutation often leads to low antioxidant ferritin and transferrin levels and increased number of transferrin receptors and may sensitize pancreatic adenocarcinomas to ferroptosis [33,73]. Still, given that KRas mutant tumors are hardly druggable, these results are quite auspicious [33,74,75].
The role of nuclear factor (erythroid-derived 2)-like 2 (Nrf-2) in ferroptosis is still a matter of debate. Normally, Nrf2 is kept inactivated by Kelch-like ECH-associated protein 1 (Keap-1). Under increased oxidative stress conditions, Nrf2 dissociates from Keap1, translocates into the nucleus, and starts the transcription of the so-called antioxidant responsive element (ARE)-dependent genes [76,77,78]. Most of the Nrf2 target genes are involved in the maintenance of redox homeostasis [79,80], including the regulation of system Xc− [81,82,83], and also in iron and heme homeostasis. They regulate heme-oxygenase 1 (HO-1), ferroportin, and light chain and heavy chain of ferritin (FTL/FTH1) [76,84,85]. In other words, Nrf2 activation could be considered a negative ferroptosis regulator since it endorses antioxidant elements and iron storage, and limits cellular ROS production [86]. Furthermore, since it has been shown that ferroptosis inducers capable of activating Nrf2 pathway promote cellular adaptation and survival and render cancer cells less sensitive to ferroptosis induction themselves [87,88,89], it could be thought that they cannot be considered good candidates for anticancer therapy. However, the activation of Nrf2 pathway could promote ferroptotic cell death. Shifting the focus from the antioxidant properties of Nrf2 effectors to their ability in increasing intracellular iron content, that evidence is not surprising. For instance, HO-1 is responsible for heme catabolism, which produces iron, monoxide, and biliverdin. Thus, it is plausible assuming that the Nrf2 antioxidant response cannot balance the strong iron production, which leads cells to ferroptosis [90]. Accordingly, Kwon et al. [90] demonstrated that hemin, the most prevalent heme metabolite originated by HO-1 catabolism, induced lipid peroxidation as a consequence of iron increase [90]. The opposite role of Nrf2 in ferroptosis seems to be cell-type specific [91], since the activation of Nrf2 pathway protected hepatocellular carcinoma cells against ferroptosis [87], while it promoted ferroptosis in neuroblastoma [54]. Taken together, those results support the hypothesis that Nrf2 could act as a double-edge sword. Even if further studies are needed to disentangle this knot, artesunate supports this hypothesis inducing different effects in different cell lines.
In HNC cells, but not in human oral keratinocytes and fibroblasts, artesunate decreased GSH intracellular levels and increased lipid ROS production and led to ferroptosis [33] (Table 1). However, in HNC cells and cisplatin-resistant HNC cells, artesunate activated the Nrf2 pathway [33] (Table 1), favoring the onset of ferroptosis resistance. As a matter of fact, Keap1 silencing decreased cancer cells’ sensitivity towards artesunate-mediated ferroptosis in both resistant and non-resistant cells, while Nrf2 silencing restored the ability of inducing ferroptosis [33]. In Panc-1 pancreatic cancer cells, induction of ferroptosis by artesunate was accompanied by an increase of HO-1 protein expression [36] (Table 1), which authors associated with the ability of artesunate to increase ROS levels, that in turn activates Nrf2-mediated antioxidant response. Hence, it could be postulated that artesunate induces ferroptosis in pancreatic cancer cells through the HO-1-mediated enhancement of intracellular labile iron (LIP) (i.e., ionic Fe complexes that are redox active). Promotion of ferroptosis by artesunate has been reported also in vivo (Table S2). In Burkitt’s lymphoma xenograft model, it suppressed tumor growth by inducing lipid peroxidation [31] (Table S2).
Withaferin A (WA) is a naturally occurring steroidal lactone derived from Withania somnifera, a medicinal plant used in Ayurvedic medicine [92]. In a variety of cancer cells, WA showed to exhibit anticancer activity through a plethora of mechanisms, including proteasome and cell-cycle inhibition, modulation of oxidative stress, and induction of apoptosis [92]. In neuroblastoma cells, WA promoted ferroptosis through a dual mechanism: at high dose (10 μM), WA directly binds and inactivates GPX4, thus inducing canonical ferroptosis; at lower dose (1 μM), WA targets Keap1 and activates the Nrf2 pathway, leading to an excessive upregulation of HO-1 and a subsequent LIP increase [54] (Table 1). Through these two mechanisms, WA also promoted ferroptosis and eradicated neuroblastoma xenografts in BALB/c mice [54] (Table S2). Of note, WA outperformed the full-blown chemotherapeutic agent etoposide both in vitro and in vivo. In vitro, WA efficiently killed a panel of high-risk and etoposide-resistant neuroblastoma cells by inducing ferroptosis [54] (Table 1). In vivo, WA intratumoral administration showed the same efficacy of etoposide in suppressing tumor growth [54] (Table S2). Most importantly, in contrast to etoposide, WA treatment also repressed neuroblastoma relapse rates in four out of five mice [54] (Table S2). Hence, taking into account that WA-induced cell death was associated with CD45-positive immune cells infiltration in tumor tissue [54], we could speculate that WA could have activated the immune system, thus inducing an anticancer vaccine effect (Table S2). This result may itself be a further step in demonstrating that ferroptosis possesses an immunogenic nature, especially in light of the recent confirmation that ferroptosis could promote antitumor immunity [93]. The only sore point of the study, together with the small number of animals used in the experimentation, was the toxic effect of WA observed in vivo. Since upon WA-systemic injection severe weight loss–related adverse effects were detected and given the scarce water solubility of WA, authors formulated WA-encapsulated nanoparticles (WA-NPs) [54]. WA-NPs showed the same efficacy of non-encapsulated WA in vitro (Table 1) and in vivo (Table S2), constraining systemic side-effects induced by WA, thus allowing systemic application and an effective tumor targeting of WA [54].
3. Necroptosis
The term necroptosis was coined in 2005 when Degterev et al. discovered that necrostatin-1 was able to inhibit tumor necrosis factor (TNF)-induced necrosis by blocking receptor-interacting serine/threonine-protein kinase 1 (RIP1) activity [94]. Even if necroptosis is a finely cellular death mechanism, it shares the morphological features of necrosis, such as cellular rounding and swelling, cytoplasmic granulation, and plasma membrane rupture [95]. Moreover, although necroptosis is a caspase-independent cell death mechanism, it shares some initiating factors with the extrinsic apoptotic pathway [96].
The best characterized type of necroptosis is the TNFα/TNF receptor (TNFR) signaling pathway, considered as a prototype mechanism of necroptosis induction [97]. TNFα binds and activates TNFR1, which recruits TNF receptor-associated death domain (TRADD), cellular inhibitor of apoptosis 1 and 2 (cIAP1 and cIAP2), TNFR-associated factor 1 and 2 (TRAF1 and TRAF2) and RIP1 to create a membrane-signaling complex, called complex I [98,99] (Figure 2). In this complex, cIAP1/2 induces Lys63-linked polyubiquitination of RIP1, which consequently leads to the activation of canonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway and, eventually, cell survival [100]. Conversely, inhibition of cIAPs activity by the second mitochondrial activator of caspase (Smac)/Diablo proteins or the Smac mimetic compounds promotes the deubiquitination of RIP1 by the deubiquitinating enzymes cylindromatosis (CYLD) and A20, which both hydrolyze Lys63-linked ubiquitin chains [100]. Subsequently, RIP1 dissociates from complex I to form either cytosolic complex IIa or complex IIb, depending on the proteins content: complex IIa is formed by TRADD, RIP1, Fas-associated death domain (FADD), and caspase-8; complex IIb is formed by RIP1, FADD, and caspase-8, but it does not contain TRADD. While in the complex IIa activation of caspase-8 is independent from RIP1 kinase activity, in complex IIb, where TRADD is not present, RIP1 kinase activity is required for caspase-8 activation and induction of RIP1-dependent apoptosis [100]. However, complex IIa and IIb are both capable of inducing apoptosis or necrosis depending on cell status. Indeed, when caspase-8 is inhibited, RIP1 interacts with and activates by autophosphorylation [101] RIP3, leading to the formation of a protein complex called necrosome [102] (Figure 2). RIP3, beside its activation through RIP1, could be directly activated also by other stimuli, as lipopolysaccharides (LPS), double-stranded (dsRNA), and DNA-dependent activator of interferon-regulatory factor [100]. The formation of necrosome induces the activation and phosphorylation of both RIP1 and RIP3, which subsequently phosphorylate mixed lineage kinase domain-like (MLKL) [103,104,105] (Figure 2). Then, phosphorylation of MLKL induces its oligomerization and translocation to plasma membrane, which is crucial for necroptosis execution [105,106,107] (Figure 2). To date, the effector mechanism by which necroptosis is executed is still controversial. Some studies report that oligomerized MLKL could interact with negatively charged phospholipids and create pore structures into the plasma membrane [108,109]. In contrast, others report that MLKL could induce a dysregulation of ionic fluxes in the plasma membrane [104,107].
Natural Compounds as Necroptosis Inducers
Several natural compounds promote necroptosis in cancer cells both in vitro (Table 2 and Table S3) and in vivo (Table S4).
Among all, shikonin is definitely the most characterized necroptosis inducer of natural origin. Shikonin is a naphtoquinone isolated from the root of Lithospermum erythrorhizon Sieb. et Zucc, Arnebia euchroma (Royle) Johnst, or Arnebia guttata Bunge [152]. It promotes necroptosis in a wide range of cancer cells, including pancreatic [134], nasopharyngeal [135,144], gastric [145], lung [136], breast [133,147,148], osteosarcoma [138], lymphoma [139], multiple myeloma [137], and glioma [140,141,142,143] (Table 2). In AGS gastric cancer cells, shikonin induced necroptosis or apoptosis in a time-dependent manner: with equal concentrations (1, 2, and 4 μM), the short-time treatment (6 h) led to necroptosis induction, while longer time treatment (24 h) led to apoptotic cell death [145]. In MCF-7 breast cancer cells, shikonin promoted necroptosis when the apoptotic machinery was inhibited [147] (Table 2). Interestingly, most of natural compounds illustrated in Table 2 induce both necroptosis and apoptosis, confirming the existing interrelation between the two cell death mechanisms. Indeed, cell fate (apoptosis versus necroptosis) is primarily affected by available caspase-8 and cIAP1, cIAP2, XIAP (X-linked inhibitor of apoptosis protein). Their deficiency favors necroptosis induction by suppressing RIP/RIP3 proteolytic cleavage or ubiquitination of RIP1 [153,154,155].
Besides the activation of RIP1 and RIP3 and the promotion of necrosome complex formation, the crucial event involved in shikonin-induced necroptosis is the production of ROS. Shikonin induced oxidative stress in nasopharyngeal [135], glioma [141,142,143], gastric [145], and breast cancer cells [147] (Table 2) and the increase in ROS levels was linked to necroptosis induction in some of those models. In glioma cancer cells, shikonin boosted ROS and mitochondrial superoxide generation [141,142,143] (Table 2). Inhibition of RIP1 and RIP3 reduced ROS, mitochondrial superoxide production, and cell death. Alongside, ROS increased RIP1 and RIP3 levels, showing that oxidative stress is a regulative factor in shikonin-mediated necroptosis [141] (Table 2). Moreover, in glioma cancer cells, oxidative stress triggered by shikonin led to the collapse of mitochondrial membrane potential, promoting the cytoplasmic release and the nuclear translocation of AIF (apoptosis-inducing factor) [142] (Table 2). Activated MLKL seems to be responsible for shikonin-induced mitochondria collapse, since its inhibition reduced ROS and superoxide production and AIF mitochondrial release [143] (Table 2). This hypothesis is further supported by the observed accumulation of MLKL in mitochondria and the enhanced expression of both mitochondrial and activated MLKL [143] (Table 2). Indeed, MLKL could boost the catalytic activity of PGAM5 (mitochondrial serine/threonine protein phosphatase family member 5) and bind mitochondrial-specific lipid cardiolipin [109], leading to mitochondrial fragmentation [105,156]. However, whether mitochondria are essential in necroptotic cell death is not clear yet. In mitochondria-deficient cells, as well as in cells from PGAM5−/− mice, necroptosis still occurred [157,158]. Interestingly, the overproduction of ROS and/or the loss of mitochondrial potential are strictly involved not only in shikonin-induced necroptosis but also in many other natural-derived necroptosis inducers, including 2-methoxy-6-acetyl-7-methyljuglone [110,111], arctigenin [116], columbianadin [121], deoxypodophyllotoxin [122], matrine [126], pristimerin [129], resibufogenin [131], and tanshinol A [149] (Table 2), thus further confirming their pivotal role in necroptosis induction.
Shikonin confirmed its ability to promote necroptosis also in many different in vivo experimental models [135,136,138,142,143,144]. In female nude mice (authors did not state the species) [135], and BALB/c nude mouse xenograft models of human nasopharyngeal [135,144], or lung cancer [136], shikonin reduced tumor growth and increased tumor cell necrosis [135,136,144], which, in the latter model, have been associated with an increase in RIP1 expression in the tumor tissue [136] (Table S4). In BALB/c nude mouse xenograft model of human glioma, shikonin induced the binding of MLKL with mitochondria and the subsequent release of AIF and promoted necroptosis [143] (Table S4). In the same model, shikonin caused DNA damage [142] (Table S4), as observed in several cancer cells in vitro [142,159,160] (Table 2), thus configuring itself as a possible mutagen compound. This aspect is certainly to be taken into account in the evaluation of the toxicological profile of shikonin, even if it is worth noting that the antitumor activity of several anticancer drugs is based on DNA-damage induction [161]. In BALB/c nude mouse xenograft model of human osteosarcoma, shikonin reduced tumor growth, increased RIP1/3 expression, and reduced lung metastasis thus suggesting an antimetastatic activity for shikonin [138] (Table S4). However, attention must be paid since the role of necroptosis in cancer metastatization is controversial. Strilic et al. reported that tumor cells-induced necroptosis of endothelial cells promotes cancer cells extravasation and metastatization through interaction with DR6 (death receptor 6) [162], hence showing that necroptosis could promote cancer cell metastatization [161]
Berberine is the major component of different plants belonging to Berberis species, and many other plants including, among all, Coptis chinensis Franch., and Hydrastis canadensis L. [163]. Besides its widely documented apoptotic anticancer activity [163], berberine promoted necroptosis in ovarian cancer cells and in three patient-derived primary ovarian cancer cell lines (POCCLs) by activating RIP3 and MLKL [118] (Table 2). Berberine triggered necroptosis also in diffuse large B-cell lymphoma (DLBCL) cancer cells, where the necroptotic mechanism has been deeply investigated [119] (Table 2). In DLBCL cells, berberine promoted mitophagy-dependent necroptosis by inducing the formation of the RIP1/RIP3/MLKL necrosome complex and mRNA degradation of PCYT1A (phosphate cytidylyltransferase 1 alpha), thus reducing its expression in cancer cells [119] (Table 2). PCYT1A is an isoform of the CTP (choline phosphate cytidylyltransferase) enzyme, which is crucial for PC (phosphatidylcoline) synthesis [164]. The authors of the study showed that PCYT1A was overexpressed in 44% of the analyzed DLBCL patients and that PCYT1A overexpression occurred in parallel with the enhanced gene and protein expression of MYC [119], an oncogene mostly involved in lymphoma cell chemoresistance [165]. Moreover, MYC-induced overexpression of PCYT1A led to inhibition of necroptotic cell death in DLBCL cells [119] (Table 2). In this context, berberine effectively suppressed DLBCL cancer cells growth by inhibiting the MYC-driven downstream effector PCYT1A, and inducing mitophagy-dependent necroptosis [119], thus being eventually considered as a promising anticancer agent to treat MYC-overexpressing lymphomas.
4. Pyroptosis
The term pyroptosis was coined by Cookson and Brennan to describe a peculiar caspase-1-dependent, pro-inflammatory regulated form of cell death involved in Salmonella-infected macrophages [166]. The term pyroptosis has been drawn from the two ancient Greek words pyro, and ptosis, which respectively mean fire or fever, and collapse or demise [166]. Pyroptosis is involved in innate immune defense against pathogenic infections or endogenous risk signals through the recruitment of immune cells by pro-inflammatory cytokines [167]. Its overactivation or dysregulation can lead to autoimmune and autoinflammatory diseases [168]. Pyroptosis is closely linked to cancer, where it acts as a double-edged sword. Indeed, as an inflammatory cell death process, pyroptosis could promote tumor cell growth by different pro-tumorigenic mechanisms [168,169]; conversely, it could suppress tumors development [168] also by enhancing anti-tumor immunity [170,171,172].
Pyroptosis shares with apoptosis some morphological and mechanistic features, including DNA damage and caspase activation. For instance, caspase-1/4/5 but also caspase-3 are involved in pyroptotic cell death [173,174,175]. Morphologically, pyroptotic cells display DNA fragmentation and chromatin condensation, but in contrast to apoptosis their nucleus remains intact; moreover, pyroptotic cells are characterized by the formation of large bubbles at the plasma membrane resulting in cell swelling, and consequent plasma membrane permeabilization together with cellular osmotic lysis [176].
Depending on the different stimuli and inflammatory mediators, pyroptosis falls into a canonical or non-canonical cell death mechanism, which converge into the same effector system, i.e., the activation of one member of the gasdermin protein (GSDM) family [177]. In canonical pyroptosis, specifics pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs) or homeostasis-altering molecular processes (HAMPs) are recognized by inflammasome sensors [178] (Figure 3). An inflammasome is a multiprotein complex formed by (1) a sensor named PRR (pattern recognition receptor), (2) an adaptor protein apoptosis-associated speck-like protein (ASC), which contains a caspase-recruitment domain, and (3) caspase-1 [179]. Different types of PRRs are involved in pyroptosis including the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), the absent in melanoma 2 (AIM2)-like receptors (ALRs), and pyrin proteins [180] (Figure 3). The most characterized NLRs in canonical pyroptosis is NLRP3 (NLR family pyrin domain-containing 3). A wide range of stimuli, such as pore-forming toxins, extracellular RNA, ROS, and mitochondrial DAMPs can trigger the NLRP3 cascade [181,182,183,184,185,186,187]. In turn, the activated-inflammasome sensors lead to the recruitment, directly or via ASC, of caspase-1 to form the full-blown inflammasome and drive caspase-1 activation [188,189]. Then, activated caspase-1 fosters the proteolytic maturation of pro-inflammatory precursors like pro-interleukin-1 beta (IL-1β) and pro-interleukin-18 (IL-18) and activation of gasdermin (GSDM) D (GSDMD) [190] (Figure 3). GSDMD could also be activated by bacterial intracellular lypopolisaccaride (LPS), without inflammasome involvement [169,191]. In the latter case, GSDMD is cleaved by caspases 4/5, which are the human caspase-11 murine orthologue. GSDMD cleavage leads to the release of the N-terminal fragment (GSDMD-NT) [186] (Figure 3). After its release, GSDMD-NT oligomerizes to form pores on the inner leaflet of the plasma membrane [192] causing osmotic cell swelling and the rupture of the plasma membrane with the spillage of the cellular content into the extracellular space, including the inflammatory cytokines IL-1β and IL-18 [193] (Figure 3). Besides, some pro-apoptotic chemotherapy drugs and molecular-targeted therapies promote pyroptosis through the caspase-3-dependent cleavage of GSDM E (GSDME) [175,194,195] (Figure 3). Cleavage of GSDME leads to the release of GSDME-NT (Figure 3), which possesses pore-forming activity as GSDMD-NT [175,194,195].
As mentioned above, pyroptosis and apoptosis are closely intertwined. For instance, NF-kB pathway is commonly referred to as apoptosis regulator [196,197], and has been found to trigger pyroptosis as well [198]. Indeed, as in the case of NF-kB, the same pro-apoptotic stimulus could, in some circumstances, provoke different cell death pathways [199]. The discriminating factor in triggering apoptosis, pyroptosis or both PCDs is the expression of GSDM in tumor cells. In tumors with low levels of GSDME, activated caspase-3 elicits apoptosis, while if the tumor expresses high levels of GSDME, caspase-3 switches its downstream pathway from apoptosis to pyroptosis or apoptosis and pyroptosis [175,200]. Of note, GSDME levels differ depending on the tumor type: low levels are detected in gastric and skin cancer, high levels in lung cancer, colorectal cancer, neuroblastoma, and melanoma. Thus, pyroptosis could be considered a tumor-type specific cell death [168,175].
Natural Compounds as Inducers of Pyroptosis
Several natural compounds and their derivatives or analogues were found to induce pyroptosis in different cancer models, both in vitro (Table 3) and in vivo (Table S5).
Galangin is a natural flavonoid found in different plants including Alpinia officinarum Hance [214]. In glioblastoma multiforme cell lines (U251 and U87MG), galangin induced apoptosis, autophagy, and GSDME-mediated pyroptosis [207] (Table 3). Interestingly, it has been found that inhibition of autophagy enhances pyroptosis and apoptosis induction. Autophagy, promoting cell survival and blocking inflammation, could suppress inflammasome activation [215], thus limiting pyroptotic cell death. For this reason, the inhibition of autophagy can represent a strategy to favor pyroptosis, as observed in cells treated with galangin, the Citrus flavanoid nobiletin or alpinumisoflavone [202,210] (Table 3).
Polyphyllin VI (PPVI) is a steroidal saponin isolated from the ethyl acetate fraction of Trillium tschonoskii Maxim [199]. PPVI displayed anticancer effects against lung cancer cells and in an athymic nude mouse xenograft model of lung cancer through the induction of apoptosis, autophagy [198,199], and pyroptosis [198] (Table 3 and Table S5). PPVI provoked pyroptosis by activating the NLRP3 inflammasome, responsible for GSDMD cleavage and caspase-1-dependent maturation and secretion of IL-1β and IL-18 [198] (Table 3). The PPVI-induced pyroptosis was associated with ROS generation and activation of the NF-kB pathway [198] (Table 3). Indeed, the increased expression of NLRP3 facilitates the NF-kB-mediated effective assembly of the inflammasome [216]. Hence, it could be supposed that the activation of NF-kB pathway by PPVI participates into the assembly of NLRP3 inflammasome and that PPVI-mediated ROS generation in turn activates NLRP3. Interestingly, ROS generation promoted PPVI-induced apoptosis [199], thus suggesting that the same cell death stimulus could activate different PCD pathways.
Notably, all the natural compounds illustrated in the Table 3 induced both pyroptosis and apoptosis, hence endorsing that the crosstalk between these two cell death pathways is really tight, as mentioned above. In PPVI-induced pyroptosis, the relationship between pyroptosis and apoptosis has been found to be the ROS-mediated activation of the NF-kB signaling pathway [198]. Another important observation is that the natural saponin dioscin induced apoptosis by activating the c-Jun N-terminal kinase (JNK)/p38 signaling pathway [206]. This means that dioscin-induced pyroptosis could be activated through the same pro-apoptotic upstream pathway triggering caspase-3 activation Thus, certain compounds’ ability to elicit pyroptosis in addition to apoptosis could be considered a potentially effective strategy to synergize their anticancer efficacy.
Although pyroptosis inducers can have an interesting role in the oncological field, pyroptosis induction should be carefully sought since it could have also a cancer promotion effect. Indeed, to treat certain tumors, such as skin cancer, inhibition of pyroptosis could be pursued. A persistent inflammatory status or alterations in inflammatory activity are implicated in skin tumorigenesis, together with the modulation of cancer progression and invasiveness by cytokines [217]. For instance, two natural compounds such as epigallocatechin-3-gallate and thymoquinone suppressed growth and migration of melanoma cells by inhibiting NLRP1 inflammasome, IL-1β-mediated secretion, and NLRP3 inflammasome, respectively [218,219]. Hence, inhibition of pyroptosis, instead of its induction, could be a potential antitumor strategy in skin cancer treatment, as in other tumor diseases where inflammation plays a key role in tumor progression.
5. Selective Activity of Natural Inducers of Non-Canonical Cell Death towards Cancer Cells
One of the main drawbacks of current anticancer chemotherapy is the non-selective cytotoxicity towards cancer cells, which is associated with the appearance of systemic toxicity and significant side effects [220]. However, only a few studies explored the impact of the previously described natural compounds on non-transformed cells, and often the results obtained in different studies are conflicting.
Regarding all the natural inducers of ferroptosis described in Table 1, controversial data arose about artesunate and WA.
Although several studies indicate that artesunate selectively kills cancer cells, many others showed cytotoxicity versus normal cells. The IC50 on human bronchial epithelial HBE cells after 24 h treatment was 1.38 times higher (212.48 μM) than that observed in A549 lung adenocarcinoma cells (153.54 μM) [221]. The IC50 on human osteosarcoma cells treated with artesunate for 48 h was about four times higher (206.3 μM) than that observed on the non-transformed counterpart hFOB1.19 human osteoblast (52.8 μM) [222]. On normal human urothelial SV-HUC-1 cells, the IC50 after 48 h artesunate treatment (1149.6 μM) was about one order of magnitude higher compared to those obtained in T24 and RT4 bladder cancer cells (129.7 μM and 103.2 μM, respectively), showing a remarkable selectivity of action [223]. Rho et al. compared artesunate cytotoxicity on both HNC cancer cells and normal oral keratinocytes (HOK) and fibroblasts (HOF), founding that all HNC cells succumbed to artesunate 100 μM, while almost all HOK and HOF cells survived to artesunate 50 μM. However, no data is available for 100 µM treatment [33]. After 24 and 48 h, artesunate at 33–521 μM exhibited a slight citoxicity on normal retina hTERT-RPE1 cells compared to retinoblastoma RB-Y79 cells (at least eight or nine times lower in hTERT-RPE1 versus RB-Y79 cells) [224]. Furthermore, artesunate showed cytotoxic effects on normal human and mouse/rat liver cells [225,226]. Indeed, exposure to artesunate 100 μM for 24, 48, and 60 h induced a significant cytotoxic effect on both human hepatocellular carcinoma cells (HepG2, Huh-7, and Hep3B) and normal hepatocytes (L02) [225]. Its cytotoxic effect was kept even at lower concentrations (0.5–10 μM), as shown on normal rat liver BRL-3A cells and mouse liver AML12 cells (24 and 48 h treatments) [226]. However, the authors of both studies [225,226] did not explicitly quantify the entity of these cytotoxic effects. Ishikava and colleagues reported that cell viability of HTLV-1 (human T lymphotropic virus type 1)-infected T-cell lines (MT-2, MT-4, and HUT-102) decreased time- and dose-dependently after artesunate exposure (20–60 μM for 24 and 48 h), but PBMCs (together with Jurkat and CEM leukemia cells) treated for 24 h with artesunate 5 and 10 μM were relatively resistant [32]. Once more, the authors of the study did not quantify this effect. Taken these studies together, we could conclude that artesunate is tumour-selective in an organ or cell-type way. However, the lack of objective and quantitative data of many studies makes it difficult to draw reliable conclusions.
WA revealed a similar tumour-type-dependent selectivity. It was cytotoxic at 2–10 μM on different human colon cancer cell lines: HCT-116 (IC50: 5.33 μM), SW-480 (IC50: 3.56 μM), and SW-620 (IC50: 5.0 μM) at 24 h treatment; however, no significant cytotoxic effect was found on normal colon epithelial FHC cells, even if the highest tested concentration was 6 μM [227]. Cell viability of Ca9–22 and HSC-3 human oral cancer cells treated for 24 h with WA 1 μM was 83.4% and 79.4%, respectively, while no cytotoxicity was recorded in HGF-1 normal oral cells [228]. Moreover, most of human fibroblasts (TIG-1 and KD) treated with WA 2 μM remained viable up to 96 h treatment, while DU-145 and LNCaP prostate cancer cells almost completely succumbed after 24 and 72 h, respectively [229]. Lastly, the IC50 on WI-38 normal lung cells after 24 h WA treatment was > 50 μM, versus ~10 μM recorded for A549 cells [230]. Furthermore, WA showed a considerable safe profile on PBMCs. The IC50 after 24 h was > 50 μM [230], and no major cytotoxic effect was recorded on both PBMCs and hematopoietic progenitor cells up to 48 h exposure at 30 μM [231]. At the same treatment time, instead, the IC50 on MOLT-4, Jurkat, REH, and K-562 leukemia cells was 1.52, 1.62, 3.09, and 0.58 μM, respectively [231]. In contrast, both U2OS osteosarcoma cells and TIG-3 normal fibroblast were killed by WA at doses equal to or less than 1.1 μM (treatment time was not specified) [232].
Dihydroisotanshinone I 10 and 20 μM after 48 h treatment significantly inhibited the proliferation of H460 (IC50: 19.4 μM) and A549 (IC50: 15.5 μM) small lung cancer cells [233]. However, cell proliferation of normal human lung fibroblasts (IMR-90) was only slightly inhibited after 24 h treatment with dihydroisotanshinone I 5 and 10 μM (no indication of IC50) [233].
The natural necroptosis inducer pristimerin showed a dubious selectivity of action. The IC50 on MCF-7 (breast carcinoma), HCT116, HepG2 (human hepatocellular carcinoma), SCC-4 and HSC-3 (human oral squamous cell carcinoma), and B16-F10 (mouse melanoma) cells after 72 h treatment with pristimerin was 7.9 μM, 9.4 μM, 7.8 μM, 12.7 μM, 2.9 μM, and 6.3 μM, respectively [234]. Instead, at the same conditions, the IC50 on normal lung MRC-5 fibroblast was 3.5 μM, showing that normal cells are even more sensitive to pristimerin activity than most tumour types [234]. Very similar results were obtained comparing the cytotoxic effect of pristimerin (72 h) on HL-60 cells (IC50: 1.31 μM) and K-562 leukemic cells (IC50: 3.2 μM) with that on PBMCs (IC50: 0.88 μM) [235]. Rodrigues et al. confirmed this trend showing an even more pronounced sensitivity of normal cells: pristimerin was cytotoxic on both HL-60 and K-562 leukemic cells (IC50 at 72 h: 8.8 μM and 13.6 μM, respectively) and markedly cytotoxic on PBMCs (IC50 at 72 h: 0.6 μM) [234]. In contrast, human breast epithelial MCF-10A cells were 2 to 3 times higher resistant to pristimerin than MDA-MB-231 breast cancer cells, in particular at 24 h [236]. To note, 1.5 to 12 h pristimerin exposure triggered necroptosis in glioma C6 cells at 2.5 μM and in U251 cells at 4.5 μM [129]. Even if Zhao and colleagues [129] did not test pristimerin cytotoxicity on the non-transformed counterpart, the cytotoxic concentrations of pristimerin for glioma C6 cells are higher and the treatment times shorter than those responsible for the cytotoxic effect on PMBCs and MRC-5 normal cells [234].
Among all the natural inducers of pyroptosis described in Table 3, only for some of them their selectivity of action towards cancer cells has been well established. Among them, the selectivity of dioscin towards tumour cells is still controversial. Treatment with dioscin 5.8 μM for 24 h reduced cell viability to about 70% in normal human pancreatic ductal epithelial cells (HPDE6-C7) compared to the 40% observed in ASPC-1 and PANC-1 pancreatic cancer cells [237]. At the same dose and treatment time (5.8 μM for 24 h), dioscin reduced normal nasopharyngeal NP69 cells’ viability to 73% compared to 40% and 33% of Panc-1- and ASPC-1-treated cells [238]. On normal cervical epithelial H8 cells treated with dioscin 5.8 μM for 24 h, the cell viability inhibition rate compared to untreated cells was 25% versus 80% and 54% observed on HeLa and SiHa cervical cancer cells, at the same experimental conditions [239]. Moreover, the IC50 on L02 hepatocytes after 48 h treatment with dioscin (13.23 μM) was more than six times higher than that observed in HepG2 cancer cells (2.38 μM) [240]. Lastly, the IC50 (the authors did not specify the treatment time) on NOZ and SGC996 gallbladder cancer cells was 4.47 μM and 5.05 μM, respectively [241], while on human kidney epithelial cells (293 T), dioscin was not toxic even at the higher tested dose (8 μM) [241]. However, Ma et al. reported that dioscin at doses over 10 μM (48 h treatment) inhibited cell proliferation of both gastric cancer (HGC-27, MGC803, and SGC7901) and normal gastric GES-1 cells [242], even if they did not explicitly quantify the entity of this antiproliferative effect.
Questionable data were also found for galangin. Despite galangin’s ability to induce different types of cell death, its active concentration on all the three glioma cell lines tested is quite high (150 µM) [207], and usually high doses are not selectivite towards tumor cells [243,244]. The analysis of galangin effects on normal human astrocytes (NHA) viability showed cytotoxic effects at double the active dose in tumor cells. Indeed, the IC50 on NHA after 24 h galangin treatment was >450 μM, whereas in U251, U87MG, and A172 glioma cells the IC50 was 221.8, 262.5 and 273.9 μM, respectively [207]. However, 24 h galangin treatment (at concentrations >50 μM) suppressed cell proliferation in NIH3T3 mouse fibroblasts to the same extent as for B16F10 murine melanoma cells. For the latter cell line, the IC50 after 24 h treatment was 145 μM, while no data are available for fibroblasts [245].
The cytotoxicity of the pyroptosis inducer osthole was explored on normal cervical fibroblasts and HeLa cervical cancer cells. On HeLa cells, the IC50 was 64.9 μM compared to 168 μM on normal cervical fibroblasts (24 h treatment) [246]. Moreover, the IC50 on HL-60 after 12 h osthole treatment was 100 μM, compared to 164 μM on PBMCs [247]. Consistently, no significant cytotoxicity was observed in PBMCs up to 72 h osthole treatment at 1.84 μM [248]. Furthermore, osthole treatment for 24 and 48 h at 200 μM did not induce any significant cytotoxic effect on normal ovarian IOSE80 cells [249]. Conversely, almost all A2780 and OV2008 ovarian cancer cells succumbed to osthole treatment at 200 μM for 24 and 48 h [249]. In this regard, the IC50 on A2780 and OVCAR3 ovarian cancer cells, i.e., the in vitro cell models where osthole promoted pyroptosis, was 73.6 μM and 75.2 μM, respectively [211]. This means that the active concentrations of osthole are abundantly lower than those toxic for normal ovarian cells.
On the whole, the majority of the studies listed above are encouraging on the, at least a partial, tumour selectivity of non-canonical cell death inducers, but data are far from be conclusive or substantial. One point to consider is that, as for many natural anticancer agents, both activity and selectivity of non-canonical cell death inducers depend on the cell-type and organ targeted. This, together with the lack of extensive studies, does not allow to draw firm conclusions. Thus, since the selective activity of anticancer agents is considered one of the most critical aspects in defining their pharmaco-toxicological profiles, a case-by-case analysis is recommended.
6. Conclusions
The ability of cancer cells of evading apoptosis is one of the hallmarks of cancers [250]. Given that anticancer activity of most anticancer drugs currently in use is based on their pro-apoptotic activity [251], it becomes clear how the discovery and characterization of non-apoptotic, also called non-canonical, cell death pathways represent a new promising approach to overcome the challenges of current anticancer therapies. As showed in this review, natural products can definitely suit this role, as promising non-canonical cell death inducers.
All PCD modalities—apoptosis, necroptosis, ferroptosis and pyroptosis—are strictly connected in both molecular and functional terms, and, depending on the cell status or eventually mutations carried by cells, the mode of cell death could switch from one to another [5]. For example, necroptosis occurs when the apoptotic cell death is impaired by caspase-8 inhibition [252]; conversely, within massive inflammasome activation, cells lacking caspase-1 or GSDMD could be unable to trigger pyroptosis but still die by apoptosis thanks to the presence of active caspase-8 [253]. Moreover, activation of apoptotic caspase-3 cleaves GSDME and could trigger both pyroptosis and apoptosis [174,199]. Additionally, RIP3 could activate NLRP3 inflammasome in the absence of MLKL [254] together with the RIP3-MLKL-NLPR3-caspase-1 axis, thus resulting in IL-1β maturation, independently of GSDMD cleavage [255,256]. Hence, all these pieces of evidence show that the different PCDs frequently share the same molecular actors, which could activate different cell death modalities depending on the factors described above. Therefore, we could actually consider all these pathways as many musicians who take part of the same orchestra, and the more musicians play, the marrier is the symphony. In other words, triggering more than one type of PCDs clearly enhances the chances of cancer cells eradication.
Another significant outcome deriving from the concomitant activation of apoptosis and the non-canonical cell deaths is the elicitation of the antitumor immune response, which would allow a switch from a mostly immune-silent or tolerogenic cell death (apoptosis) into an immunogenic one [257,258,259]. For instance, pyroptosis induction commuted the immune-silent cisplatin-mediated apoptosis into immunogenic [260]. Indeed, in different models, GSDME activation promotes tumor suppression by increasing the anticancer properties of tumor-infiltrating natural killer (NK) cells and CD4+ and CD8+ T lymphocytes, together with an antitumor vaccination effect, triggering both innate and adaptive antitumor immunity [170,171,259]. Similarly, necroptotic cancer cells are, without any doubt, immunogenic. These dying cells promoted antitumor immunity by inducing DC (dendritic cells) maturation, cross-priming and proliferation of CD8+ T cells and NK cells in vitro and in vivo and successfully created an antitumor vaccine effect in different tumor mice models [261,262,263,264]. Regarding ferroptosis, many hints have been produced about the interaction between ferroptotic cells and the immune system. For instance, ferroptotic cells release HMGB-1 (high mobility group box 1) [265], while the activation of CD8+ T cells synergizes ferroptosis [266]. However, only recently the antitumor immunogenicity of ferroptosis has been validated. Indeed, it has been demonstrated that early, but not late, ferroptotic cells promote the phenotypic maturation of bone-marrow derived dendritic cells (BMDCs) and elicit an antitumor vaccination effect in the well-accepted prophylactic tumor vaccination model of immune competent C57BL/6 J mice [93]. Those results definitely confirm that ferroptosis could promote antitumor immunity. Still, the coexistence of apoptosis and non-canonical cell deaths could be regarded as a new remarkable strategy to neutralize apoptosis resistance and, thanks to the adaptive immune stimulation, lessen the incidence of metastases and relapses.
Nonetheless, this apparently idyllic scenario displays different problems. Induction of necroptosis and pyroptosis is strictly related to the expression of their molecular mediators, which is cancer-type-dependent. For necroptosis, decreased RIP1/RIP3/MLKL expression has been found in AML, melanoma, and breast, colorectal, gastric, ovarian, head and neck squamous cell, and cervical squamous cell carcinomas [96]. Regarding pyroptosis-related mediators, GSDMD expression was found to be decreased in gastric cancer [168], while GSDME expression is low in gastric and skin cancer [168,175]. Thus, the presence or absence of crucial mediators dictates whether cells can go through that specific PCD or not. However, to overcome this limitation and exploit natural compounds’ great potential to induce non-canonical cell death, nanotechnologies can come to the aid. Several nanomaterials demonstrated to counteract the specific pitfalls of every single type of cell death that usually limit their therapeutical use, such as GSDMs silencing for pyroptosis [260] or RIP1/RIP3/MLKL low levels for necroptosis [267], restoring the capability of pursuing that PCD in those resistant models.
Although a huge number of natural compounds has been identified as inducers of non-canonical cell death, only few of them have been deeply characterized for the underpinned molecular networks involved in their antitumor activity. Furthermore, very few studies have investigated the selective activity towards cancer cells together with the drawing of a toxicological profile. This is a critical issue since thee three mentioned non-canonical cell deaths are pro-inflammatory and in some circumstances could promote tumor progression [268,269,270,271,272]. Overall, natural products antitumor potential should be evaluated on a case-by-case basis.
In conclusion, natural products have proven to be interesting and promising non-canonical cell death inducers. However, taking into account all the issues mentioned above, further studies are needed to better characterize their antitumor activity and, especially, to investigate their toxicological profile in order to define their antitumor potential and pave the way for clinical studies.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2072-6694/13/2/304/s1, Table S1: In vitro induction of ferroptosis by natural products used in association; Table S2: Natural products as in vivo inducers of ferroptosis; Table S3: In vitro induction of necroptosis by natural products used in association; Table S4: Natural products as in vivo inducers of necroptosis; Table S5: Natural products as in vivo inducers of pyroptosis.
Author Contributions
Writing–original draft preparation, G.G.; writing–review and editing, E.C., C.F.; supervision, C.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Ichim, G.; Green, D.R. Die Another Way—Non-Apoptotic Mechanisms of Cell Death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, M.; Cerella, C. Non-Canonical Programmed Cell Death Mechanisms Triggered by Natural Compounds. Semin. Cancer Biol. 2016, 40–41, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Guaman-Ortiz, L.; Orellana, M.; Ratovitski, E. Natural Compounds as Modulators of Non-Apoptotic Cell Death in Cancer Cells. Curr. Genom. 2017, 18, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging Connectivity of Programmed Cell Death Pathways and Its Physiological Implications. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Gezici, S.; Şekeroğlu, N. Current Perspectives in the Application of Medicinal Plants against Cancer: Novel Therapeutic Agents. Anticancer. Agents Med. Chem. 2019, 19, 101–111. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pr. 2016, 25, 41–59. [Google Scholar] [CrossRef]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 Control Cell Fate in Response to Extensive DNA Damage via TNF-α Feedforward Signaling. Cell 2011, 145, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, Necroptosis, and Pyroptosis in Anticancer Immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System Xc− Cystine/Glutamate Antiporter: An Update on Molecular Pharmacology and Roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Long, Y.C. Crosstalk between Cystine and Glutathione Is Critical for the Regulation of Amino Acid Signaling Pathways and Ferroptosis. Sci. Rep. 2016, 6, 30033. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological Inhibition of Cystine-Glutamate Exchange Induces Endoplasmic Reticulum Stress and Ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Sato, H. The Oxidative Stress-Inducible Cystine/Glutamate Antiporter, System x (c) (-): Cystine Supplier and Beyond. Amino. Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione Peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved Mysteries: How Does Lipid Peroxidation Cause Ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Gao, Z.; Deng, G.; Li, Y.; Huang, H.; Sun, X.; Shi, H.; Yao, X.; Gao, L.; Ju, Y.; Luo, M. Actinidia Chinensis Planch Prevents Proliferation and Migration of Gastric Cancer Associated with Apoptosis, Ferroptosis Activation and Mesenchymal Phenotype Suppression. Biomed. Pharmacother. 2020, 126, 110092. [Google Scholar] [CrossRef]
- Wei, G.; Sun, J.; Luan, W.; Hou, Z.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Natural Product Albiziabioside A Conjugated with Pyruvate Dehydrogenase Kinase Inhibitor Dichloroacetate to Induce Apoptosis-Ferroptosis-M2-TAMs Polarization for Combined Cancer Therapy. J. Med. Chem. 2019, 62, 8760–8772. [Google Scholar] [CrossRef]
- Chen, Y.; Li, N.; Wang, H.; Wang, N.; Peng, H.; Wang, J.; Li, Y.; Liu, M.; Li, H.; Zhang, Y.; et al. Amentoflavone Suppresses Cell Proliferation and Induces Cell Death through Triggering Autophagy-Dependent Ferroptosis in Human Glioma. Life Sci. 2020, 247, 117425. [Google Scholar] [CrossRef]
- Mbaveng, A.T.; Ndontsa, B.L.; Kuete, V.; Nguekeu, Y.M.M.; Çelik, İ.; Mbouangouere, R.; Tane, P.; Efferth, T. A Naturally Occuring Triterpene Saponin Ardisiacrispin B Displayed Cytotoxic Effects in Multi-Factorial Drug Resistant Cancer Cells via Ferroptotic and Apoptotic Cell Death. Phytomedicine 2018, 43, 78–85. [Google Scholar] [CrossRef]
- Mbaveng, A.T.; Chi, G.F.; Bonsou, I.N.; Abdelfatah, S.; Tamfu, A.N.; Yeboah, E.M.O.; Kuete, V.; Efferth, T. N-Acetylglycoside of Oleanolic Acid (Aridanin) Displays Promising Cytotoxicity towards Human and Animal Cancer Cells, Inducing Apoptotic, Ferroptotic and Necroptotic Cell Death. Phytomedicine 2020, 76, 153261. [Google Scholar] [CrossRef]
- Ooko, E.; Saeed, M.E.M.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin Derivatives Induce Iron-Dependent Cell Death (Ferroptosis) in Tumor Cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Wang, N.; Zeng, G.-Z.; Yin, J.-L.; Bian, Z.-X. Artesunate Activates the ATF4-CHOP-CHAC1 Pathway and Affects Ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, C.; Senba, M.; Mori, N. Evaluation of Artesunate for the Treatment of Adult T-Cell Leukemia/Lymphoma. Eur. J. Pharmacol. 2020, 872, 172953. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.-L.; Kim, E.H.; Jang, H.; Shin, D. Nrf2 Inhibition Reverses the Resistance of Cisplatin-Resistant Head and Neck Cancer Cells to Artesunate-Induced Ferroptosis. Redox Biol. 2017, 11, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Z.; Wang, M.; Cao, X.; Qi, J.; Wang, D.; Gong, A.; Zhu, H. Role of GRP78 Inhibiting Artesunate-Induced Ferroptosis in KRAS Mutant Pancreatic Cancer Cells. Drug Des. Dev. 2019, 13, 2135–2144. [Google Scholar] [CrossRef] [Green Version]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of Reactive Oxygen Species to Ovarian Cancer Cell Growth Arrest and Killing by the Anti-Malarial Drug Artesunate: Impact of Artesunate on Ovarian Cancer. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of Artesunate as a Specific Activator of Ferroptosis in Pancreatic Cancer Cells. Oncoscience 2015, 2, 517. [Google Scholar] [CrossRef] [Green Version]
- Malfa, G.A.; Tomasello, B.; Acquaviva, R.; Genovese, C.; La Mantia, A.; Cammarata, F.P.; Ragusa, M.; Renis, M.; Di Giacomo, C. Betula Etnensis Raf. (Betulaceae) Extract Induced HO-1 Expression and Ferroptosis Cell Death in Human Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2773. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Sun, J.; Hou, Z.; Luan, W.; Wang, S.; Cui, S.; Cheng, M.; Liu, Y. Novel Antitumor Compound Optimized from Natural Saponin Albiziabioside A Induced Caspase-Dependent Apoptosis and Ferroptosis as a P53 Activator through the Mitochondrial Pathway. Eur. J. Med. Chem. 2018, 157, 759–772. [Google Scholar] [CrossRef]
- Du, J.; Wang, T.; Li, Y.; Zhou, Y.; Wang, X.; Yu, X.; Ren, X.; An, Y.; Wu, Y.; Sun, W.; et al. DHA Inhibits Proliferation and Induces Ferroptosis of Leukemia Cells through Autophagy Dependent Degradation of Ferritin. Free Radic. Biol. Med. 2019, 131, 356–369. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Y.; Zhang, X.; Ma, Q.; Song, Y.; Zhang, L.; Wang, D.; Xing, J.; Hou, B.; Li, H.; et al. Dihydroartemisinin-Induced Unfolded Protein Response Feedback Attenuates Ferroptosis via PERK/ATF4/HSPA5 Pathway in Glioma Cells. J. Exp. Clin. Cancer Res. 2019, 38, 402. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Shen, Y.-C.; Wu, C.-Y.; Tsai, Y.-Y.; Yang, Y.-H.; Lin, Y.-Y.; Kuan, F.-C.; Lu, C.-N.; Chang, G.-H.; Tsai, M.-S.; et al. Danshen Improves Survival of Patients with Breast Cancer and Dihydroisotanshinone I Induces Ferroptosis and Apoptosis of Breast Cancer Cells. Front. Pharmacol. 2019, 10, 1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbaveng, A.T.; Fotso, G.W.; Ngnintedo, D.; Kuete, V.; Ngadjui, B.T.; Keumedjio, F.; Andrae-Marobela, K.; Efferth, T. Cytotoxicity of Epunctanone and Four Other Phytochemicals Isolated from the Medicinal Plants Garcinia Epunctata and Ptycholobium Contortum towards Multi-Factorial Drug Resistant Cancer Cells. Phytomedicine 2018, 48, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wu, Q.; Feng, J.; Yan, L.; Sun, Y.; Liu, S.; Xiang, Y.; Zhang, M.; Pan, T.; Chen, X.; et al. Erianin, a Novel Dibenzyl Compound in Dendrobium Extract, Inhibits Lung Cancer Cell Growth and Migration via Calcium/Calmodulin-Dependent Ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Llabani, E.; Hicklin, R.W.; Lee, H.Y.; Motika, S.E.; Crawford, L.A.; Weerapana, E.; Hergenrother, P.J. Diverse Compounds from Pleuromutilin Lead to a Thioredoxin Inhibitor and Inducer of Ferroptosis. Nat. Chem. 2019, 11, 521–532. [Google Scholar] [CrossRef]
- Tang, H.M.; Cheung, P.C.K. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins 2019, 11, 492. [Google Scholar] [CrossRef] [Green Version]
- Khorsandi, K.; Kianmehr, Z.; hosseinmardi, Z.; Hosseinzadeh, R. Anti-Cancer Effect of Gallic Acid in Presence of Low Level Laser Irradiation: ROS Production and Induction of Apoptosis and Ferroptosis. Cancer Cell Int. 2020, 20, 18. [Google Scholar] [CrossRef]
- Niu, Y.; Zhang, J.; Tong, Y.; Li, J.; Liu, B. Physcion 8-O-β-Glucopyranoside Induced Ferroptosis via Regulating MiR-103a-3p/GLS2 Axis in Gastric Cancer. Life Sci. 2019, 237, 116893. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kasukabe, T.; Kumakura, S. Piperlongumine Rapidly Induces the Death of Human Pancreatic Cancer Cells Mainly through the Induction of Ferroptosis. Int J. Oncol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Mbaveng, A.T.; Chi, G.F.; Nguenang, G.S.; Abdelfatah, S.; Sop, R.V.T.; Ngadjui, B.T.; Kuete, V.; Efferth, T. Cytotoxicity of a Naturally Occuring Spirostanol Saponin, Progenin III, towards a Broad Range of Cancer Cell Lines by Induction of Apoptosis, Autophagy and Necroptosis. Chem. Biol. Interact. 2020, 326, 109141. [Google Scholar] [CrossRef]
- Song, Z.; Xiang, X.; Li, J.; Deng, J.; Fang, Z.; Zhang, L.; Xiong, J. Ruscogenin Induces Ferroptosis in Pancreatic Cancer Cells. Oncol. Rep. 2019, 43, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Shi, C.; Li, T.; Wu, Y.; Hu, C.; Huang, G. Solasonine Promotes Ferroptosis of Hepatoma Carcinoma Cells via Glutathione Peroxidase 4-Induced Destruction of the Glutathione Redox System. Biomed. Pharmacol. 2020, 129, 110282. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-Y.; Huang, Z.-X.; Chen, G.-Q.; Sheng, F.; Zheng, Y.-S. Typhaneoside Prevents Acute Myeloid Leukemia (AML) through Suppressing Proliferation and Inducing Ferroptosis Associated with Autophagy. Biochem. Biophys. Res. Commun. 2019, 516, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Mbaveng, A.T.; Bitchagno, G.T.M.; Kuete, V.; Tane, P.; Efferth, T. Cytotoxicity of Ungeremine towards Multi-Factorial Drug Resistant Cancer Cells and Induction of Apoptosis, Ferroptosis, Necroptosis and Autophagy. Phytomedicine 2019, 60, 152832. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yan, H.; Zhang, L.; Shan, M.; Chen, P.; Ding, A.; Li, S. A Review on the Phytochemistry, Pharmacology, and Pharmacokinetics of Amentoflavone, a Naturally-Occurring Biflavonoid. Molecules 2017, 22, 299. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Lee, M.S.; Oh, W.K.; Sul, J.Y. Fatty Acid Synthase Inhibition by Amentoflavone Induces Apoptosis and Antiproliferation in Human Breast Cancer Cells. Biol. Pharm. Bull. 2009, 32, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-C.; Tsai, J.-J.; Tseng, C.-W.; Kuo, Y.-C.; Chuang, Y.-C.; Lin, S.-S.; Hsu, F.-T. Amentoflavone Inhibits ERK-Modulated Tumor Progression in Hepatocellular Carcinoma in Vitro. Vivo 2018, 32, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.-H.; Yeh, C.-Y.; Chung, J.G.; Chiang, I.-T.; Hsu, F.-T. Amentoflavone Induces Apoptosis and Reduces Expression of Anti-Apoptotic and Metastasis-Associated Proteins in Bladder Cancer. Anticancer Res. 2019, 39, 3641–3649. [Google Scholar] [CrossRef]
- Park, H.-J.; Kim, M.-M. Amentoflavone Induces Autophagy and Modulates P53. Cell J. 2018, 21, 27–34. [Google Scholar] [CrossRef]
- Pei, J.-S.; Liu, C.-C.; Hsu, Y.-N.; Lin, L.-L.; Wang, S.-C.; Chung, J.-G.; Bau, D.-T.; Lin, S.-S. Amentoflavone Induces Cell-Cycle Arrest and Apoptosis in MCF-7 Human Breast Cancer Cells via Mitochondria-Dependent Pathway. Vivo 2012, 26, 963–970. [Google Scholar]
- Siveen, K.S.; Kuttan, G. Effect of Amentoflavone, a Phenolic Component from Biophytum Sensitivum, on Cell Cycling and Apoptosis of B16F-10 Melanoma Cells. J. Environ. Pathol. Toxicol. Oncol. 2011, 30, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of Lipid Peroxidation, Iron and Ferritinophagy. Biochim. Biophys. Acta 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis Is an Autophagic Cell Death Process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Chen, G.-Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.-X.; Jiang, X. Artemisinin Compounds Sensitize Cancer Cells to Ferroptosis by Regulating Iron Homeostasis. Cell Death Differ. 2020, 27, 242–254. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER Stress and the Unfolded Protein Response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Xu, C. Endoplasmic Reticulum Stress: Cell Life and Death Decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Tikoo, S.; Maity, S.; Sengupta, S.; Sengupta, S.; Kaur, A.; Kumar Bachhawat, A. Mammalian Proapoptotic Factor ChaC1 and Its Homologues Function as Γ-glutamyl Cyclotransferases Acting Specifically on Glutathione. EMBO Rep. 2012, 13, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and MRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Kimata, Y.; Kohno, K. Endoplasmic Reticulum Stress-Sensing Mechanisms in Yeast and Mammalian Cells. Curr. Opin. Cell Biol. 2011, 23, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Lee, A.S. Role of the Unfolded Protein Response, GRP78 and GRP94 in Organ Homeostasis. J. Cell Physiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, A.D.; Piperdi, B. KRAS Mutation in Colon Cancer: A Marker of Resistance to EGFR-I Therapy. Ann. Surg Oncol. 2010, 17, 1168–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shady, W.; Petre, E.N.; Vakiani, E.; Ziv, E.; Gonen, M.; Brown, K.T.; Kemeny, N.E.; Solomon, S.B.; Solit, D.B.; Sofocleous, C.T. Kras Mutation Is a Marker of Worse Oncologic Outcomes after Percutaneous Radiofrequency Ablation of Colorectal Liver Metastases. Oncotarget 2017, 8, 66117–66127. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress–Induced DPP3–KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D. Mechanistic Studies of the Nrf2-Keap1 Signaling Pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Lau, A.; Villeneuve, N.; Sun, Z.; Wong, P.; Zhang, D. Dual Roles of Nrf2 in Cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Gañán-Gómez, I.; Wei, Y.; Yang, H.; Boyano-Adánez, M.C.; García-Manero, G. Oncogenic Functions of the Transcription Factor Nrf2. Free Radic. Biol. Med. 2013, 65, 750–764. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.-X.; Singh, G. Expression of XCT and Activity of System Xc− Are Regulated by NRF2 in Human Breast Cancer Cells in Response to Oxidative Stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Albrecht, P.; Tien, M.-L.T.; Henke, N.; Karumbayaram, S.; Kornblum, H.I.; Wiedau-Pazos, M.; Schubert, D.; Maher, P.; Methner, A. Induction of Nrf2 and XCT Are Involved in the Action of the Neuroprotective Antibiotic Ceftriaxone in Vitro. J. Neurochem. 2009, 111, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-Dependent Upregulation of XCT Modulates the Sensitivity of T24 Bladder Carcinoma Cells to Proteasome Inhibition. Mol. Cell Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and Proteomic Profiling of KEAP1 Disrupted and Sulforaphane-Treated Human Breast Epithelial Cells Reveals Common Expression Profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 Regulates Ferroportin 1-Mediated Iron Efflux and Counteracts Lipopolysaccharide-Induced Ferroportin 1 MRNA Suppression in Macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular Mechanisms of Ferroptosis and Its Role in Cancer Therapy. J. Cell Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the P62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells: Hepatobiliary Malignancies. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Fan, Z.; Wirth, A.-K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme Oxygenase-1 Accelerates Erastin-Induced Ferroptotic Cell Death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Bebber, C.M.; Müller, F.; Prieto Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassannia, B.; Logie, E.; Vandenabeele, P.; Vanden Berghe, T.; Vanden Berghe, W. Withaferin A: From Ayurvedic Folk Medicine to Preclinical Anti-Cancer Drug. Biochem. Pharmacol. 2020, 173, 113602. [Google Scholar] [CrossRef] [PubMed]
- Efimova, I.; Catanzaro, E.; Van der Meeren, L.; Turubanova, V.D.; Hammad, H.; Mishchenko, T.A.; Vedunova, M.V.; Fimognari, C.; Bachert, C.; Coppieters, F.; et al. Vaccination with Early Ferroptotic Cancer Cells Induces Efficient Antitumor Immunity. J. Immunother. Cancer 2020, 8, e001369. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical Inhibitor of Nonapoptotic Cell Death with Therapeutic Potential for Ischemic Brain Injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Berghe, T.V.; Vanlangenakker, N.; Buettner, S.; Eisenberg, T.; Vandenabeele, P.; Madeo, F.; Kroemer, G. Programmed Necrosis from Molecules to Health and Disease. Int. Rev. Cell Mol. Biol. 2011, 289, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The Role of Necroptosis in Cancer Biology and Therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S. The Mechanism of Necroptosis in Normal and Cancer Cells. Cancer Biol. Ther. 2013, 14, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Vanlangenakker, N.; Bertrand, M.J.M.; Bogaert, P.; Vandenabeele, P.; Vanden Berghe, T. TNF-Induced Necroptosis in L929 Cells Is Tightly Regulated by Multiple TNFR1 Complex I and II Members. Cell Death Dis. 2011, 2, e230. [Google Scholar] [CrossRef]
- Walsh, C.M. Grand Challenges in Cell Death and Survival: Apoptosis vs. Necroptosis. Front. Cell Dev. Biol. 2014, 2. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 Kinase as a Specific Cellular Target of Necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, T.; Cho, Y.; Chan, F.K.-M. Positive and Negative Phosphorylation Regulates RIP1- and RIP3-Induced Programmed Necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhou, Z.; Li, L.; Zhong, C.-Q.; Zheng, X.; Wu, X.; Zhang, Y.; Ma, H.; Huang, D.; Li, W.; et al. Diverse Sequence Determinants Control Human and Mouse Receptor Interacting Protein 3 (RIP3) and Mixed Lineage Kinase Domain-like (MLKL) Interaction in Necroptotic Signaling. J. Biol. Chem. 2013, 288, 16247–16261. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma Membrane Translocation of Trimerized MLKL Protein Is Required for TNF-Induced Necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Bao, J.; Lin, W.; Gao, H.; Zhao, W.; Zhang, Q.; Leung, C.-H.; Ma, D.-L.; Lu, J.; Chen, X. 2-Methoxy-6-Acetyl-7-Methyljuglone (MAM), a Natural Naphthoquinone, Induces NO-Dependent Apoptosis and Necroptosis by H 2 O 2 -Dependent JNK Activation in Cancer Cells. Free Radic. Biol. Med. 2016, 92, 61–77. [Google Scholar] [CrossRef]
- Sun, W.; Yu, J.; Gao, H.; Wu, X.; Wang, S.; Hou, Y.; Lu, J.-J.; Chen, X. Inhibition of Lung Cancer by 2-Methoxy-6-Acetyl-7-Methyljuglone through Induction of Necroptosis by Targeting Receptor-Interacting Protein 1. Antioxid. Redox Signal. 2019, 31, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhong, B.; Jin, L.; Hou, Y.; Ai, N.; Ge, W.; Li, L.; Liu, S.; Lu, J.-J.; Chen, X. 2-Methoxy-6-Acetyl-7-Methyljuglone (MAM) Induced Programmed Necrosis in Glioblastoma by Targeting NAD(P)H: Quinone Oxidoreductase 1 (NQO1). Free Radic. Biol. Med. 2020, 152, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Tao, H.-R.; Fang, L.; Kong, X.-Q.; Han, L.-N.; Li, N.; Xu, Y.-X.; Li, L.-Y.; Yu, M.; Zhang, H. 11-Methoxytabersonine Induces Necroptosis with Autophagy through AMPK/MTOR and JNK Pathways in Human Lung Cancer Cells. Chem. Pharm. Bull. 2020, 68, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balhamar, S.O.M.S.; Panicker, N.G.; Akhlaq, S.; Qureshi, M.M.; Ahmad, W.; Rehman, N.U.; Ali, L.; Al-Harrasi, A.; Hussain, J.; Mustafa, F. Differential Cytotoxic Potential of Acridocarpus Orientalis Leaf and Stem Extracts with the Ability to Induce Multiple Cell Death Pathways. Molecules 2019, 24, 3976. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-J.; Nam, H.-S.; Cho, M.-K.; Lee, S.-H. Arctigenin Induces Necroptosis through Mitochondrial Dysfunction with CCN1 Upregulation in Prostate Cancer Cells under Lactic Acidosis. Mol. Cell. Biochem. 2020, 467, 45–56. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Park, K.-S.; Baek, B.J.; Lee, K.-A.; Lee, S.-H. Apoptosis and Necroptosis-Inducing Effects of Arctigenin on Nasal Septum Carcinoma RPMI-2650 Cells in 2D and 3D Culture. Mol. Cell. Toxicol. 2020, 16, 1–11. [Google Scholar] [CrossRef]
- Button, R.W.; Lin, F.; Ercolano, E.; Vincent, J.H.; Hu, B.; Hanemann, C.O.; Luo, S. Artesunate Induces Necrotic Cell Death in Schwannoma Cells. Cell Death Dis. 2014, 5, e1466. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Fan, J.; Ai, G.; Liu, J.; Luo, N.; Li, C.; Cheng, Z. Berberine in Combination with Cisplatin Induces Necroptosis and Apoptosis in Ovarian Cancer Cells. Biol. Res. 2019, 52, 37. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Wang, L.; Fei, X.-C.; Jiang, X.-F.; Zheng, Z.; Zhao, Y.; Wang, C.-F.; Li, B.; Chen, S.-J.; Janin, A.; et al. MYC Is a Positive Regulator of Choline Metabolism and Impedes Mitophagy-Dependent Necroptosis in Diffuse Large B-Cell Lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Zhang, W.; Yang, H.; Bi, J.; Xie, Y.; Cheng, B.; Wang, Y.; Chen, S. Celastrol Induces Necroptosis and Ameliorates Inflammation via Targeting Biglycan in Human Gastric Carcinoma. Int. J. Mol. Sci. 2019, 20, 5716. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.I.; Hong, J.-Y.; Choi, J.S.; Lee, S.K. Columbianadin Inhibits Cell Proliferation by Inducing Apoptosis and Necroptosis in HCT116 Colon Cancer Cells. Biomol. Ther. 2016, 24, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Jiang, Z.; Duan, H.; Sun, L.; Zhang, S.; Chen, M.; Wang, Y.; Gao, Q.; Song, Y.; Zhu, X.; et al. Deoxypodophyllotoxin Triggers Necroptosis in Human Non-Small Cell Lung Cancer NCI-H460 Cells. Biomed. Pharmacolther. 2013, 67, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, G.; Han, G.; Feng, S.; Liu, Y.; Chen, J.; Liu, C.; Zhao, L.; Jin, F. Emodin Induced Necroptosis in the Glioma Cell Line U251 via the TNF-α/RIP1/RIP3 Pathway. Investig. New Drugs 2020, 38, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Moon, H.-I.; Kim, S.; Quynh, N.; Yu, J.; Sandag, Z.; Le, D.-D.; Lee, H.; Lee, H.; Lee, M.-S. Anticancer Activity of Gomisin J from Schisandra Chinensis Fruit. Oncol. Rep. 2018, 41, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.-M.; Li, Y.-Q.; Xu, K.-Q.; Zhang, Y.-Y.; Tan, S.-M.; Zhang, Q.; Peng, J.; Luo, X.-J. Jujuboside B Promotes the Death of Acute Leukemia Cell in a RIPK1/RIPK3/MLKL Pathway-Dependent Manner. Eur. J. Pharmacol. 2020, 876, 173041. [Google Scholar] [CrossRef]
- Xu, B.; Xu, M.; Tian, Y.; Yu, Q.; Zhao, Y.; Chen, X.; Mi, P.; Cao, H.; Zhang, B.; Song, G.; et al. Matrine Induces RIP3-Dependent Necroptosis in Cholangiocarcinoma Cells. Cell Death Discov. 2017, 3, 16096. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Yu, X.; Xiao, L.; Hu, Z.; Luo, X.; Tao, Y.; Yang, L.; Liu, X.; Chen, H.; Ding, Z.; et al. Neoalbaconol Induces Energy Depletion and Multiple Cell Death in Cancer Cells by Targeting PDK1-PI3-K/Akt Signaling Pathway. Cell Death Dis. 2013, 4, e804. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Wu, C.; Zhu, M.; Song, W.; Wang, H.; Wang, J.; Guo, J.; Li, N.; Liu, J.; Li, Y.; et al. Ophiopogonin D’ Induces RIPK1-dependent Necroptosis in Androgen-dependent LNCaP Prostate Cancer Cells. Int. J. Oncol. 2019, 56, 439–447. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, C.; Lu, B.; Zhou, Z.; Jin, Y.; Wang, Z.; Zheng, L.; Liu, K.; Luo, T.; Zhu, D.; et al. Pristimerin Triggers AIF-Dependent Programmed Necrosis in Glioma Cells via Activation of JNK. Cancer Lett. 2016, 374, 136–148. [Google Scholar] [CrossRef]
- Khorsandi, L.; Orazizadeh, M.; Niazvand, F.; Abbaspour, M.R.; Mansouri, E.; Khodadadi, A. Quercetin Induces Apoptosis and Necroptosis in MCF-7 Breast Cancer Cells. Bratisl. Med. J. 2017, 118, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Ma, Y.; Wang, H.; Dai, Y.; Chen, C.; Liu, Y.; Jing, L.; Sun, X. Resibufogenin Suppresses Colorectal Cancer Growth and Metastasis through RIP3-Mediated Necroptosis. J. Transl. Med. 2018, 16, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerová, J.; Uldrijan, S.; Táborská, E.; Vaculová, A.H.; Slaninová, I. Necroptosis Modulated by Autophagy Is a Predominant Form of Melanoma Cell Death Induced by Sanguilutine. Biol. Chem. 2012, 393, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S. Shikonin Induced Necroptosis via Reactive Oxygen Species in the T-47D Breast Cancer Cell Line. Asian Pac. J. Cancer Prev. 2015, 16, 7261–7266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Xiao, W.; Huang, L.; Yu, G.; Ni, J.; Yang, L.; Wan, R.; Hu, G. Shikonin Induces Apoptosis and Necroptosis in Pancreatic Cancer via Regulating the Expression of RIP1/RIP3 and Synergizes the Activity of Gemcitabine. Am. J. Transl. Res. 2017, 9, 5507–5517. [Google Scholar] [PubMed]
- Zhang, Z.; Zhang, Z.; Li, Q.; Jiao, H.; Chong, D.; Sun, X.; Zhang, P.; Huo, Q.; Liu, H. Shikonin Induces Necroptosis by Reactive Oxygen Species Activation in Nasopharyngeal Carcinoma Cell Line CNE-2Z. J. Bioenerg. Biomembr. 2017, 49, 265–272. [Google Scholar] [CrossRef]
- Kim, H.-J.; Hwang, K.-E.; Park, D.-S.; Oh, S.-H.; Jun, H.Y.; Yoon, K.-H.; Jeong, E.-T.; Kim, H.-R.; Kim, Y.-S. Shikonin-Induced Necroptosis Is Enhanced by the Inhibition of Autophagy in Non-Small Cell Lung Cancer Cells. J. Transl. Med. 2017, 15, 123. [Google Scholar] [CrossRef]
- Wada, N.; Kawano, Y.; Fujiwara, S.; Kikukawa, Y.; Okuno, Y.; Tasaki, M.; Ueda, M.; Ando, Y.; Yoshinaga, K.; Ri, M.; et al. Shikonin, Dually Functions as a Proteasome Inhibitor and a Necroptosis Inducer in Multiple Myeloma Cells. Int. J. Oncol. 2015, 46, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The Anti-Tumor Effect of Shikonin on Osteosarcoma by Inducing RIP1 and RIP3 Dependent Necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef] [Green Version]
- Piao, J.-L.; Cui, Z.-G.; Furusawa, Y.; Ahmed, K.; Rehman, M.U.; Tabuchi, Y.; Kadowaki, M.; Kondo, T. The Molecular Mechanisms and Gene Expression Profiling for Shikonin-Induced Apoptotic and Necroptotic Cell Death in U937 Cells. Chem. Biol. Interact. 2013, 205, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Luo, Y.; Zhao, J.; Yang, F.; Zhao, H.; Fan, W.; Ge, P. Shikonin Kills Glioma Cells through Necroptosis Mediated by RIP-1. PLoS ONE 2013, 8, e66326. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Gong, X.; Wang, Z.; Ding, Y.; Wang, C.; Luo, T.; Piao, M.; Meng, F.; Chi, G.; Luo, Y.; et al. Shikonin Induces Glioma Cell Necroptosis in Vitro by ROS Overproduction and Promoting RIP1/RIP3 Necrosome Formation. Acta Pharmacol. Sin. 2017, 38, 1543–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Lu, B.; Wang, C.; Wang, Z.; Luo, T.; Piao, M.; Meng, F.; Chi, G.; Luo, Y.; Ge, P. RIP1 and RIP3 Contribute to Shikonin-Induced DNA Double-Strand Breaks in Glioma Cells via Increase of Intracellular Reactive Oxygen Species. Cancer Lett. 2017, 390, 77–90. [Google Scholar] [CrossRef]
- Ding, Y.; He, C.; Lu, S.; Wang, X.; Wang, C.; Wang, L.; Zhang, J.; Piao, M.; Chi, G.; Luo, Y.; et al. MLKL Contributes to Shikonin-Induced Glioma Cell Necroptosis via Promotion of Chromatinolysis. Cancer Lett. 2019, 467, 58–71. [Google Scholar] [CrossRef]
- Liu, T.; Sun, X.; Cao, Z. Shikonin-Induced Necroptosis in Nasopharyngeal Carcinoma Cells via ROS Overproduction and Upregulation of RIPK1/RIPK3/MLKL Expression. Onco Targets Ther. 2019, 12, 2605–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-J.; Kao, S.-H.; Hunag, J.-E.; Sheu, G.-T.; Yeh, C.-W.; Hseu, Y.-C.; Wang, C.-J.; Hsu, L.-S. Shikonin Time-Dependently Induced Necrosis or Apoptosis in Gastric Cancer Cells via Generation of Reactive Oxygen Species. Chem Biol. Interact. 2014, 211, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shin, H.; Cho, Y. Shikonin Induces Programmed Necrosis-like Cell Death through the Formation of Receptor Interacting Protein 1 and 3 Complex. Food Chem. Toxicol. 2013, 55, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S. Targeting Cell Necroptosis and Apoptosis Induced by Shikonin via Receptor Interacting Protein Kinases in Estrogen Receptor Positive Breast Cancer Cell Line, MCF-7. Anticancer Agents Med. Chem. 2018, 18, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S.; Ghasemzadeh, M. RIP1K and RIP3K Provoked by Shikonin Induce Cell Cycle Arrest in the Triple Negative Breast Cancer Cell Line, MDA-MB-468: Necroptosis as a Desperate Programmed Suicide Pathway. Tumor Biol. 2016, 37, 4479–4491. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Gao, H.; Hou, Y.; Lu, J.; Feng, Y.; Xu, Q.; Liu, B.; Chen, X. Induction of an MLKL Mediated Non-Canonical Necroptosis through Reactive Oxygen Species by Tanshinol A in Lung Cancer Cells. Biochem. Pharmacol. 2020, 171, 113684. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Chang, T.-W.; Hsieh, W.-H.; Hung, M.-C.; Lin, I.-H.; Lai, S.-C.; Tzeng, Y.-J. Simultaneous Induction of Apoptosis and Necroptosis by Tanshinone IIA in Human Hepatocellular Carcinoma HepG2 Cells. Cell Death Discov. 2016, 2, 16065. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hu, X.; Liu, L.; Liang, X.; Xiao, J. Extracts Derived from a Traditional Chinese Herbal Formula Triggers Necroptosis in Ectocervical Ect1/E6E7 Cells through Activation of RIP1 Kinase. J. Ethnopharmacol. 2019, 239, 111922. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Vong, C.T.; Chen, H.; Gao, Y.; Lyu, P.; Qiu, L.; Zhao, M.; Liu, Q.; Cheng, Z.; Zou, J.; et al. Naturally Occurring Anti-Cancer Compounds: Shining from Chinese Herbal Medicine. Chin. Med. 2019, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 Inactivates Its Caspase-Independent Apoptosis Pathway by Removal of Kinase Domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. CIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by CFLIP Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a Signaling Platform That Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.G.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread Mitochondrial Depletion via Mitophagy Does Not Compromise Necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K.M. Differential Roles of RIPK1 and RIPK3 in TNF-Induced Necroptosis and Chemotherapeutic Agent-Induced Cell Death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.-M.; Qiu, Y.-K.; Wu, Z.; Zhao, Y.-F. DNA Damage Induced by Shikonin in the Presence of Cu(II) Ions: Potential Mechanism of Its Activity to Apoptotic Cell Death. J. Asian Nat. Prod. Res. 2011, 13, 12–19. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, L.-J.; Li, L.-H.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Shikonin Regulates HeLa Cell Death via Caspase-3 Activation and Blockage of DNA Synthesis. J. Asian Nat. Prod. Res. 2004, 6, 155–166. [Google Scholar] [CrossRef]
- Reuvers, T.G.A.; Kanaar, R.; Nonnekens, J. DNA Damage-Inducing Anticancer Therapies: From Global to Precision Damage. Cancers 2020, 12, 2098. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-Cell-Induced Endothelial Cell Necroptosis via Death Receptor 6 Promotes Metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Du, X.; Ma, H.; Yao, J. The Anti-Cancer Mechanisms of Berberine: A Review. Cancer Manag. Res. 2020, 12, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, A.; Wei, Y.-C.; Lim, K.; Barbosa, A.D.; Liu, C.-H.; Weber, U.; Mlodzik, M.; Oras, K.; Collier, S.; Hussain, M.M.; et al. PCYT1A Regulates Phosphatidylcholine Homeostasis from the Inner Nuclear Membrane in Response to Membrane Stored Curvature Elastic Stress. Dev. Cell 2018, 45, 481–495.e8. [Google Scholar] [CrossRef] [Green Version]
- Lwin, T.; Zhao, X.; Cheng, F.; Zhang, X.; Huang, A.; Shah, B.; Zhang, Y.; Moscinski, L.C.; Choi, Y.S.; Kozikowski, A.P.; et al. A Microenvironment-Mediated c-Myc/MiR-548m/HDAC6 Amplification Loop in Non-Hodgkin B Cell Lymphomas. J. Clin. Investig. 2013, 123, 4612–4626. [Google Scholar] [CrossRef] [Green Version]
- Cookson, B.T.; Brennan, M.A. Pro-Inflammatory Programmed Cell Death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Liu, X.; Lieberman, J. A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 135, pp. 81–117. ISBN 978-0-12-812405-5. [Google Scholar]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The Role of Pyroptosis in Cancer: Pro-Cancer or pro-“Host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Jiang, S.; Zhang, Y.; Li, P.; Wang, K. The Multifaceted Roles of Pyroptotic Cell Death Pathways in Cancer. Cancers 2019, 11, 1313. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E Suppresses Tumour Growth by Activating Anti-Tumour Immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Ding, J.; Wang, C.; Zhou, X.; Gao, W.; Huang, H.; Shao, F.; Liu, Z. A Bioorthogonal System Reveals Antitumour Immune Function of Pyroptosis. Nature 2020, 579, 421–426. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, M.; Chen, M.; Chen, Z.; Peng, X.; Zhou, F.; Song, J.; Qu, J. Programming Cell Pyroptosis with Biomimetic Nanoparticles for Solid Tumor Immunotherapy. Biomaterials 2020, 254, 120142. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.A.; Cookson, B.T. Salmonella Induces Macrophage Death by Caspase-1-Dependent Necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Kolb, R.; Liu, G.-H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in Cancer: A Double-Edged Sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, W.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and Its Morphology Is Different from MLKL Channel-Mediated Necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Masters, S.L. Homeostasis-Altering Molecular Processes as Mechanisms of Inflammasome Activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Xue, Y.; Enosi Tuipulotu, D.; Tan, W.H.; Kay, C.; Man, S.M. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 2019, 40, 1035–1052. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.-D. The Cell Biology of Inflammasomes: Mechanisms of Inflammasome Activation and Regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Scott, M.J. Caspase-1 as a Multifunctional Inflammatory Mediator: Noncytokine Maturation Roles. J. Leukoc. Biol. 2016, 100, 961–967. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.K.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.-D. Regulation of Inflammasome Activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica Crystals and Aluminum Salts Activate the NALP3 Inflammasome through Phagosomal Destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Poyet, J.-L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD Protein ASC Is an Activating Adaptor for Caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [Green Version]
- de Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrín, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Brough, D.; Rothwell, N.J. Caspase-1-Dependent Processing of pro-Interleukin-1beta Is Cytosolic and Precedes Cell Death. J. Cell Sci. 2007, 120, 772–781. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis—A Cell Death Modality of Its Kind? Eur. J. Immunol. 2010, 40, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by Caspase-3 Determines Lobaplatin-Induced Pyroptosis in Colon Cancer Cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Zhang, S.; Wu, J.; Chen, M.; Cai, M.-C.; Fu, Y.; Li, W.; Wang, J.; Zhao, X.; Yu, Z.; et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin. Cancer Res. 2018, 24, 6066–6077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-ΚB Activation Pathways, Emerging Molecular Targets for Cancer Prevention and Therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.-F.; Mei, Q.-B.; Zhou, X.-G.; Tang, Y.; Xiong, R.; Qiu, W.-Q.; Pan, R.; Law, B.Y.-K.; Wong, V.K.-W.; Yu, C.-L.; et al. Polyphyllin VI Induces Caspase-1-Mediated Pyroptosis via the Induction of ROS/NF-ΚB/NLRP3/GSDMD Signal Axis in Non-Small Cell Lung Cancer. Cancers 2020, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.-F.; Qin, D.-L.; Mei, Q.-B.; Qiu, W.-Q.; Pan, R.; Xiong, R.; Zhao, Y.; Law, B.Y.-K.; Wong, V.K.-W.; Tang, Y.; et al. Polyphyllin VI, a Saponin from Trillium Tschonoskii Maxim. Induces Apoptotic and Autophagic Cell Death via the ROS Triggered MTOR Signaling Pathway in Non-Small Cell Lung Cancer. Pharmacol. Res. 2019, 147, 104396. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME Mediates Caspase-3-Dependent Pyroptosis in Gastric Cancer. Biochem. Biophys. Res. Commun. 2018, 495, 1418–1425. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, W.; Tian, H.; Zhang, H. Alpinumisoflavone Triggers GSDME-dependent Pyroptosis in Esophageal Squamous Cell Carcinomas. Anat. Rec. 2020, ar.24414. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Sun, M.; He, T.; Liu, Y.; Yang, X.; Shi, X.; Liu, X. Alpinumisoflavone Suppresses Hepatocellular Carcinoma Cell Growth and Metastasis via NLRP3 Inflammasome-Mediated Pyroptosis. Pharmacol. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yue, E.; Tuguzbaeva, G.; Chen, X.; Qin, Y.; Li, A.; Sun, X.; Dong, C.; Liu, Y.; Yu, Y.; Zahra, S.M.; et al. Anthocyanin Is Involved in the Activation of Pyroptosis in Oral Squamous Cell Carcinoma. Phytomedicine 2019, 56, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Jiang, Y.; Zhang, W.; Xu, C.; Du, W.; Tuguzbaeva, G.; Qin, Y.; Li, A.; Zhang, L.; Sun, G.; et al. Pyroptosis Is Involved in the Pathogenesis of Human Hepatocellular Carcinoma. Oncotarget 2016, 7, 84658–84665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, R.; Yan, X.; Tang, G. Casticin Elicits Inflammasome-Induced Pyroptosis through Activating PKR/JNK/NF-ΚB Signal in 5-8F Cells. Biomed. Pharmacol. 2020, 123, 109576. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, W.; Cheng, C.; Mo, F.; Chen, L.; Peng, G.; Cai, X.; Wang, J.; Yang, S.; Liu, X. Dioscin Inhibits the Growth of Human Osteosarcoma by Inducing G2/M-Phase Arrest, Apoptosis, and GSDME-Dependent Cell Death in Vitro and in Vivo. J. Cell Physiol. 2020, 235, 2911–2924. [Google Scholar] [CrossRef]
- Kong, Y.; Feng, Z.; Chen, A.; Qi, Q.; Han, M.; Wang, S.; Zhang, Y.; Zhang, X.; Yang, N.; Wang, J.; et al. The Natural Flavonoid Galangin Elicits Apoptosis, Pyroptosis, and Autophagy in Glioblastoma. Front. Oncol. 2019, 9, 942. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Zhuan, B.; Wang, H.; Wang, Y.; Wang, X.; Yuan, Q.; Yang, Z. Huaier Extract Suppresses Non-small Cell Lung Cancer Progression through Activating NLRP3-dependent Pyroptosis. Anat. Rec. 2019, ar.24307. [Google Scholar] [CrossRef]
- Li, Q.; Chen, L.; Dong, Z.; Zhao, Y.; Deng, H.; Wu, J.; Wu, X.; Li, W. Piperlongumine Analogue L50377 Induces Pyroptosis via ROS Mediated NF-ΚB Suppression in Non-Small-Cell Lung Cancer. Chem. Biol. Interact. 2019, 313, 108820. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, J.; Mao, L.; Guo, Y.; Hao, Y.; Deng, Y.; Han, X.; Li, Q.; Liao, W.; Yuan, M. Nobiletin Triggers Reactive Oxygen Species-Mediated Pyroptosis through Regulating Autophagy in Ovarian Cancer Cells. J. Agric. Food Chem. 2020, 68, 1326–1336. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, J.; Xu, Y.; Huang, X.; Wang, X.; Huang, W.; Li, H. Osthole Inhibits Ovarian Carcinoma Cells through LC3-Mediated Autophagy and GSDME-Dependent Pyroptosis except for Apoptosis. Eur. J. Pharmacol. 2020, 874, 172990. [Google Scholar] [CrossRef]
- Zhang, C.; Li, C.; Wang, Y.; Xu, L.; He, X.; Zeng, Q.; Zeng, C.; Mai, F.; Hu, B.; Ouyang, D. Chemotherapeutic Paclitaxel and Cisplatin Differentially Induce Pyroptosis in A549 Lung Cancer Cells via Caspase-3/GSDME Activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Guo, J.; Yang, C. Tanshinone II A Enhances Pyroptosis and Represses Cell Proliferation of HeLa Cells by Regulating MiR-145/GSDMD Signaling Pathway. Biosci. Rep. 2020, 40, BSR20200259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lin, B.; Li, H.; Lan, L.; Yu, H.; Wu, S.; Wu, J.; Zhang, H. Galangin Suppresses Hepatocellular Carcinoma Cell Proliferation by Reversing the Warburg Effect. Biomed. Pharmacol. 2017, 95, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy Balances Inflammation in Innate Immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef] [Green Version]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Giurgea, I.; Amselem, S.; Karabina, S.-A. Photoaging and Skin Cancer: Is the Inflammasome the Missing Link? Mech. Ageing Dev. 2018, 172, 131–137. [Google Scholar] [CrossRef]
- Ellis, L.Z.; Liu, W.; Luo, Y.; Okamoto, M.; Qu, D.; Dunn, J.H.; Fujita, M. Green Tea Polyphenol Epigallocatechin-3-Gallate Suppresses Melanoma Growth by Inhibiting Inflammasome and IL-1β Secretion. Biochem. Biophys. Res. Commun. 2011, 414, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Muneer, K.M.; Tamimi, I.A.; Chang, M.E.; Ata, M.O.; Yusuf, N. Thymoquinone Suppresses Metastasis of Melanoma Cells by Inhibition of NLRP3 Inflammasome. Toxicol. Appl. Pharmacol. 2013, 270, 70–76. [Google Scholar] [CrossRef]
- Remesh, A. Toxicities of Anticancer Drugs and Its Management. Int. J. Basic Clin. Pharmacol. 2012, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gu, S.; Sun, D.; Dai, H.; Chen, H.; Zhang, Z. The Selectivity of Artemisinin-Based Drugs on Human Lung Normal and Cancer Cells. Environ. Toxicol Pharmacol. 2018, 57, 86–94. [Google Scholar] [CrossRef]
- Xu, Q.; Li, Z.; Peng, H.; Sun, Z.; Cheng, R.; Ye, Z.; Li, W. Artesunate Inhibits Growth and Induces Apoptosis in Human Osteosarcoma HOS Cell Line in Vitro and in Vivo. J. Zhejiang Univ. Sci. B 2011, 12, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, W.; Wang, Z.-Z.; Xue, J. Artesunate Induces Apoptosis of Bladder Cancer Cells by MiR-16 Regulation of COX-2 Expression. Int. J. Mol. Sci. 2014, 15, 14298–14312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Wang, H.; Kunda, P.; Chen, X.; Liu, Q.-L.; Liu, T. Artesunate Exerts Specific Cytotoxicity in Retinoblastoma Cells via CD71. Oncol. Rep. 2013, 30, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Qin, G.; Wu, L.; Wang, X.; Chen, T. Artesunate Induces ROS-Dependent Apoptosis via a Bax-Mediated Intrinsic Pathway in Huh-7 and Hep3B Cells. Exp. Cell Res. 2016, 347, 251–260. [Google Scholar] [CrossRef]
- Yin, S.; Yang, H.; Zhao, X.; Wei, S.; Tao, Y.; Liu, M.; Bo, R.; Li, J. Antimalarial Agent Artesunate Induces G0/G1 Cell Cycle Arrest and Apoptosis via Increasing Intracellular ROS Levels in Normal Liver Cells. Hum. Exp. Toxicol. 2020, 39, 1681–1689. [Google Scholar] [CrossRef]
- Koduru, S.; Kumar, R.; Srinivasan, S.; Evers, M.B.; Damodaran, C. Notch-1 Inhibition by Withaferin-A: A Therapeutic Target against Colon Carcinogenesis. Mol. Cancer Ther. 2010, 9, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.-Y.; Wang, Y.-Y.; Lan, T.-H.; Lin, L.-C.; Yuan, S.-S.F.; Tang, J.-Y.; Chang, H.-W. Low Dose Combined Treatment with Ultraviolet-C and Withaferin a Enhances Selective Killing of Oral Cancer Cells. Antioxidants 2020, 9, 1120. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Okuzaki, D.; Fukushima, K.; Mukai, S.; Ohno, S.; Ozaki, Y.; Yabuta, N.; Nojima, H. Withaferin A Induces Cell Death Selectively in Androgen-Independent Prostate Cancer Cells but Not in Normal Fibroblast Cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Liu, X.; Chen, L.; Liang, T.; Tian, X.-D.; Liu, Y.; Zhang, T. Withaferin A Induces Mitochondrial-Dependent Apoptosis in Non-Small Cell Lung Cancer Cells via Generation of Reactive Oxygen Species. J. Buon 2017, 22, 244–250. [Google Scholar]
- Mandal, C.; Dutta, A.; Mallick, A.; Chandra, S.; Misra, L.; Sangwan, R.S.; Mandal, C. Withaferin A Induces Apoptosis by Activating P38 Mitogen-Activated Protein Kinase Signaling Cascade in Leukemic Cells of Lymphoid and Myeloid Origin through Mitochondrial Death Cascade. Apoptosis 2008, 13, 1450–1464. [Google Scholar] [CrossRef]
- Vaishnavi, K.; Saxena, N.; Shah, N.; Singh, R.; Manjunath, K.; Uthayakumar, M.; Kanaujia, S.P.; Kaul, S.C.; Sekar, K.; Wadhwa, R. Differential Activities of the Two Closely Related Withanolides, Withaferin A and Withanone: Bioinformatics and Experimental Evidences. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-Y.; Cherng, J.-Y.; Yang, Y.-H.; Lin, C.-L.; Kuan, F.-C.; Lin, Y.-Y.; Lin, Y.-S.; Shu, L.-H.; Cheng, Y.-C.; Liu, H.T.; et al. Danshen Improves Survival of Patients with Advanced Lung Cancer and Targeting the Relationship between Macrophages and Lung Cancer Cells. Oncotarget 2017, 8, 90925–90947. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.B.D.C.; de Oliveira, F.P.; Dias, R.B.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; Costa, E.V.; da Silva, F.M.A.; Rocha, W.C.; Koolen, H.H.F.; et al. In Vitro and in Vivo Anti-Leukemia Activity of the Stem Bark of Salacia Impressifolia (Miers) A. C. Smith (Celastraceae). J. Ethnopharmacol. 2019, 231, 516–524. [Google Scholar] [CrossRef] [PubMed]
- da Costa, P.M.; Ferreira, P.M.P.; da Silva Bolzani, V.; Furlan, M.; de Freitas Formenton Macedo dos Santos, V.A.; Corsino, J.; de Moraes, M.O.; Costa-Lotufo, L.V.; Montenegro, R.C.; Pessoa, C. Antiproliferative Activity of Pristimerin Isolated from Maytenus Ilicifolia (Celastraceae) in Human HL-60 Cells. Toxicol Vitr. 2008, 22, 854–863. [Google Scholar] [CrossRef]
- Wu, C.-C.; Chan, M.-L.; Chen, W.-Y.; Tsai, C.-Y.; Chang, F.-R.; Wu, Y.-C. Pristimerin Induces Caspase-Dependent Apoptosis in MDA-MB-231 Cells via Direct Effects on Mitochondria. Mol. Cancer Ther. 2005, 4, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Xu, L.; Yin, L.; Qi, Y.; Han, X.; Xu, Y.; Zhao, Y.; Liu, K.; Peng, J. Potent Effects of Dioscin against Pancreatic Cancer via MiR-149-3P-mediated Inhibition of the Akt1 Signalling Pathway. Br. J. Pharmacol. 2017, 174, 553–568. [Google Scholar] [CrossRef]
- Si, L.; Zheng, L.; Xu, L.; Yin, L.; Han, X.; Qi, Y.; Xu, Y.; Wang, C.; Peng, J. Dioscin Suppresses Human Laryngeal Cancer Cells Growth via Induction of Cell-Cycle Arrest and MAPK-Mediated Mitochondrial-Derived Apoptosis and Inhibition of Tumor Invasion. Eur. J. Pharmacol. 2016, 774, 105–117. [Google Scholar] [CrossRef]
- Zhao, X.; Tao, X.; Xu, L.; Yin, L.; Qi, Y.; Xu, Y.; Han, X.; Peng, J. Dioscin Induces Apoptosis in Human Cervical Carcinoma HeLa and SiHa Cells through ROS-Mediated DNA Damage and the Mitochondrial Signaling Pathway. Molecules 2016, 21, 730. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.T.; Zheng, L.H.; Bao, Y.L.; Yu, C.L.; Wu, Y.; Meng, X.Y.; Li, Y.X. Reversal Effect of Dioscin on Multidrug Resistance in Human Hepatoma HepG2/Adriamycin Cells. Eur. J. Pharmacol. 2011, 654, 129–134. [Google Scholar] [CrossRef]
- Song, X.; Wang, Z.; Liang, H.; Zhang, W.; Ye, Y.; Li, H.; Hu, Y.; Zhang, Y.; Weng, H.; Lu, J.; et al. Dioscin Induces Gallbladder Cancer Apoptosis by Inhibiting ROS-Mediated PI3K/AKT Signalling. Int. J. Biol. Sci. 2017, 13, 782–793. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Wang, R.; Zou, X. Dioscin Inhibits Gastric Tumor Growth through Regulating the Expression Level of LncRNA HOTAIR. BMC Compl. Altern. Med. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T. Targeted Cancer Therapy: The next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.J.; Alhadlaq, H.A.; Kumar, S.; Alrokayan, S.A.; Ahamed, M. Selective Cancer-Killing Ability of Metal-Based Nanoparticles: Implications for Cancer Therapy. Arch. Toxicol. 2015, 89, 1895–1907. [Google Scholar] [CrossRef]
- Zhang, W.; Lan, Y.; Huang, Q.; Hua, Z. Galangin Induces B16F10 Melanoma Cell Apoptosis via Mitochondrial Pathway and Sustained Activation of P38 MAPK. Cytotechnology 2013, 65, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.-Y.; Hsu, C.-S.; Wang, K.-T.; Wang, M.-C.; Wang, C.-C. Antitumor Effects of Osthol from Cnidium Monnieri: An in Vitro Andin Vivo Study. Phytother. Res. 2007, 21, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-L.; Wang, M.-C.; Chen, L.-G.; Wang, C.-C. Cytotoxic Activity of Coumarins from the Fruits of Cnidium Monnieri on Leukemia Cell Lines. Planta Med. 2003, 69, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Karolina Kordulewska, N.; Kostyra, E.; Matysiewicz, M.; Cieślińska, A.; Jarmołowska, B. Impact of Fexofenadine, Osthole and Histamine on Peripheral Blood Mononuclear Cell Proliferation and Cytokine Secretion. Eur. J. Pharmacol. 2015, 761, 254–261. [Google Scholar] [CrossRef]
- Jiang, G.; Liu, J.; Ren, B.; Tang, Y.; Owusu, L.; Li, M.; Zhang, J.; Liu, L.; Li, W. Anti-Tumor Effects of Osthole on Ovarian Cancer Cells in Vitro. J. Ethnopharmacol. 2016, 193, 368–376. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Attardi, L.D. The Role of Apoptosis in Cancer Development and Treatment Response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Berghe, T.V.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Taabazuing, C.Y.; Okondo, M.C.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Spall, S.K.; Anderton, H.; Masters, S.L.; et al. RIPK3 Promotes Cell Death and NLRP3 Inflammasome Activation in the Absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL Triggers the NLRP3 Inflammasome in a Cell-Intrinsic Manner. Proc. Natl. Acad Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.G.; Gale, M.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1β Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.V.; D’Herde, K.; Vandenabeele, P. Clearance of Apoptotic and Necrotic Cells and Its Immunological Consequences. Apoptosis 2006, 11, 1709–1726. [Google Scholar] [CrossRef]
- Krysko, O.; Aaes, T.L.; Kagan, V.E.; D’Herde, K.; Bachert, C.; Leybaert, L.; Vandenabeele, P.; Krysko, D.V. Necroptotic Cell Death in Anti-Cancer Therapy. Immunol. Rev. 2017, 280, 207–219. [Google Scholar] [CrossRef]
- Shen, C.; Pandey, A.; Man, S.M. Gasdermins Deliver a Deadly Punch to Cancer. Cell Res. 2020, 30, 463–464. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.-X.; Deng, R.-H.; Wang, H.; Liu, X.-H.; Wang, X.-N.; Qin, R.; Jin, X.; Lei, T.-R.; Zheng, D.; Zhou, P.-H.; et al. Epigenetics-Based Tumor Cells Pyroptosis for Enhancing the Immunological Effect of Chemotherapeutic Nanocarriers. Nano Lett. 2019, 19, 8049–8058. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [Green Version]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; da Silva, R.B.; e Sousa, C.R.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-KB Signaling in Dying Cells Determines Cross-Priming of CD8+ T Cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral Activation of the Necroptotic Pathway Components RIPK1 and RIPK3 Potentiates Antitumor Immunity. Sci. Immunol. 2019, 4, eaaw2004. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Huang, Y.; Zhu, Q.; Cheng, H.; Pei, Y.; Feng, J.; Xu, M.; Jiang, G.; Song, Q.; Jiang, T.; et al. Necroptotic Cancer Cells-Mimicry Nanovaccine Boosts Anti-Tumor Immunity with Tailored Immune-Stimulatory Modality. Biomaterials 2018, 164, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The Release and Activity of HMGB1 in Ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T Cells Regulate Tumour Ferroptosis during Cancer Immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with MRNA Coding for the Necroptosis Mediator MLKL Induces Antitumor Immunity Directed against Neo-Epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef] [Green Version]
- Philipp, S.; Sosna, J.; Adam, D. Cancer and Necroptosis: Friend or Foe? Cell. Mol. Life Sci. 2016, 73, 2183–2193. [Google Scholar] [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Ly, N.N.G.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The Necrosome Promotes Pancreatic Oncogenesis via CXCL1 and Mincle-Induced Immune Suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.; Vince, J.E. Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef]
- Jayakumar, A.; Bothwell, A.L.M. RIPK3-Induced Inflammation by I-MDSCs Promotes Intestinal Tumors. Cancer Res. 2019, 79, 1587–1599. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of ferroptotic cell death pathway. Glu: Glutamate; GSH: Glutathione; GSSG: Oxidized glutathione; GPX4: Glutathione peroxidase 4; LOH: Lipid alcohols; LOOH: Lipid hydroperoxides; Nrf2: Nuclear factor (erythroid-derived 2)-like 2; Trf1: Transferrin receptor 1; LIP: Labile iron pool; FTH1: Ferritin heavy chain 1; FTL: Ferritin light chain; NCOA4: Nuclear receptor coactivator.
Figure 1.
Schematic representation of ferroptotic cell death pathway. Glu: Glutamate; GSH: Glutathione; GSSG: Oxidized glutathione; GPX4: Glutathione peroxidase 4; LOH: Lipid alcohols; LOOH: Lipid hydroperoxides; Nrf2: Nuclear factor (erythroid-derived 2)-like 2; Trf1: Transferrin receptor 1; LIP: Labile iron pool; FTH1: Ferritin heavy chain 1; FTL: Ferritin light chain; NCOA4: Nuclear receptor coactivator.

Figure 2.
Schematic representation of necroptotic cell death pathway. cIAP1/2: Cellular inhibitors apoptosis proteins; CYL: Cylindromatosis; FADD: Fas-associated death domain; MLKL: Mixed lineage kinase domain-like; Nf-kB: Nuclear factor kappa-light-chain-enhancer of activated B cells; RIP1: Receptor-interacting protein 1; RIP3: Receptor-interacting protein 3; TNFα: Tumor necrosis factor alfa; TNFR1: Tumor necrosis factor receptor 1; TRADD: TNF Receptor-associated death domain; TRAF1/2: TNFR-associated factors 1/2.
Figure 2.
Schematic representation of necroptotic cell death pathway. cIAP1/2: Cellular inhibitors apoptosis proteins; CYL: Cylindromatosis; FADD: Fas-associated death domain; MLKL: Mixed lineage kinase domain-like; Nf-kB: Nuclear factor kappa-light-chain-enhancer of activated B cells; RIP1: Receptor-interacting protein 1; RIP3: Receptor-interacting protein 3; TNFα: Tumor necrosis factor alfa; TNFR1: Tumor necrosis factor receptor 1; TRADD: TNF Receptor-associated death domain; TRAF1/2: TNFR-associated factors 1/2.

Figure 3.
Schematic representation of canonical, non-canonical, and caspase-3-dependent pyroptotic cell death pathway. AIM2: Absent in melanoma 2; ASC: Apoptosis-associated speck-like protein containing a CARD (caspase activation and recruitment domain); DAMPs: Damage-associated molecular patterns; GSDMD: Gasdermin D; GSDMD-NT: N-terminal fragment of GSDMD; GSDME: Gasdermin E; GSDME-NT: N-terminal fragment of GSDME; HAMPs: Homeostasis-altering molecular processes; IL-1β: Interleukin-1 beta; IL-18: Interleukin-18; LPS: Lypopolisaccaride; NLRC4: NLR (nucleotide-binding oligomerization domain (NOD)-like receptor) family CARD domain-containing protein 4; NLRP3: NLR family pyrin domain-containing 3; NLRP1: NLR family pyrin domain-containing 1; PAMPs: Pathogen-associated molecular patterns.
Figure 3.
Schematic representation of canonical, non-canonical, and caspase-3-dependent pyroptotic cell death pathway. AIM2: Absent in melanoma 2; ASC: Apoptosis-associated speck-like protein containing a CARD (caspase activation and recruitment domain); DAMPs: Damage-associated molecular patterns; GSDMD: Gasdermin D; GSDMD-NT: N-terminal fragment of GSDMD; GSDME: Gasdermin E; GSDME-NT: N-terminal fragment of GSDME; HAMPs: Homeostasis-altering molecular processes; IL-1β: Interleukin-1 beta; IL-18: Interleukin-18; LPS: Lypopolisaccaride; NLRC4: NLR (nucleotide-binding oligomerization domain (NOD)-like receptor) family CARD domain-containing protein 4; NLRP3: NLR family pyrin domain-containing 3; NLRP1: NLR family pyrin domain-containing 1; PAMPs: Pathogen-associated molecular patterns.


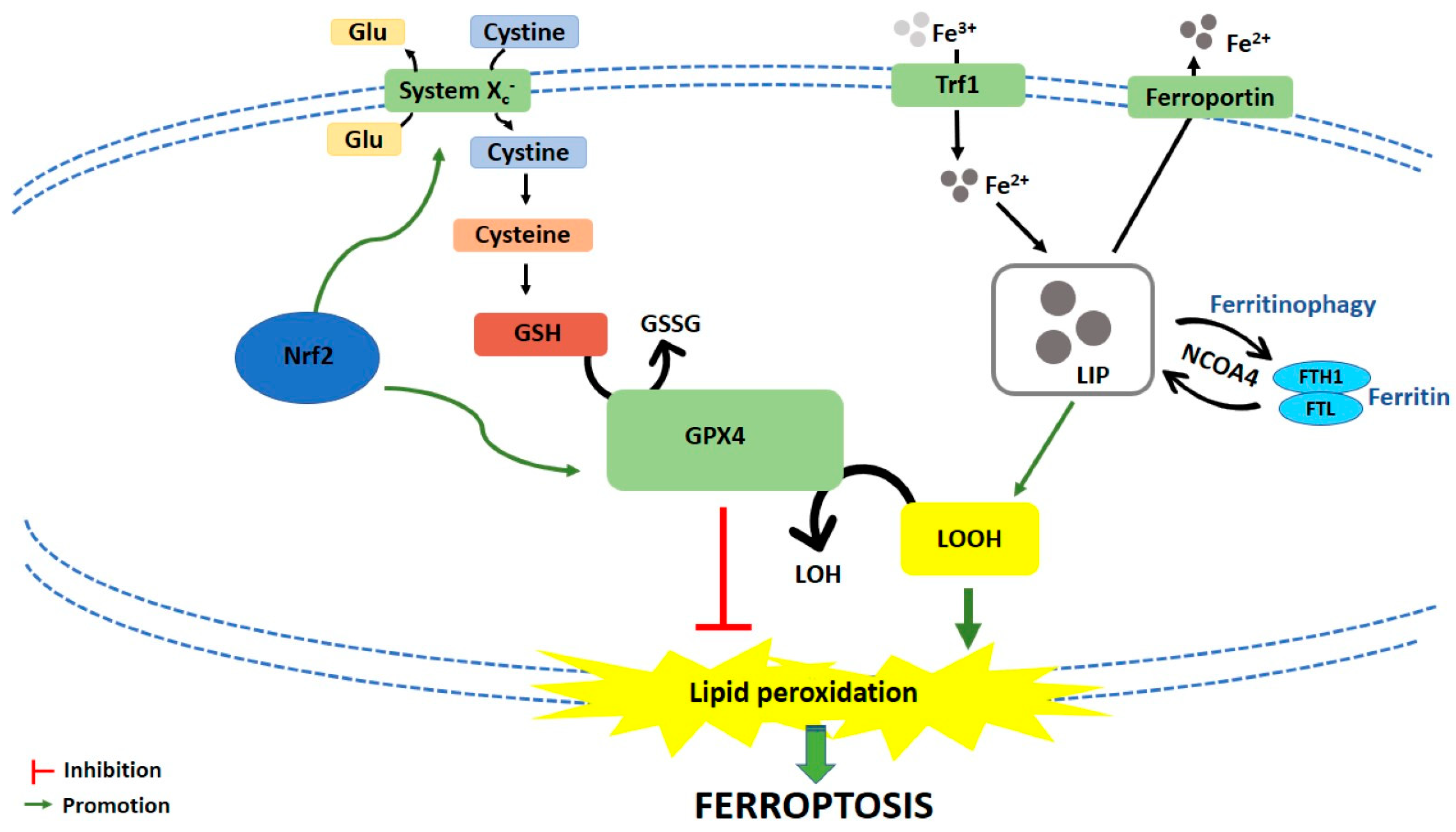{kind=link}
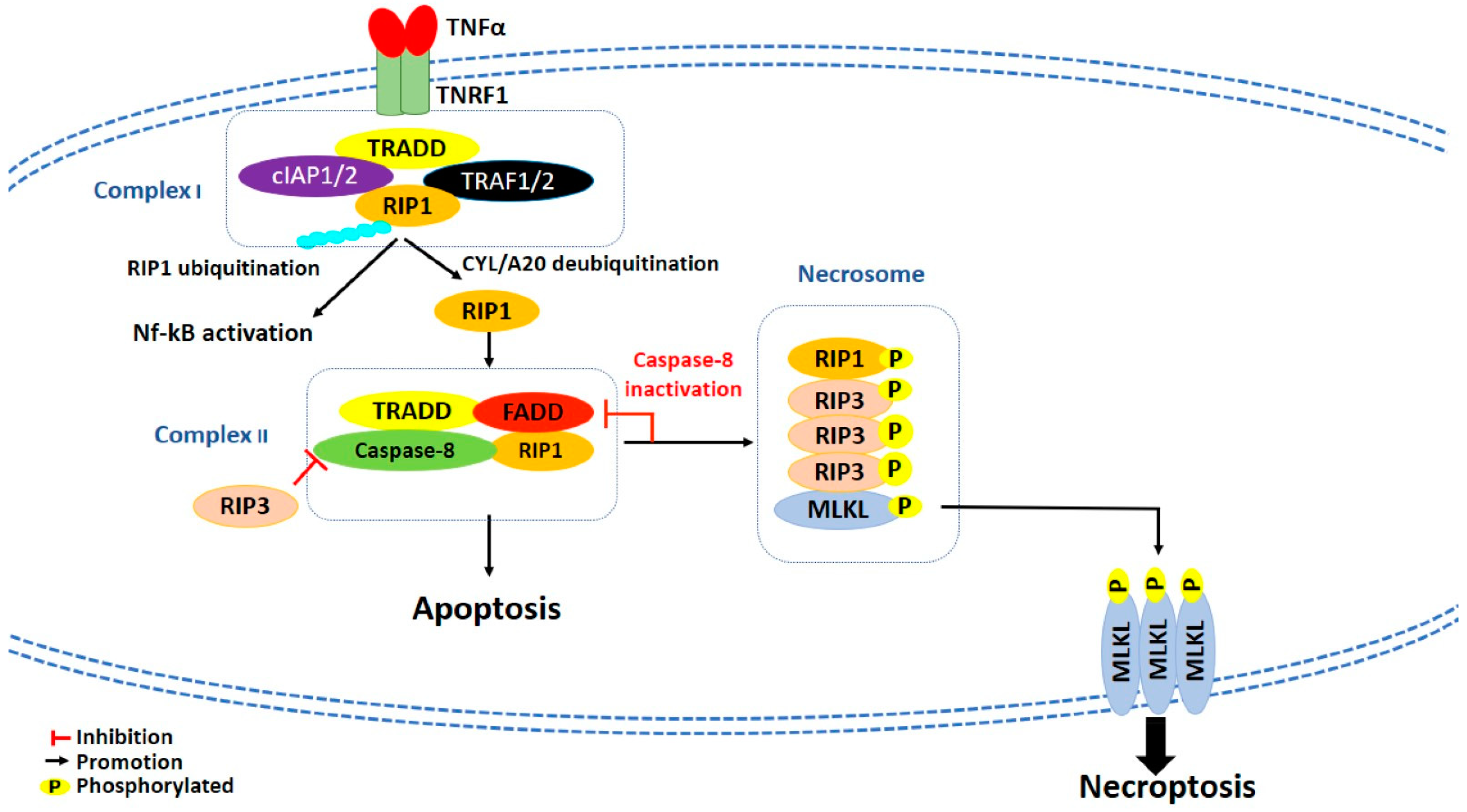{kind=link}
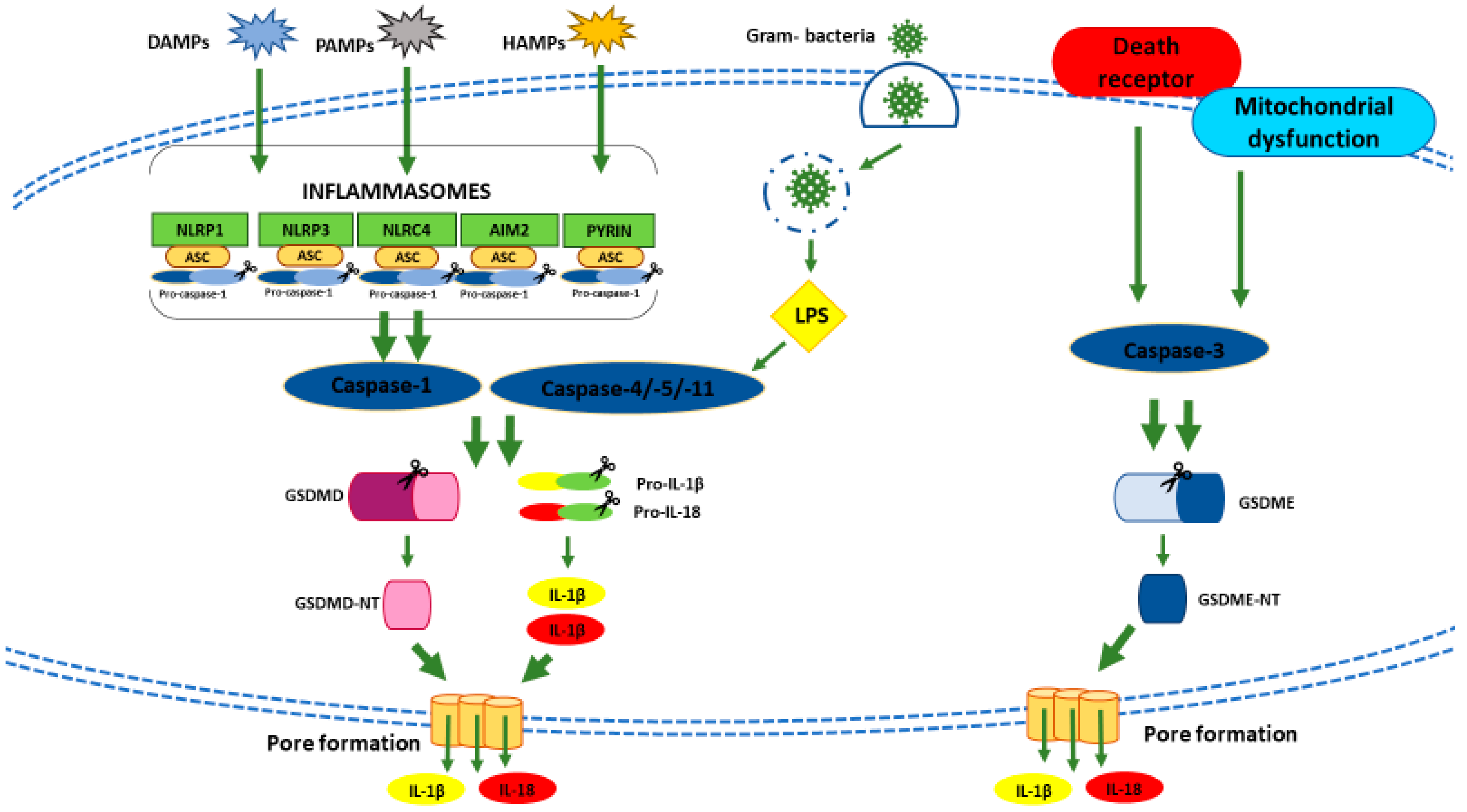{kind=link}
Table 1.
Natural products as in vitro inducers of ferroptosis.
| Compound | Compound Source | Cell Line(s) | Concentrations (Where Specified) | Time (Where Specified) | Ferroptosis Markers | Supplementary Effects | Reference |
|---|---|---|---|---|---|---|---|
| Actinia chinensis (Planch), drug-containing rat serum | Actinia chinensis Planch | HGC-27 | 90, 180 and 360 mg/mL | 48 h | ↓ Cell proliferation | [25] | |
| 24 and 48 h | ↓ Cell migration | ||||||
| 180 mg/mL | 48 h | ↑ ROS | ↓ after Ferr-1 treatment | ||||
| 90, 180 and 360 mg/mL | ↓ GPX4 | ||||||
| ↓ xCT | |||||||
| Albiziabioside A | Albizia inundata Mart. | MCF-7 | 10 μM | 24 h | ↑ Cytotoxicity | ↑ after Fe2+ treatment | [26] |
| ↓ after Ferr-1 treatment | |||||||
| ↓ after DFO treatment | |||||||
| ↓ after vitamin E treatment | |||||||
| / | ↑ ROS | ||||||
| 24 h | ↓ GSH/GSSG ratio | ||||||
| 48 h | ↓ GPX4 protein expression | ||||||
| / | ↑ MDA | ||||||
| ↑ Lipid peroxides | |||||||
| Amentoflavone | Selaginella spp. and other plants | U251, U373 | 10 and 20 μM | / | ↑ Fe2+ | [27] | |
| ↓ FTH | ↑ after ATG7 knockdown | ||||||
| ↑ MDA | ↓ after FTH overexpression | ||||||
| ↑ Lipid ROS | ↓ after FTH overexpression | ||||||
| ↓ after BafA1 treatment | |||||||
| ↓ after ATG7 knockdown | |||||||
| ↓ GSH | ↓ after FTH overexpression | ||||||
| ↓ after BafA1 treatment | |||||||
| ↓ after ATG7 knockdown | |||||||
| 20 μM | ↑ Cell death ratio (%) | ↓ after Ferr-1 treatment | |||||
| ↓ after DFO treatment | |||||||
| ↓ after FTH overexpression | |||||||
| ↓ after BafA1 treatment | |||||||
| ↓ after ATG7 knockdown | |||||||
| Ardisiacrispin B | Ardisia kivuensis Taton | CCRF-CEM | 0.59, 0.93, 2.33, 4.66, 9.32, 18.64 and 37.28 μM | 24 h | ↑ Cytotoxicity | ↓ after Ferr-1 treatment | [28] |
| ↓ after DFO treatment | |||||||
| 0.3, 0.6, 1.2 and 2.4 μM | ↑ ROS | ||||||
| Aridanin | Tetrapleura tetraptera (Schum. & Thonn) Taub. | CCRF-CEM | 1, 2, 4, 8, 15, 30 and 61 μM | 24 h | ↓ Cell viability | ↓ after Ferr-1 treatment | [29] |
| ↓ after DFO treatment | |||||||
| Artenimol (artemisinin semi-syntethic derivative) | Artemisia annua L. | CCRF-CEM | 0.01, 0.1, 1, 10 and 100 μM | / | ↓ Cell viability | ↓ after Ferr-1 treatment | [30] |
| ↓ after DFO treatment | |||||||
| Artesunate (artemisin semi-synthetic derivative) | Artemisia annua L. | DAUDI, CA-46 | 4 and 20 μM | 48 h | ↓ Cell viability | ↑ after DFO treatment | [31] |
| ↑ after Ferr-1 treatment | |||||||
| ↑ after Lip-1 treatment | |||||||
| ↑ after down-regulation of CHAC1 expression | |||||||
| 5, 10 and 20 μM | 24 and 48 h | ↑ ROS | |||||
| ↑ Lipid peroxidation | ↓ after down-regulation of CHAC-1 expression | ||||||
| 5, 10 and 20 μM | 24 h | ↑ CHAC1, ↑ ATF4, ↑ CHOP protein expression | |||||
| MT-2 | 50 μM | 24 h | ↑ ROS | [32] | |||
| 0.4, 2 and 10 μM | ↑ Cytotoxicity | ↓ after DFO treatment | |||||
| 2 and 10 μM | ↓ after Ferr-1 treatment | ||||||
| HUT-102 | 50 μM | 24 h | ↑ ROS | ↓ after NAC treatment | |||
| 2 and 10 μM | ↑ Cytotoxicity | ↓ after DFO treatment | |||||
| 10 and 50 μM | ↓ after Ferr-1 treatment | ||||||
| HN9 | 50 μM | 72 h | ↓ Cell viability | ↓ after HTF treatment | [33] | ||
| ↑ after DFO treatment | |||||||
| ↑ after Trolox treatment | |||||||
| 2.5 and 5 μM | ↑ after Keap1 knockdown | ||||||
| ↑ after Nrf2 knockdown | |||||||
| 50 μM | 24 h | ↑ ROS | ↓ after Ferr-1 treatment | ||||
| ↓ after Trolox treatment | |||||||
| ↑ Lipid ROS | ↓ after Ferr-1 treatment | ||||||
| ↓ after Trolox treatment | |||||||
| HN9, HN9-cisR | 10, 25 and 50 μM | 24 h | ↑ Nrf2 protein expression | ||||
| ↓ xCT, ↓ RAD51, ↓ Keap1 protein expression | |||||||
| HN9-cisR, HN3-cisR, HN4-cisR | 10, 25 and 50 μM | 24 h | ↑ Nrf2, ↑ HO-1, ↑ NQO1 protein expression | ||||
| ↓ Keap1 protein expression | |||||||
| 50 μM | ↑ Nrf2, ↑ HO-1, ↑ NQO1 mRNA levels | ||||||
| HN3-cisR | 25 and 50 μM | 24 h | ↓ GSH | ↓ after trigonellin treatment | |||
| ↑ after Trolox treatment | |||||||
| ↑ after Nrf2 knockdown | |||||||
| ↑ ROS | ↓ after Trolox treatment | ||||||
| ↑ after Nrf2 knockdown | |||||||
| ↑ Lipid ROS | ↓ after trigonellin treatment | ||||||
| ↓ Cell viability | ↓ after Nrf2 knockdown | ||||||
| ↓ after HO-1 knockdown | |||||||
| ↑ after Trolox treatment | |||||||
| PaTU8988, AsPC-1 | 20 μM | 24 h | ↓ Cell viability | ↑ after Ferr-1 treatment | [34] | ||
| ↑ after GRP78 overexpression | |||||||
| ↓ after GRP78 knockdown | |||||||
| ↑ MDA | ↓ after DFO treatment | ||||||
| ↓ after Ferr-1 treatment | |||||||
| ↓ after GRP78 overexpression | |||||||
| ↑ after GRP78 knockdown | |||||||
| ↑ Lipid peroxidation | ↓ after Ferr-1 treatment | ||||||
| 10, 20 and 40 μM | ↑ GRP78 mRNA levels | ||||||
| ↑ GRP78 protein expression | |||||||
| HEY1 | 25 and 50 μM | 48 h | ↑ Cell death | ↓ after Ferr-1 treatment | [35] | ||
| ↓ after DFO treatment | |||||||
| ↑ after HT treatment | |||||||
| HEY2 | 100 μM | ↑ Cell death | ↓ after Ferr-1 treatment | ||||
| HEY2, SKOV3 | 50 and 100 μM | ↑ Cell death | ↓ after DFO treatment | ||||
| ↑ after HT treatment | |||||||
| HEY1, HEY2, SKOV-3 | 10, 25, 50 and 100 μM | 24 h | ↑ ROS | ↓ after GSH treatment | |||
| HEY1, HEY2, SKOV-3, OVCAR8, TOV-112D, TOV-21G | 25, 50 and 100 μM | 48 h | ↑ Cell death | ↓ after GSH treatment | |||
| Panc-1 | 50 μM | 24 h | ↑ ROS | ↓ after Trolox treatment | [36] | ||
| ↓ Colony formation | ↑ after DFO treatment | ||||||
| ↑ after Trolox treatment | |||||||
| ↑ after Ferr-1 treatment | |||||||
| ↓ after HTF treatment | |||||||
| ↑ HO-1 protein expression | |||||||
| ↑ Lipid peroxidation | ↓ after Trolox treatment | ||||||
| ↓ after Ferr-1 treatment | |||||||
| Panc-1, COLO-357 | 48 h | ↑ Cell death | ↓ after Ferr-1 treatment | ||||
| BxPC-3, Panc-1 | 24 and 48 h | ↑ Cell death | ↓ after DFO treatment | ||||
| BxPC-3, Panc-1, AsPC-1 | ↑ Cell death | ↑ after HTF treatment | |||||
| Betula etnensis Raf. methanolic extract | Betula etnensis Raf. | CaCo2 | 5, 50, 250 and 500 μg/mL | 72 h | ↓ Cell viability | [37] | |
| 5, 50 and 250 μg/mL | ↑ LDH release | ||||||
| ↑ ROS | |||||||
| ↑ LOOH | |||||||
| ↓ RSH | |||||||
| 5 and 50 μg/mL | ↓ HO-1 levels | ||||||
| 250 μg/mL | ↑ HO-1 levels | ||||||
| D13 (albiziabioside A derivative) | Albizia inundata Mart. | HCT116 | 0.31, 1.25 and 5 μM | / | ↑ Cytotoxicity | ↓ after Ferr-1 treatment | [38] |
| ↓ after DFO treatment | |||||||
| ↑ after Fe2+ treatment | |||||||
| ↑ after Fe3+ treatment | |||||||
| 48 h | ↓ GPX4 protein expression | ||||||
| / | ↑ MDA | ||||||
| Dihydroartemisinin (artemisin semi-synthetic derivative) | Artemisia annua L. | THP-1 | 5, 10 and 15 μM | 12 h | ↓ Cell viability | [39] | |
| ↑ ROS | |||||||
| HL-60 | 5, 10 and 15 μM | 12 h | ↓ Cell viability | ↑ after Ferr-1 treatment | |||
| ↑ after DFO treatment | |||||||
| ↑ after NAC treatment | |||||||
| ↑ after BafA1 treatment | |||||||
| ↑ after 3-MA treatment | |||||||
| ↑ after ATG7 knockdown | |||||||
| ↑ after FTH overexpression | |||||||
| ↑ after ISCU overexpression | |||||||
| ↑ Lipid ROS | ↓ after ATG7 knockdown | ||||||
| ↓ after FTH overexpression | |||||||
| ↓ GSH | ↑ after ISCU overexpression | ||||||
| ↑ ROS | ↓ after DFO treatment | ||||||
| ↓ after NAC treatment | |||||||
| ↓ after ISCU overexpression | |||||||
| ↑ IRP2 protein expression | |||||||
| ↓ FTH, ↓ GPX4 protein expression | ↑ after ISCU overexpression | ||||||
| ↑ after BafA1 treatment | |||||||
| G0101, G0107 | 10, 20, 40, 80 and 160 μM | 24 h | ↑ ROS | [40] | |||
| 20, 40, 80 and 160 μM | ↑ Lipid ROS | ||||||
| ↑ MDA | |||||||
| ↓ GSH | |||||||
| ↑ GSSG | |||||||
| ↑ Cell death | ↓ after DFO treatment | ||||||
| ↓ after Ferr-1 treatment | |||||||
| ↓ after Lip-1 treatment | |||||||
| U251 U373 | 5, 10, 20 and 40 μM 20, 40, 80 and 160 μM | 24 h | ↓ GSH | ||||
| U251 U373 | 2.5, 5, 10, 20 and 40 μM 10, 20, 40, 80 and 160 μM | 24 and 48 h | ↑ ROS | ↓ after DFO treatment | |||
| ↑ after PERKi treatment | |||||||
| ↑ after ATF4 siRNA treatment | |||||||
| ↑ after HSPA5 siRNA treatment | |||||||
| U251 U373 | 2.5, 5, 10 and 20 μM 10, 20, 40, 80 and 160 μM | 3, 6, 12, 24 and 48 h | ↑ Lipid ROS | ↑ after ATF4 siRNA treatment | |||
| ↑ after HSPA5 siRNA treatment | |||||||
| U251 U373 | 5, 10, 20 and 40 μM 80 μM | 3, 6, 12, 24 and 48 h | ↑ MDA | ↑ after PERKi treatment | |||
| ↑ after ATF4 siRNA treatment | |||||||
| ↑ after HSPA5 siRNA treatment | |||||||
| U251 U373 | 10, 20 and 40 μM 40, 80 and 160 μM | 48 h | ↑ Cell death | ↓ after DFO treatment | |||
| ↓ after Ferr-1 treatment | |||||||
| ↓ after Lip-1 treatment | |||||||
| ↑ after PERKi treatment | |||||||
| ↑ after ATF4 siRNA treatment | |||||||
| ↑ after HSA5 siRNA treatment | |||||||
| Dihydroisotanshinone I | Salvia miltiorrhiza Bunge | MCF-7 | 5 and 10 μM | 24 h | ↓ GPX4 activity | [41] | |
| MCF-7, MDA-MB231 | 10 μM | ↓ GPX4 protein expression | |||||
| 5 and 10 μM | ↑ MDA | ||||||
| 10 μM | ↓ GSH/GSSG ratio | ||||||
| Epunctanone | Garcinia epunctata Stapf. | CCRF-CEM | 1.04, 1.66, 4.14, 8.28, 16.56, 33.11 and 66.23 μM | 24 h | ↑ Cytotoxicity | ↓ after Ferr-1 treatment | [42] |
| ↓ after DFO treatment | |||||||
| 2.95, 5.91, 11.81 and 23.63 μM | ↑ ROS | ||||||
| Erianin | Dendrobium chrysotoxum Lindl | H460, H1299 | 50 and 100 nM | 24 h | ↑ Cell death | ↓ after NAC treatment | [43] |
| ↓ after Ferr-1 treatment | |||||||
| ↓ after Lip-1 treatment | |||||||
| ↓ after GSH treatment | |||||||
| 50 and 100 nM | / | ↑ ROS | |||||
| ↓ GSH | |||||||
| 12.5, 25, 50 and 100 nM | ↑ MDA | ||||||
| ↑ HO-1, ↑ transferrin protein expression | |||||||
| ↓ GPX4, ↓ CHAC2, ↓ SLC40A1, ↓ SLC7A11 protein expression | |||||||
| 5, 10 and 25 μM | 24 h | ↑ Ca2+ levels | |||||
| ↑ Calmodulin protein expression | |||||||
| Ferroptocide (pleuromutilin semi-syntetic derivative) | Pleurotus passeckerianus; Drosophila subatrata; Clitopilus scyphoides, and others spp. | ES-2 | 5, 10 and 25 μM | 1 h | ↑ ROS | ↓ after DFO treatment | [44] |
| 10 and 25 μM | ↑ Mitochondrial ROS | ||||||
| 10 μM | ↑ Lipid ROS | ↓ after DFO treatment | |||||
| 5, 10 and 25 μM | 14 h | ↑ Cell death | ↓ after Ferr-1 treatment | ||||
| ↓ after DFO treatment | |||||||
| ↓ after Trolox treatment | |||||||
| HCT116 | 5, 10 and 25 μM | 10, 24 and 48 h | ↑ Cell death | ↓ after DFO treatment | |||
| ↓ after Trolox treatment | |||||||
| ↓ after NAC-1 treatment | |||||||
| ↑ after TXN knockdown | |||||||
| ↓ after Ferr-1 treatment | |||||||
| 5, 10 and 25 μM | 1.5 and 72 h | ↑ ROS | ↑ after TXN knockdown | ||||
| 10 μM | 2 and 72 h | ↑ Lipid ROS | ↑ after TXN knockdown | ||||
| ↓ after DFO treatment | |||||||
| 4T1 | 5, 10 and 25 μM | 18 h | ↑ Cell death | ↓ after DFO treatment | |||
| ↓ after Ferr-1 treatment | |||||||
| 10 μM | 2 h | ↑ Lipid ROS | ↓ after Ferr-1 treatment | ||||
| ↓ after DFO treatment | |||||||
| HT-29 | 5, 10 and 25 μM | 12 h | ↑ Cell death | ↓ after DFO treatment | |||
| ↓ after Trolox treatment | |||||||
| ↓ after NAC treatment | |||||||
| ↓ after Ferr-1 treatment | |||||||
| Gallic Acid | Natural polyhydroxy phenolic compound, found in various foods | HeLa | 50 μg/mL | 12 h | ↑ Lipid peroxidation | [45] | |
| HeLa, H446, SHSY-5Y | 36 h | ↑ Cell death | ↓ after DFO treatment | ||||
| A375, MDA-MB-231 | 10, 25, 50, 100 and 200 μg/mL | 24 h | ↓ Cell viability | [46] | |||
| MDA-MB-231 | 25 μg/mL | ↑ ROS | |||||
| A375 | 50 μg/mL | ||||||
| MDA-MB-231 | / | / | ↓ GPX4 activity | ||||
| A375, MDA-MB-231 | / | / | ↑ MDA | ||||
| Physcion 8-O-β-glucopyranoside | Rumex japonicus Houtt. | MGC-803, MKN-45 | 10, 20, 30, 40 and 50 μM | 24, 48, 72 and 96 h | ↓ Cell viability | ↑ after Ferr-1 treatment | [47] |
| ↑ after GPNA treatment | |||||||
| ↑ after 968 treatment | |||||||
| ↓ after GLS2 knockdown | |||||||
| / | / | ↓ Cell proliferation | ↑ after miR-103a-3p overexpression | ||||
| / | 24 h | ↓ Cell invasion | ↑ after miR-103a-3p overexpression | ||||
| ↓ Cell migration | ↑ after miR-103a-3p overexpression | ||||||
| / | / | ↑ Lipid ROS | ↓ after GPNA treatment | ||||
| ↓ after 968 treatment | |||||||
| ↓ after GLS2 knockdown | |||||||
| ↓ after miR-103a-3p overexpression | |||||||
| ↑ MDA | ↓ after GPNA treatment | ||||||
| ↓ after 968 treatment | |||||||
| ↓ after GLS2 knockdown | |||||||
| ↓ after miR-103a-3p overexpression | |||||||
| ↑ Fe2+ | ↓ after GPNA treatment | ||||||
| ↓ after 968 treatment | |||||||
| ↓ after GLS2 knockdown | |||||||
| ↓ after miR-103a-3p overexpression | |||||||
| ↓ miR-103a-3p expression | |||||||
| ↑ GLS2 protein levels | ↓ after miR-103-3p transfection | ||||||
| Piperlongumine | Piper Longum L. | Panc-1 | 4, 6, 8, 10, 12 and 14 μM | 16 h | ↓ Cell viability | ↑ after NAC treatment | [48] |
| ↑ after Ferr-1 treatment | |||||||
| ↑ after Lip-1 treatment | |||||||
| ↑ after DFO treatment | |||||||
| MIAPaCa-2 | 10 μM | 16 h | ↓ Cell viability | ↑ after CPX treatment | |||
| ↑ after PD146176 treatment | |||||||
| 4 h | ↓ GSH | ||||||
| Progenin III | Raphia vinifera P. Beauv | CCRF-CEM | 2, 3, 7, 14 and 55 μM | 24 h | ↓ Cell viability | ↑ after Ferr-1 treatment | [49] |
| ↑ after DFO treatment | |||||||
| 1.59 and 3.18 μM | ↑ ROS | ||||||
| Ruscogenin | Ruscus aculeatus L. Radix Ophiopogon japonicas (Thunb.) Ker Gawl. | BxPC-3, SW1990 | 7 μM | / | ↑ Cell death | ↑ after FAC treatment | [50] |
| ↓ after DFO treatment | |||||||
| / | 6 h | ↑ Cell death | ↓ after transferrin knockdown | ||||
| ↓ after ferroportin overexpression | |||||||
| 3 and 7 μM | 12 and 24 h | ↑ Fe2+ | ↓ after DFO treatment | ||||
| 1, 2, 4, 6 and 24 h | ↑ ROS | ↓ after DFO treatment | |||||
| 6 and 12 μM | 24 h | ↑ Transferrin | |||||
| ↓ Ferroportin | |||||||
| Solasonine | Solanum melongena L. | HepG2 | 15 ng/mL | 24 h | ↑ Cell death | ↓ after Ferr-1 treatment | [51] |
| ↓ after DFO treatment | |||||||
| ↑ Lipid ROS | ↓ after Ferr-1 treatment | ||||||
| ↓ after DFO treatment | |||||||
| ↓ GSS, ↓ GPX4 mRNA levels | |||||||
| ↓ GSS, ↓ GPX4 protein expression | |||||||
| Typhaneoside | Pollen Typhae | Kas-1, HL-60, NB4 | 40 μM | 24 h | ↓ Cell viability | ↑ after Ferr-1 treatment | [52] |
| ↑ after DFO treatment | |||||||
| ↑ after 3-MA treatment | |||||||
| ↑ after BafA1 treatment | |||||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ after rapamycin treatment | |||||||
| ↑ after ATG7 knockdown | |||||||
| 20, 30 and 40 μM | ↑ ROS | ↓ after DFO treatment | |||||
| ↓ after NAC treatment | |||||||
| ↓ GSH | |||||||
| ↑ Lipid ROS | ↓ after ATG7 knockdown | ||||||
| ↓ after BafA1 treatment | |||||||
| ↓ GPX4, ↓ FTH mRNA levels | |||||||
| ↑ IRP2 mRNA levels | |||||||
| Ungeremine | Crinum zeylanicum L. | CCRF-CEM | 2.37, 3.76, 9.40, 18.79, 37.58, 75.17 and 150.33 μM | 24 h | ↓ Cell proliferation | ↑ after Ferr-1 treatment | [53] |
| ↑ after DFO treatment | |||||||
| 1.22, 2.45, 4.89 and 9.78 μM | ↑ ROS | ||||||
| Whitaferin A | Withania somnifera (L.) Dunal | IMR-32 | / | / | ↑ ROS | [54] | |
| 1 and 10 μM | 2, 4, 8, 12 and 24 h | ↓ GPX4 expression | |||||
| 10 μM | 3 and 5 h | ↓ GPX4 activity | |||||
| / | / | ↑ Lipid peroxidation | ↓ after DFO treatment | ||||
| 1 μM | 4, 8 and 12 h | ↑ Fe2+ | ↑ after hemin treatment | ||||
| 1, 2, 4, 8, 12 and 24 h | ↑ HO-1, ↑ Keap1, ↑ Nrf2 protein expression | ||||||
| 6, 8, 12 and 16 h | ↑ Cell death | ↓ after GPX4 overexpression | |||||
| ↓ after ZnPP treatment | |||||||
| ↓ HO-1 knockdown | |||||||
| ↑ after hemin treatment | |||||||
| IMR-32, SK-N-SH | 1 and 10 μM | 6, 8, 12 and 16 h | ↑ Cell death | ↓ after Ferr-1 treatment | |||
| ↓ after CPX treatment | |||||||
| ↓ after α-tocopherol treatment | |||||||
| ↓ after UOI26 treatment | |||||||
| ↓ after Flt3 inhibitor treatment | |||||||
| 1 μM | / | Nrf2 pathway activation | |||||
| / | ↑ FTH1, ↑ HO-1 gene expression | ||||||
| 1, 2, 4, 8, 12 and 24 h | ↑ FTH1, ↑ HO-1 mRNA levels | ||||||
| WA-NPs | Withania somnifera L. Dunal | IMR-32 | 1 and 10 μM | 8, 10, 12, 16, 20 and 24 h | ↑ Cell death | [54] |
Abbreviations: ↑: Increase; ↓: Decrease; 3-MA:3-methyladenine; 968: Compound 968, GLS2 inhibitor;; ATF4: Activating transcription factor 4; ATG7: Autophagy related 7; BafA1: Bafilomycin 1; CHAC1: Glutathione-specific Gamma-glutamylcyclotransferase 1; CHOP: CCAAT/enhancer-binding protein homologous protein; CPX: Ciclopirox, intracellular iron chelator; DFO: Deferoxamine; FAC: Ferric ammonium citrate; Fe2+: Ferrous ion; Fe3+: Ferric ion; Ferr-1: Ferrostatin-1; Flt3: Receptor tyrosine kinase fms-like tyrosine kinase 3; FTH: Ferritin heavy chain; FTH1: Ferritin heavy chain 1; GLS2: Glutaminase 2; GPNA: Glutamine transporter inhibitor; GPX4: Glutathione peroxidase IV; GSH: Glutathione; GSS: Glutathione synthetase; GSSG: Oxidized glutathione; HO-1: Heme oxygenase 1; HN3-cisR: Cisplatin-resistant HN3 cells; HN4-cisR: Cisplatin-resistant HN4 cells; HN9-cisR: Cisplatin-resistant HN9 cells; HSPA5: Heat shock protein family A (Hsp70) member 5; HTF: Holo-transferrin; IRP2: Iron regulator protein 2; ISCU: Iron-sulfur cluster assembly enzyme; Keap1: Kelch-like ECH-associated protein 1; Lip-1: Liproxstatin-1; MDA: Malondialdehyde; NAC: N-acetylcysteine; NQO1: NAD(P)H quinone dehydrogenase 1; Nrf2: Nuclear factor erythroid 2–related factor 2; PD146176: Lypoxygenase inhibitor; PERKi: PERK inhibitor I (GSK2606414); ROS: Reactive oxygen species; RSH: Thiols; Spp.: Species; TXN: Thioredoxin; WA-NPs: Whitaferin A nanoparticles; xCT: Cystine/glutamate antiporter; ZnPP: Zinc protoporphyrin, HO-1 inhibitor; Z-VAD-FMK: Pan-caspase inhibitor.
Table 2.
Natural compounds as in vitro inducers of necroptosis.
| Compound | Compound Source | Cell Line(s) | Concentrations (Where Specified) | Time (Where Specified) | Necroptosis Markers | Supplementary Effects | Reference |
|---|---|---|---|---|---|---|---|
| 2-methoxy-6-acetyl-7-methyljuglone | Polygonum cuspidatum Sieb. et Zucc. | A549 | 5 μM | 24 h | Intact nuclear envelope | [110] | |
| Mitochondrial swelling | |||||||
| Loss of mitochondrial matrix | |||||||
| Cytoplasm vacuolization | |||||||
| 2.5, 5 and 7.5 μM | ↑ LDH release | ||||||
| 2.5, 5 and 7.5 μM | 16 h | No caspase-3/-7 activation | |||||
| 5 and 7.5 μM | 24 h | ↓ Cell viability | ↑ after Nec-1 treatment | ||||
| ↑ after SP600125 treatment | |||||||
| ↑ after JNK knockdown | |||||||
| ↑ after NAC treatment | |||||||
| ↑ after GSH treatment | |||||||
| ↑ after CAT treatment | |||||||
| ↑ after DTT treatment | |||||||
| ↑ after Hgb treatment | |||||||
| ↑ after iNOS knockdown | |||||||
| 7.5 μM | 1, 3, 6 and 16 h | ↑ p-JNK protein expression | ↓ after SP600125 treatment | ||||
| ↓ after CAT treatment | |||||||
| ↑ p-P38 protein expression | ↓ after SB203580 treatment | ||||||
| / | 1, 2 and 4 h | ↑ ROS | ↓ after NAC treatment | ||||
| ↓ after GSH treatment | |||||||
| 7.5 μM | 1 h | ↓ GSH/GSSG ratio | ↑ after GSH treatment | ||||
| 1, 2 and 4 h | ↑ H2O2 | ||||||
| 2 and 4 h | ↑ NO | ↓ after SP600125 treatment | |||||
| ↑ after JNK knockdown | |||||||
| ↑ HROS | |||||||
| ↑ O2− | |||||||
| 7.5 μM | 1, 3, 6, 16 and 24 h | ↑ iNOS protein expression | |||||
| 5 and 7.5 μM | 6 h | ↑ NOS activity | ↓ after L-NMMA treatment | ||||
| 2.5, 5 and 7.5 μM | 24 h | ↓ p-IκBα, ↓ NF-kB protein expression | |||||
| 10 μM | 1, 2 and 4 h | ↑ Lipid peroxidation | |||||
| A549 | 7.5 μM | 8 h | Swollen mitochondria | [111] | |||
| Damaged cell membrane | |||||||
| A549 H1299 | 7.5 μM 2.5 μM | 1, 2, 4 and 8 h | No caspase-3/-8/-9 cleavage | ||||
| No PARP cleavage | |||||||
| ↑ p-RIP1, ↑ p-RIP3, ↑ p-MLKL protein expression | |||||||
| A549 H1299 | / | 8 h 4 h | RIP1-RIP3 interaction | ↓ after Nec-1s treatment | |||
| A549 H1299 | 7.5 μM 2.5 μM | 1 h | ↑ ROS | ↓ after Nec-1s treatment | |||
| A549 H1299 | 7.5 μM 2.5 μM | 4 h 2 and 4 h | ↑ Ca2+ | ↓ after Nec-1s treatment | |||
| ↓ after SP600125 treatment | |||||||
| ↓ after BAPTA-AM treatment | |||||||
| ↓ after CAT treatment | |||||||
| A549 H1299 | 7.5 μM 2.5 μM | 4 h | ↑ JNK1/2, ↑ p-JNK1/2 protein expression | ↓ after Nec-1s treatment | |||
| ↓ after SP600125 treatment | |||||||
| ↓ after BAPTA-AM treatment | |||||||
| ↓ after CAT treatment | |||||||
| A549 H1299 | 7.5 μM 2.5 μM | 1 and 2 h | Lysosomal membrane permeabilization | ↓ after Nec-1s treatment | |||
| ↓ after SP600125 treatment | |||||||
| ↓ after BAPTA-AM treatment | |||||||
| ↓ after CAT treatment | |||||||
| ↓ after Hgb treatment | |||||||
| A549 | 7.5 μM | 4 h | ↑ Mitochondrial ROS | ↓ after Nec-1s treatment | |||
| ↓ after SP600125 treatment | |||||||
| ↓ after BAPTA-AM treatment | |||||||
| ↓ after CAT treatment | |||||||
| ↓ after MnSOD overexpression | |||||||
| H1299 | 2.5 μM | 4 h | ↑ Mitochondrial ROS | ↓ after Nec-1s treatment | |||
| ↓ after SP600125 treatment | |||||||
| ↓ after BAPTA-AM treatment | |||||||
| ↓ after CAT treatment | |||||||
| A549 H1299 | 7.5 μM 2.5 μM | 4 h | ↓ ΔΨm | ↑ after Nec-1s treatment | |||
| ↑ after SP600125 treatment | |||||||
| ↑ after BAPTA-AM treatment | |||||||
| ↑ after CAT treatment | |||||||
| H1299 | 2.5 μM | 6 h | ↓ Cell viability | ↑ after Nec-1s treatment | |||
| ↑ after BAPTA-AM treatment | |||||||
| ↑ after K45A treatment | |||||||
| ↑ after DI60N treatment | |||||||
| A549 | 7.5 μM | 8 h | ↓ Cell viability | ↑ after Nec-1s treatment | |||
| ↑ after BAPTA-AM treatment | |||||||
| ↑ after K45A treatment | |||||||
| ↑ after DI60N treatment | |||||||
| ↑ after MnSOD overexpression | |||||||
| A549/Cis | 10 μM | 72h | ↓ Cell viability | ↑ after Nec-1s treatment | |||
| Extensive vacuolation | |||||||
| Damaged and swollen mitochondria | |||||||
| Intact cell nuclei | |||||||
| U87, U251 | 5, 7.5 and 10 μM | 8 h | ↑ PI positive cells | [112] | |||
| / | 8 h | ↑ LDH release | |||||
| 5, 7.5 and 10 μM | / | No caspase-3/-7 activation | |||||
| / | 1, 2 and 4 h | ↑ O2− generation | ↓ after NAC treatment | ||||
| ↓ after DIC treatment | |||||||
| 2, 4 and 8 h | ↑ Cytosolic Ca2+ accumulation | ↓ after BAPTA-AM treatment | |||||
| ↓ after NAC treatment | |||||||
| 0.5, 1, 2, 4 and 8 h | ↑ p-CaMKII protein expression | ||||||
| 0.5, 1 and 2 h | ↑ p-JNK1/2 protein expression | ↓ after BAPTA-AM treatment | |||||
| ↓ after NAC treatment | |||||||
| 8 h | ↓ Cell viability | ↑ after NAC treatment | |||||
| ↑ after GSH treatment | |||||||
| ↑ after CAT treatment | |||||||
| ↑ after DTT treatment | |||||||
| ↑ after BAPTA-AM treatment | |||||||
| ↑ after KN93 treatment | |||||||
| ↑ after SP600125 treatment | |||||||
| ↑ after DIC treatment | |||||||
| ↑ after NQO1 knockdown | |||||||
| U251 | / | 4 h | Intracellular bubbles | ||||
| 1 and 2 h | Mitochondrial fragmentation | ||||||
| 4 h | ↓ ΔΨm | ↑ after SP600125 treatment | |||||
| ↑ after BAPTA-AM treatment | |||||||
| ↑ Mitochondrial O2− generation | ↓ after DIC treatment | ||||||
| ↓ after CAT treatment | |||||||
| ↓ after SP600125 treatment | |||||||
| ↓ after BAPTA-AM | |||||||
| 11-methoxytabersonine | Tabernaemontana bovina Lour. | A549, H157 | 1, 2.5 and 5 μM | 48 h | No caspase-3 activation | [113] | |
| 2.5 μM | 24 and 48 h | ↑ LDH release | ↑ after 3-MA treatment | ||||
| ↑ after CQ treatment | |||||||
| 2.5 μM | 16 and 24 h | RIP1-RIP3 interaction | ↑ after 3-MA treatment | ||||
| 2.5, 5, 7.5 and10 μM | 48 h | ↓ Cell viability | ↑ after Nec-1 treatment | ||||
| ↑ after 3-MA treatment | |||||||
| ↑ after CQ treatment | |||||||
| Acridocarpus orientalis dichloromethane fraction | Acridocarpus orientalis A. Juss | HeLa | 250 μg/mL | / | No caspase-3/7-8-9 activation | [114] | |
| 24 h | ↑ Cell death | ↓ after Nec-1 treatment | |||||
| ↓ after Nec-1 + Z-VAD-FMK treatment | |||||||
| Acridocarpus orientalis n-butanol fraction | Acridocarpus orientalis A. Juss | HeLa | 125 μg/mL | 24 h | ↑ Cell death | ↓ after Nec-1 treatment | [114] |
| ↓ after Nec-1 + Z-VAD-FMK treatment | |||||||
| Arctigenin | Arctium lappa L., Saussurea heteromalla | PC-3, PC-3AcT | 20 μM | 24 and 48 h | ↓ Cell viability | ↑ after Nec-1 treatment | [115] |
| ↑ after ATP treatment | |||||||
| 20 and 40 μM | 48 h | ↑ p-RIP3, ↑ p-MLKL protein expression | ↓ after Nec-1 treatment | ||||
| ↓ after ATP treatment | |||||||
| ↓ after CCN1 knockdown | |||||||
| 5, 10, 20 and 40 μM | ↑ CCN1 protein expression | ↓ after NAC treatment | |||||
| ↓ p-Akt protein expression | ↑ after NAC treatment | ||||||
| PC-3, PC-3AcT-cells derived spheroids | 20 μM | 48 h | ↓ Spheroid growth and viability | ↓ after NAC treatment | |||
| ↑ CCN1, ↑ p-RIP3, ↑ p-MLKL protein expression | |||||||
| RPMI-2650 | 5 μM | 24, 48 and 72 h | ↓ Cell viability | ↑ after Nec-1 treatment | [116] | ||
| ↑ after ATP treatment | |||||||
| 48 h | ↑ Necrotic cells | ↓ after NAC treatment | |||||
| ↑ ROS | ↓ after NAC treatment | ||||||
| ↓ ΔΨm | ↑ after NAC treatment | ||||||
| ↓ ATP levels | ↓ after Nec-1 treatment | ||||||
| / | ↑ RIP3, ↑ p-RIP3 protein expression | ||||||
| ↑ MLKL, ↑ p-MLKL protein expression | |||||||
| ↑ p-ATM protein expression | |||||||
| ↑ p-ATR protein expression | |||||||
| ↑ p-CHK1/2 protein expression | |||||||
| Aridanin | Tetrapleura tetraptera (Schum. & Thonn) Taub. | CCRF-CEM | 1, 2, 4, 8, 15, 30, and 61 μM | 24 h | ↓ Cell viability | ↑ after Nec-1 treatment | [29] |
| 3.18 and 6.36 μM | ↑ PI positive cells | ||||||
| Artesunate (artemisin semi-synthetic derivative) | Artemisia annua L. | Human Primary schwannoma cells | 200 μM | 24 h | ↓ Cell viability | ↑ after Nec-1 treatment | [117] |
| ↓ after CQ treatment (↑ after Nec-1 treatment) | |||||||
| 100 μM | 20 h | ↑ p-MLKL protein expression | |||||
| RT4 | 25 and 50 μM | 20 h | ↓ Cell viability | ↑ after Nec-1 treatment | |||
| ↑ after RIP1 knockdown | |||||||
| 25 and 50 μM | ↑ p-MLKL protein expression | ||||||
| 10, 25, 50 and 100 μM | ↑ RIP1 protein expression | ||||||
| Hela, COLO-205 | 50 μM | 20 h | ↑ p-MLKL protein expression | ||||
| Berberine | Huang Lian Chinese herb (Coptis chinesis Franch) and Hydrastis Canadensis L. | OVCAR3 | 100 μM | 24 h | Extensive vacuolation | [118] | |
| Rupture of plasma membrane | |||||||
| OVCAR3, POCCLs | ↑ RIP3, ↑ MLKL mRNA levels | ||||||
| ↑ RIP3, ↑ MLKL protein expression | |||||||
| ↑ p-RIP3, ↑ p-MLKL protein expression | |||||||
| DB, RAMOS | 6.25, 12.5, 25 and 50 μM | / | ↑ Growth inhibition | [119] | |||
| DB | 30 μM | 48 h | Formation of RIP1/RIP3/MLKL complex | ||||
| RAMOS | 20 μM | ||||||
| DB | 30 μM | 48 h | Swollen mitochondria | ||||
| Intact cell nuclei | |||||||
| 12 and 24 h | ↓ PCYT1A mRNA levels | ||||||
| 24 h | ↑ Degradation of PCYT1A mRNA | ||||||
| / | ↓ PCYT1A protein expression | ||||||
| Celastrol | Tripterygium wilfordii Hook. f. | HGC-27, AGS | 0.25, 0.5, 1 and 2 μM | 24 h | ↓ Cell viability | ↑ after RIP3 knockdown | [120] |
| ↑ after Nec-1 treatment | |||||||
| ↑ after Nec-1 + Z-VAD-FMK treatment | |||||||
| ↑ after BGN overexpression | |||||||
| ↓ after BGN knockdown | |||||||
| ↑ after NSA treatment | |||||||
| ↑ after Nec-1 treatment+ BGN overexpression | |||||||
| 0.5 μM | ↑ PI positive cells | ↓ after BGN overexpression | |||||
| 0.25, 0.5, 1 and 2 μM | ↑ RIP1, ↑ RIP3 protein expression | ||||||
| ↑ p-RIP1, ↑ p-RIP3 protein expression | ↓ after BGN overexpression | ||||||
| ↓ after Nec-1 treatment | |||||||
| ↓ BGN protein expression | |||||||
| 0.5 μM | Cell rounding and shrinkage | ↓ after BGN overexpression | |||||
| ↑ MLKL protein expression | |||||||
| ↑ p-MLKL protein expression | ↓ after BGN overexpression | ||||||
| ↑ MLKL translocation to plasma membrane | ↓ after BGN overexpression | ||||||
| ↓ TNF-α secretion | ↑ after BGN overexpression | ||||||
| ↓ IL-8 secretion | ↑ after BGN overexpression | ||||||
| Columbianadin | Angelica decursiva Fr. Et Sav | HCT116 | 50 μM | 48 h | ↑ RIP1, ↑ RIP3 protein expression | [121] | |
| ↓ Caspase-8 cleavage | |||||||
| ↑ ROS | ↓ after CAT treatment | ||||||
| ↓ CAT protein expression | |||||||
| ↓ SOD-1/2 protein expression | |||||||
| Deoxypodophyllotoxin | Pulsatilla koreana (Yabe ex Nakai) Nakai ex T. Mori | NCI-H460 | 30 nM | 24 h | Rupture of plasma membrane | ↓ after Nec-1 treatment | [122] |
| Cytoplasmatic vacuolation | ↓ after Nec-1 treatment | ||||||
| Mitochondria swelling | ↓ after Nec-1 treatment | ||||||
| Cytoskeletal degradation | ↓ after Nec-1 treatment | ||||||
| Dilation of endoplasmic reticulum elements | ↓ after Nec-1 treatment | ||||||
| ↑ PI penetration | ↓ after Nec-1 treatment | ||||||
| ↓ ΔΨm | ↑ after Nec-1 treatment | ||||||
| Emodin | Rheum palmatum L. | U251 | / | 12 h | ↑ LDH release | ↓ after Nec-1 treatment | [123] |
| ↓ after GSK-872 treatment | |||||||
| 10, 20 and 40 μM | 12 h | ↑ RIP1 protein expression | ↓ after Nec-1 treatment | ||||
| ↑ RIP3 protein expression | ↓ after GSK-872 treatment | ||||||
| ↑ TNF-α protein expression | |||||||
| Gomisin J | Schisandra chinensis (Turcz.) Baill. | MCF-7, MDA-MB-231 | 30 μg/mL | 72 h | ↓ Cell viability | [124] | |
| ↑ Extracellular CypA protein expression | |||||||
| Jujuboside B | Zizyphus jujube Mill var. spinosa (Bunge) Hu ex H. F. Chow | U937 | 40, 80 and 120 μM | 24 h | ↑ RIP1, ↑ p-RIP1 protein expression | ↓ after Nec-1 treatment | [125] |
| ↑ RIP3, ↑ p-RIP3 protein expression | ↓ after Nec-1 treatment | ||||||
| ↑ MLKL, ↑ p-MLKL protein expression | ↓ after Nec-1 treatment | ||||||
| 80 μM | ↓ Cell viability | ↓ after Nec-1 treatment | |||||
| ↓ Colony formation | ↓ after Nec-1 treatment | ||||||
| Matrine | Sophora flavescens Aiton | Mz-ChA-1, QBC939 | 1.5 mg/mL | 24 and 48 h | Extensive organelle and cell swelling | [126] | |
| Cytoplasmatic vacuolation | |||||||
| Loss of membrane integrity | |||||||
| No alterations of nuclei morphology | |||||||
| 48 h | ↑ PI positive cells | ↓ after Nec-1 treatment | |||||
| ↓ after RIP3 knockdown | |||||||
| ↓ after NSA treatment | |||||||
| ↓ after NAC treatment | |||||||
| 3, 6, 9 and 12 h | ↑ RIP3 protein expression | ||||||
| 2 h | ↑ MLKL membrane translocation | ↓ after Nec-1 treatment | |||||
| 0.25, 0.5, 1, 1.5 and 2 mg/mL | 24 h | ↑ ROS | ↓ after Nec-1 treatment | ||||
| ↓ after NSA treatment | |||||||
| Neoalbacol | Albatrellus confluens | C666-1 | 40 μM | / | Cell swelling | [127] | |
| 24 h | ↓ Cell viability | ↑ after Nec-1 treatment | |||||
| ↑ after Akt overexpression | |||||||
| ↑ after Nec-1 + 3-MA treatment | |||||||
| ↓ after SP600125 treatment | |||||||
| 20, 30 and 40 μM | / | RIP1-RIP3 interaction | ↓ after Nec-1 treatment | ||||
| 8 h | ↓ p-Akt protein expression | ↑ after Akt overexpression | |||||
| ↓ p-TSC2, ↓ p-mTOR, ↓ p-p70S6K1 protein expression | |||||||
| / | / | ↓ TNF-α, ↓ EGF, ↓ IL-6 protein expression | |||||
| 40 μM | / | ↓ GLUT1/4 mRNA levels | |||||
| ↓ HK2 mRNA levels | ↑ after Akt overexpression | ||||||
| 20, 30 and 40 μM | ↓ HK2 protein expression | ↑ after Akt overexpression | |||||
| ↓ GLUT1/4 protein expression | |||||||
| 40 μM | 2, 4, 6, 8 and 10 h | ↓ ATP levels | ↑ after Akt overexpression | ||||
| ↓ Glucose concentration | ↑ after Akt overexpression | ||||||
| / | ↑ p-JNK protein expression | ↓ after Nec-1 treatment | |||||
| HK1 | 40 μM | 24 h | ↓ Cell viability | ↑ after Nec-1 treatment | |||
| ↑ after Nec-1 + 3-MA treatment | |||||||
| ↓ after SP600125 treatment | |||||||
| 20, 30 and 40 μM | 8 h | ↓ p-Akt, ↓ p-TSC2, ↓ p-mTOR, ↓p-p70S6K1 protein expression | |||||
| 40 μM | / | ↓ GLUT1/4, ↓ HK2 mRNA levels | |||||
| Ophiopogonin D’ | Ophiopogon japonicus (Thunb.) Ker Gawl | LNCaP | 2.5 and 5 μM | 24 h | ↑ Necrotic cells | ↓ after NSA treatment | [128] |
| ↓ after Nec-1 treatment | |||||||
| ↓ after Nec-1 + NSA treatment | |||||||
| 5 μM | ↓ Cell viability | ↑ after Nec-1 treatment | |||||
| ↑ after Nec-1 + NSA treatment | |||||||
| 2.5 and 5 μM | 6 h | ↑ RIP3 protein expression | |||||
| ↑ MLKL, ↑ p-MLKL protein expression | |||||||
| 5 μM | ↑ RIP1 protein expression | ||||||
| ↑ Caspase-8, ↑ cleaved caspase-8 protein expression | |||||||
| RIP3-MLKL interaction | |||||||
| ↑ FasL protein expression | |||||||
| ↑ Soluble FasL protein expression | ↓ after Nec-1 treatment | ||||||
| ↓ Fas protein expression | |||||||
| ↑ FADD protein expression | |||||||
| ↓ Bim protein expression | |||||||
| ↓ AR, ↓ PSA protein expression | ↓ after Nec-1 treatment | ||||||
| Pristimerin | Various plant spp. of Celastraceae and Hippocrateaceae families | C6 U251 | 2.5 μM 4.5 μM | 6 h | Loss of membrane integrity | [129] | |
| Intact nuclear membrane | |||||||
| Swollen mitochondria | |||||||
| ↓ Cell viability | ↑ after AIF knockdown | ||||||
| ↑ after SP600125 treatment | |||||||
| ↑ PI positive cells | ↓ after AIF knockdown | ||||||
| ↓ after SP600125 treatment | |||||||
| ↓ after JNK knockdown | |||||||
| 1.5 and 6 h | ↓ ΔΨm | ↑ after SP600125 treatment | |||||
| ↑ after JNK knockdown | |||||||
| 1.5, 3 and 6 h | No caspase-3 activation | ||||||
| 1.5, 3, 6 and 12 h | ↑ AIF nuclear translocation | ↓ after SP600125 treatment | |||||
| ↓ after JNK knockdown | |||||||
| 1.5 and 6 h | ↑ JNK protein expression | ||||||
| ↑ p-JNK protein expression | ↓ after SP600125 treatment | ||||||
| ↓ after JNK knockdown | |||||||
| Progenin III | Raphia vinifera P. Beauv | CCRF-CEM | 2, 3, 7, 14 and 55 μM | 24 h | ↓ Cell proliferation | ↑ after Nec-1 treatment | [49] |
| 1.59 and 3.18 μM | ↑ ROS | ||||||
| Quercetin | Natural flavonoid found in many different plant spp. | MCF-7 | 50 μg/mL | 48 h | ↓ Cell viability | ↑ after Nec-1 treatment | [130] |
| ↓ Cell proliferation | ↑ after Nec-1 treatment | ||||||
| / | / | ↑ RIP1, ↑ RIP3 mRNA levels | ↓ after Nec-1 treatment | ||||
| Resibufogenin | Asiatic toad (Bufo gargarizans) | SW480 | 5, 10, 15 and 20 μM | 24 and 48 h | ↑ Necrotic cells | [131] | |
| 5, 10 and 20 μM | / | ↑ RIP3 protein expression | |||||
| 10 μM | ↑ RIP3 mRNA levels | ||||||
| HCT116 | 5, 10 and 20 μM | 24 and 48 h | ↓ Cell viability | ↑ after NAC treatment | |||
| ↑ after RIP3 knockdown | |||||||
| ↑ after NSA treatment | |||||||
| 5, 10, 15 and 20 μM | ↑ Necrotic cells | ↑ after NAC treatment | |||||
| ↑ after RIP3 knockdown | |||||||
| ↑ after NSA treatment | |||||||
| 20 μM | / | Extensive vacuolation | |||||
| Organelle and cell swelling | |||||||
| 10 and 20 μM | 24 h | ↑ LDH release | |||||
| 5, 10 and 20 μM | ↑ ROS | ||||||
| 5, 10 and 20 μM | / | ↑ RIP1 protein expression | |||||
| ↑ RIP3 protein expression | ↓ after RIP3 knockdown | ||||||
| 10 μM | ↑ RIP3 mRNA levels | ||||||
| 10 and 20 μM | ↑ MLKL, ↑ p-MLKL protein expression | ↓ after RIP3 knockdown | |||||
| 5 μM | 36 h | ↓ Cell migration | |||||
| ↓ Cell invasion | |||||||
| RIP3 +/+ MEFs | 5, 10 and 20 μM | / | ↑ PYGL, ↑ GLUL, ↑ GLUD1 protein expression | ||||
| ↑ PYGL, ↑ GLUL, ↑ GLUD1 activity | |||||||
| 10 μM | ↓ Cell migration | ||||||
| 5, 10 and 20 μM | ↑ ZO-1, ↑ E-cadherin, ↑ fibronectin, ↑ vimentin, ↑ SNAIL protein expression | ||||||
| Sanguilutine | Plant spp. of Papaveraceae, Fumariaceae, Ranunculaceae and Rutaceae families | Mel-JuSo | 0.7 μg/mL | 48 h | ↓ Cell viability | ↑ after Nec-1 treatment | [132] |
| ↓ after 3-MA treatment | |||||||
| ↓ after LY294002 treatment | |||||||
| A375 | 0.5, 0.7 and 1 μg/mL | ↓ Cell viability | ↑ after Nec-1 treatment | ||||
| ↓ after 3-MA treatment | |||||||
| ↓ after LY294002 treatment | |||||||
| ↓ after BafA1 treatment | |||||||
| ↑ after Nec-1 + 3-MA treatment | |||||||
| A375-Bcl2 | 0.7 μg/mL | ↓ Cell viability | ↑ after Nec-1 treatment | ||||
| ↓ after 3-MA treatment | |||||||
| ↓ after LY294002 treatment | |||||||
| Shikonin | Lithospermum erythrorhizon Siebold & Zucc., Arnebia euchroma (Royle) Johnst, or Arnebia guttata Bunge | F-47D | 5 μM | 12 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | [133] |
| 4 h | ↑ ROS | ||||||
| AsPC-1 | 5 and 10 μM | 24 h | ↑ Necrotic cells | ↑ after Nec-1 + Z-VAD-FMK treatment | [134] | ||
| ↓ after RIP3 knockdown | |||||||
| 5 μM | ↑ RIP3 mRNA levels | ||||||
| ↑ RIP3 protein expression | |||||||
| / | 24, 48 and 72 h | ↓ Cell proliferation | ↑ after RIP3 knockdown | ||||
| CNE-2Z | 6.4 μM | 24 h | ↑ Cell death | ↓ after Nec-1 treatment | [135] | ||
| 6.4 and 12.8 μM | ↑ RIP1, ↑ RIP3 protein expression | ↓ after Nec-1 treatment | |||||
| 6.4 and 12.8 μM | 6 h | ↑ ROS | ↓ after NAC treatment | ||||
| / | ↑ Mitochondrial ROS | ↓ after Nec-1 treatment | |||||
| 3.2, 6.4 and 12.8 μM | / | ↓ Cell viability | ↑ after Nec-1 treatment | ||||
| ↑ after NAC treatment | |||||||
| A549 | 3 and 6 μM | 3 and 6 h | ↓ Cell viability | ↑ after Nec-1 treatment | [136] | ||
| 3 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | |||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ after 3-MA treatment | |||||||
| ↑ after BafA1 treatment | |||||||
| ↑ after ATG5 siRNA treatment | |||||||
| ↑ RIP1 protein expression | ↓ after Nec-1 treatment | ||||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ after 3-MA treatment | |||||||
| ↑ after BafA1 treatment | |||||||
| ↑ after ATG5 siRNA treatment | |||||||
| KMS-12-PE, RPMI-8226, U266 | 10 and 20 μM | 7 h | ↑ Cell death | ↓ after Nec-1 treatment | [137] | ||
| RPMI-8226 | 10 μM | 2 h | Cell membrane swelling | ||||
| Translucent cytoplasm | |||||||
| 20 μM | / | No caspase-3/-8 cleavage | |||||
| No RIP1 cleavage | |||||||
| K7, K12, K7M3, U20S, 143B | 3 μM | 8 h | ↓ Cell viability | ↑ after Nec-1 treatment | [138] | ||
| K7 | 3 μM | ↑ PI positive cells | ↓ after Nec-1 treatment | ||||
| K7 | 1, 3 and 5 μM | ↑ RIP1, ↑ RIP3 protein expression | |||||
| U20S | 1, 3, 5 and 7.5 μM | ||||||
| K7 U20S | 1, 3 and 5 μM 1, 3, 5 and 7.5 μM | No PARP cleavage | |||||
| No caspase-3/-6 cleavage | |||||||
| U937 | 10 μM | 3 and 6 h | ↑ LDH release | ↓ after Nec-1 treatment | [139] | ||
| 6 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | |||||
| No caspase-3/-8 cleavage | ↑ after Nec-1 treatment | ||||||
| ↑ TNF-α gene expression | |||||||
| ↑ TNF-α mRNA levels | |||||||
| ↑ TNF-α protein expression | |||||||
| C6 U87 | 3 and 6 μM 5 and 10 μM | 1.5 and 3 h | ↓ Cell viability | ↑ after Nec-1 treatment | [140] | ||
| ↑ after NAC treatment | |||||||
| 3 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | |||||
| ↓ after NAC treatment | |||||||
| ↑ ROS | ↓ after Nec-1 treatment | ||||||
| ↓ after NAC treatment | |||||||
| ↑ RIP1 protein expression | ↓ after Nec-1 treatment | ||||||
| ↓ after NAC treatment | |||||||
| Electron-lucent cytoplasm | |||||||
| Loss of membrane integrity | |||||||
| Intact nuclear membrane | |||||||
| Swollen organelles | |||||||
| U87 C6 | 5 and 10 μM 3 and 6 μM | 3 h | ↑ LDH release | ↓ after Nec-1 treatment | [141] | ||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| ↑ ROS | ↓ after Nec-1 treatment | ||||||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| 2 h | ↑ Mitochondrial O2− | ↓ after Nec-1 treatment | |||||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| / | ↑ RIP1, ↑ RIP3 protein expression | ↓ after Nec-1 treatment | |||||
| SHG-44 U251 | 2 and 4 μM 5 and 10 μM | 3 h | ↑ LDH release | ↓ after Nec-1 treatment | |||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| ↑ after rotenone treatment | |||||||
| ↓ after rotenone + Nec-1 treatment | |||||||
| ↑ Necrotic cells | ↓ after Nec-1 treatment | ||||||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| ↑ after rotenone treatment | |||||||
| ↓ after rotenone + Nec-1 treatment | |||||||
| ↑ ROS | ↓ after Nec-1 after treatment | ||||||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| ↑ after rotenone treatment | |||||||
| ↓ after rotenone + Nec-1 treatment | |||||||
| 2 h | ↑ Mitochondrial O2− | ↓ after Nec-1 treatment | |||||
| ↓ after GSK-872 treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| ↑ after rotenone treatment | |||||||
| ↓ after rotenone + Nec-1 treatment | |||||||
| / | ↑ RIP1 protein expression | ↓ after Nec-1 treatment | |||||
| ↑ after rotenone treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| ↑ RIP3 protein expression | ↓ after GSK-782 treatment | ||||||
| ↑ after rotenone treatment | |||||||
| ↓ after MnTBAP treatment | |||||||
| RIP1-RIP3 interaction | ↑ after rotenone treatment | ||||||
| ↓ after MnTBAP treatment | |||||||
| U87 C6 | 5 and 10 μM 3 and 6 μM | 3 h | ↓ Cell viability | ↑ after Nec-1 treatment | [142] | ||
| ↑ after NAC treatment | |||||||
| 15, 30, 60 and 120 min | ↑ RIP1 protein expression | ↓ after Nec-1 treatment | |||||
| ↓ after RIP1 knockdown | |||||||
| ↑ RIP3 protein expression | ↓ after Nec-1 treatment | ||||||
| ↓ after RIP3 knockdown | |||||||
| U87 C6 | 10 μM 6 μM | 15, 30, 60 and 120 min | ↑ γ-H2AX protein expression | ||||
| ↑ p-ATM protein expression | |||||||
| ↑ ROS | ↓ after Nec-1 treatment | ||||||
| ↓ after GSK-872 treatment | |||||||
| ↓ after NAC treatment | |||||||
| 1 h | ↑ Mitochondrial O2− | ↓ after Nec-1 treatment | |||||
| ↓ after GSK-872 treatment | |||||||
| ↓ GSH | ↑ after Nec-1 treatment | ||||||
| ↑ after GSK-872 treatment | |||||||
| SHG-44 U251 | 4 μM 10 μM | 3 h | ↑ Necrotic cells | ||||
| SHG-44 U251 | 2 and 4 μM 5 and 10 μM | ↓ Cell viability | ↑ after Nec-1 treatment | ||||
| ↑ after GSK-782 treatment | |||||||
| ↑ after RIP1 knockdown | |||||||
| ↑ after RIP3 knockdown | |||||||
| ↑ after NAC treatment | |||||||
| 15, 30, 60 and 120 min | ↑ RIP1 protein expression | ↓ after Nec-1 treatment | |||||
| ↓ after RIP1 knockdown | |||||||
| ↓ after NAC treatment | |||||||
| ↑ RIP3 protein expression | ↓ after GSK-782 treatment | ||||||
| ↓ after RIP3 knockdown | |||||||
| ↓ after NAC treatment | |||||||
| SHG-44 | 4 μM | 2 h | RIP1-RIP3 interaction | ↓ after Nec-1 treatment | |||
| ↓ after NAC treatment | |||||||
| SHG-44 U251 | 4 μM 10 μM | 15, 30, 60 and 120 min | ↑ CypA protein expression | ||||
| 2 h | No caspase-3/-8 cleavage | ||||||
| ↓ TNF-α release | |||||||
| ↑ TNF-α gene expression | |||||||
| 1 h | ↑ DNA damage, ↑ DNA DSBs | ↓ after Nec-1 treatment | |||||
| ↓ after GSK-782 treatment | |||||||
| ↓ after NAC treatment | |||||||
| ↑ γ-H2AX foci | |||||||
| SHG-44 U251 | 2 and 4 μM 5 and 10 μM | 15, 30, 60 and 120 min | ↑ γ-H2AX, ↑ p-ATM protein expression | ↑ after Nec-1 treatment | |||
| ↑after GSK-782 treatment | |||||||
| ↑ after RIP1 knockdown | |||||||
| ↑ after RIP3 knockdown | |||||||
| ↓ after NAC treatment | |||||||
| SHG-44 U251 | 4 μM 10 μM | 15, 30, 60 and 120 min | ↑ ROS | ↓ after Nec-1 treatment | |||
| ↓ after GSK-872 treatment | |||||||
| ↓ after NAC treatment | |||||||
| 1 h | ↑ Mitochondrial O2− | ↓ after Nec-1 treatment | |||||
| ↓ after GSK-872 treatment | |||||||
| ↓ GSH | ↑ after Nec-1 treatment | ||||||
| ↑ after GSK-872 treatment | |||||||
| SHG-44 U251 | 2 and 4 μM 5 and 10 μM | 3 h | ↑ LDH release | ↓ after NSA treatment | [143] | ||
| ↓ after AIF knockdown | |||||||
| / | ↑ Necrotic cells | ↓ after NSA treatment | |||||
| ↓ after MLKL knockdown | |||||||
| SHG-44 U251 U87 C6 | 2 and 4 μM 5 and 10 μM 10 μM 6 μM | 15, 30, 60 and 120 min | ↑ MLKL, ↑ p-MLKL protein expression | ↓ after NSA treatment | |||
| ↓ after MLKL knockdown | |||||||
| 2 h | ↓ Mitochondrial AIF protein expression | ||||||
| SHG-44 U251 U87 C6 | 4 μM 10 μM 10 μM 6 μM | 2 h | ↑ Cytoplasmatic/nuclear AIF protein expression | ↓ after AIF knockdown | |||
| ↓ after NSA treatment | |||||||
| ↓ after MLKL knockdown | |||||||
| ↓ after MnTBAP treatment | |||||||
| SHG-44 U251 | 4 μM 10 μM | 2 h | ↓ Δψm | ↑ after NSA treatment | |||
| ↑ after MnTBAP treatment | |||||||
| / | ↑ Mitochondrial O2− | ↓ after NSA treatment | |||||
| ↓ after MnTBAP treatment | |||||||
| ↓ after MLKL knockdown | |||||||
| / | ↑ ROS | ↓ after MnTBAP treatment | |||||
| ↓ after MLKL knockdown | |||||||
| 15, 30, 60 and 120 min | ↑ Mitochondrial MLKL, ↑ Mitochondrial p-MLKL protein expression | ||||||
| SHG-44 | 4 μM | 2 h | Mitochondrial accumulation of MLKL | ||||
| 5–8F | 7.5 μM | 6 h | Ruptured plasma membrane | [144] | |||
| Increased cell volume | |||||||
| Swollen organelles | |||||||
| Loss of membrane integrity | |||||||
| ↑ Necrotic cells | ↓ after Nec-1 treatment | ||||||
| ↓ after NAC treatment | |||||||
| ↑ Caspase-3/-8 activity | ↑ after Nec-1 treatment | ||||||
| / | ↑ ROS | ↓ after NAC treatment | |||||
| / | ↑ RIP1, ↑ RIP3, ↑ MLKL protein expression | ↓ after Nec-1 treatment | |||||
| ↓ after NAC treatment | |||||||
| AGS | 1, 2 and 4 μM | 6 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | [145] | ||
| ↑ ROS | ↓ after NAC treatment | ||||||
| ↓ Δψm | ↑ after Nec-1 treatment | ||||||
| ↑ after NAC treatment | |||||||
| 2 and 4 μM | ↓ Cell viability | ↑ after Nec-1 treatment | |||||
| NIH3T3 | 0.5, 1 and 2.5 μM | 3 h | ↓ Cytotoxicity | ↓ after Nec-1 treatment | [146] | ||
| ↑ ROS | ↓ after NAC treatment | ||||||
| / | RIP1-RIP3 interaction | ↓ after Nec-1 treatment | |||||
| MCF-7 | 5 μM | 12 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | [147] | ||
| ↑ RIP1, ↑ RIP3 protein expression | |||||||
| 4 h | ↑ ROS | ||||||
| 12 h | ↓ Δψm | ↓ after Nec-1 treatment | |||||
| 24 h | No caspase-3 activation | ||||||
| ↑ Caspase-8 activity | ↑ after Nec-1 treatment | ||||||
| 12 h | Extensive vacuolation | ||||||
| Loss of membrane integrity | |||||||
| MDA-MB-468 | 5 μM | 12 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | [148] | ||
| ↑ RIP1, ↑ RIP3 protein expression | |||||||
| 4 h | ↑ ROS | ||||||
| 12 h | ↓ Δψm | ↓ after Nec-1 treatment | |||||
| 24 h | ↑ Caspase-3/-8 activity | ↑ after Nec-1 treatment | |||||
| 12 h | Extensive vacuolation | ||||||
| Loss of membrane integrity | |||||||
| Tanshinol A | Salvia miltiorrhiza Bunge | H1299 | 2, 4 and 8 μM | 8 h | No caspase-3/-7 activation | [149] | |
| 1, 2, 4 and 8 h | ↑ p-MLKL protein expression | ||||||
| 8 μM | 8 h | ↑ PI positive cells | |||||
| ↓ Cell viability | ↑ after MLKL knockdown | ||||||
| ↑ after NAC treatment | |||||||
| ↑ after CAT treatment | |||||||
| ↓ after MLKL transfection | |||||||
| ↑ MLKL membrane translocation | |||||||
| ↑ ROS | ↓ after NAC treatment | ||||||
| ↓ after CAT treatment | |||||||
| A549 | 10, 15 and 20 μM | 8 h | No caspase-3/-7 activation | ||||
| No caspase-3/-7/-8/-9 cleavage | |||||||
| 20 μM | / | ↑ PI positive cells | |||||
| 15 and 20 μM | ↑ Necrotic cells | ||||||
| 10, 15 and 20 μM | 1, 2, 4 and 8 h | ↑ p-MLKL protein expression | ↓ after MLKL knockdown | ||||
| ↓ after NAC treatment | |||||||
| ↓ after CAT treatment | |||||||
| 20 μM | 8 h | ↑ MLKL membrane translocation | ↓ after NAC treatment | ||||
| ↓ Cell viability | ↑ after MLKL knockdown | ||||||
| ↑ after NAC treatment | |||||||
| ↑ after CAT treatment | |||||||
| ↓ after MLKL transfection | |||||||
| ↑ ROS | ↓ after NAC treatment | ||||||
| ↓ after CAT treatment | |||||||
| Tanshinone IIA | Salvia miltiorrhiza Bunge | HepG2 | 5 and 10 μg/mL | 12 h | ↑ Necrotic cells | ↓ after Nec-1 treatment | [150] |
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ LDH release | ↓ after Nec-1 treatment | ||||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ CypA protein expression | ↓ after Nec-1 treatment | ||||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ HMGB1 protein expression | ↓ after Nec-1 treatment | ||||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ FLIPL, ↑ FLIPS protein expression | ↑ after Nec-1 treatment | ||||||
| ↑ Cleaved caspase-3/-8 protein expression | ↓ after Nec-1 treatment | ||||||
| ↓ after Z-VAD-FMK treatment | |||||||
| ↓ RIP1 protein expression | ↑ after Z-VAD-FMK treatment | ||||||
| ↑ Cleaved RIP1 protein expression | ↓ after Nec-1 treatment | ||||||
| ↓after Z-VAD-FMK treatment | |||||||
| Formation of RIP1/RIP3/FADD/FLIPs complex | |||||||
| ↓ MLKL monomer protein expression | ↑ after Nec-1 treatment | ||||||
| ↑ after Z-VAD-FMK treatment | |||||||
| ↑ Cleaved MLKL protein expression | ↓ after Nec-1 treatment | ||||||
| ↓ after Z-VAD-FMK treatment | |||||||
| Ungeremine | Crinum zeylanicum L. | CCRF-CEM | 2.37, 3.76, 9.40, 18.79, 37.58, 75.17 and 150.33 μM | 24 h | ↓ Cell proliferation | ↑ after Nec-1 treatment | [53] |
| 1.22, 2.45, 4.89 and 9.78 μM | ↑ ROS | ||||||
| 4.89 and 9.78 μM | ↑ RIP3 protein expression | ||||||
| Youdujing extract | Traditional Chinese herbal formula Youdujing | Ect1/E6E7 | 1, 2 and 4 mg/mL | 12 h | No caspase-3/-7 activation | [151] | |
| 2 and 4 mg/mL | / | ↑ RIP1 protein expression | ↓ after RIP1 knockdown | ||||
| ↑ after RIP1 overexpression | |||||||
| 4 mg/mL | RIP1-MLKL interaction | ||||||
| 3 h | ↓ Cell viability | ↑ after Nec-1 treatment | |||||
| ↑ after RIP1 knockdown | |||||||
| ↓ after RIP1 overexpression |
Abbreviations: ↑: Increase; ↓: Decrease; 3-MA: 3-methyladenine; A549/Cis: cisplatin resistant A549 cells; A375-Bcl2: Bcl-2 transfected A-375 cells; AIF: Apoptosis-inducing factor; Akt: Protein kinase B; AR: Androgen receptor; ATG5: Autophagy-related gene 5; ATP: Adenosine triphosphate; BafA1: Bafilomycin-A1; BAPTA-AM: Calcium chelator; BGN: Biglycan; Ca2+: Calcium; CaMKII: Calcium-calmodulin dependent protein kinase II dihydroethidium; CAT: Catalase; CCN1: Cell communication network factor 1; CQ: Chloroquine; CypA: Cyclophilin A; DI60N: Kinase dead RIP3; DIC: Dicoumarol; DSBs: Double strand-breaks; DTT: Dithiothreitol; EGF: Epidermal growth factor; FADD: FAS-associated death domain; FasL: FAS ligand; FLIPL: Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein isoform L; FLIPS: Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein isoform S; GLUD1: Glutamate dehydrogenase; GLUL: Glutamine synthetase; GLUT1/4: Glucose transporter 1/4; GSH: Glutathione; GSK-872: RIP3 inhibitor; GSSG: Glutathione disulfide; H2O2: Hydrogen peroxide; Hgb: Hemoglobin; HK2: Hexokinase 2; HROS: Highly Reactive oxygen species; IL-6: Interleukin-6; IL-8: Interleukin-8; iNOS: Inducible nitric oxide synthase; JNK1/2: c-Jun N-terminal kinase 1/2; K45A: Kinase dead RIP1; KN93: CaMKII inhibitor; L-NMMA: NG-monomethyl L- arginine, pan-NOS inhibitor; LDH: Lactate dehydrogenase; LY294002: Autophagy inhibitor; MLKL: Mixed lineage kinase domain like pseudokinase; MnSOD: Manganese superoxide dismutase; MnTBAP: Superoxide dismutase mimetic and peroxynitrite scavenger; NAC: N-acetyl-L-cysteine; Nec-1: Necrostatin-1; Nec-1s: Necrostatin-1s, 7-Cl-O-Nec-1; NF-kB: Nuclear factor kappa-light-chain-enhancer of activated B cells; NO: Nitric oxide; NQO1: NAD(P)H: quinone oxidoreductase 1; NSA: Necrosulfonamide; O2−: Superoxide; p-Akt: Phospho-protein kinase B; p-ATM: Phospho-ataxia telangiectasia mutated kinase; p-ATR: Phospho-ataxia telangiectasia and Rad3-related kinase; p-CHK1/2: Phospho-checkpoint kinase 1/2; p-IκBα: Phospho-inhibitor of nuclear factor kappa B; p-MKLK: Phospho-mixed lineage kinase domain like pseudokinase; p-mTOR: Phospho-mammalian target of rapamycin; p-p70S6K1: Phospho-p70 ribosomal protein S6 kinase 1; p-RIP1: Phospho-receptor-interacting serine/threonine-protein kinase 1; p-RIP3: Phospho-receptor-interacting serine/threonine-protein kinase 3; p-TSC2: Phospho-tuberous sclerosis complex 2; PARP: Poly ADP (adenosine diphosphate)-ribose polymerase; PCYT1A: Phosphate cytidylyltransferase 1 alpha; PI: Propidium iodide; POCCLs: Patient-derived primary ovarian cancer cell lines; PSA: Prostate specific antigen; PYGL: Glycogen phosphorylase; RIP1: Receptor-interacting serine/threonine-protein kinase 1; RIP3: Receptor-interacting serine/threonine-protein kinase 3; ROS: Reactive oxygen species; SB203580: P38 inhibitor; SOD-1/2: Superoxide dismutase 1/2; Spp.: species; SP600125: JNK inhibitor; TNF-α: Tumor necrosis factor-α; z-LLY-fmk: Calpain inhibitor; Z-VAD-FMK: pan-caspase inhibitor; ZO-1: Zonula occludens-1; γ-H2AX: Phospho-H2A histone family member X; ΔΨm: Mitochondrial membrane potential.
Table 3.
Natural products as in vitro inducers of pyroptosis.
| Compound | Compound Source | Cell Line (s) | Concentrations (Where Specified) | Time (Where Specified) | Pyroptosis Markers | Supplementary Effects | Reference |
|---|---|---|---|---|---|---|---|
| Alpinumisoflavone | Derris eriocarpa F.C | KYSE30, KYSE510 | 5, 10 and 20 μM | 24 and 48 h | ↓ Cell viability | ↓ after GSDME knockdown | [201] |
| 10 and 20 μM | / | ↓ Colony formation | |||||
| Cell swelling and bubble at plasma membrane | |||||||
| ↑ LDH release | ↓ after Z-DEVD-FMK treatment | ||||||
| ↓ after caspase-3 knockdown | |||||||
| ↓ after GSDME knockdown | |||||||
| ↑ Cleaved GSDME protein expression | ↓ after Z-DEVD-FMK treatment | ||||||
| ↓ after caspase-3 knockdown | |||||||
| Huh7, MMC 7721 | 2, 5, 10 and 20 μM | 48 h | ↓ Cell viability | ↓ after MCC950 treatment | [202] | ||
| ↓ after NLPR3 knockdown | |||||||
| 10 and 20 μM | 14 days | ↓ Colony formation | |||||
| 24 h | ↓ Cell invasion | ↑ after MCC950 treatment | |||||
| ↑ after NLPR3 knockdown | |||||||
| ↓ Cell migration | ↑ after MCC950 treatment | ||||||
| ↑ after NLPR3 knockdown | |||||||
| 48 h | ↑ LDH release | ↑ after CQ treatment | |||||
| ↑ after ATG5 knockdown | |||||||
| ↑ NLRP3, ↑ cleaved caspase-1, ↑ cleaved IL-1β, ↑ cleaved IL-18 mRNA levels | |||||||
| ↑ NLRP3, ↑ cleaved caspase-1, ↑ cleaved IL-1β, ↑ cleaved IL-18, ↑ cleaved GSDMD protein expression | ↓ after MCC950 treatment | ||||||
| ↓ after NLPR3 knockdown | |||||||
| ↑ after CQ treatment | |||||||
| ↑ after ATG5 knockdown | |||||||
| Anthocyanin | Flavonoid found in different plant spp. | Tca8113, SCC15 | 250 μg/mL | 24, 48 and 72 h | ↓ Cell viability | ↑ after AC-YVAD-CMK treatment | [203] |
| 48 h | ↓ Cell migration | ↑ after AC-YVAD-CMK treatment | |||||
| 48 h | ↓ Cell invasion | ↑ after AC-YVAD-CMK treatment | |||||
| 48 h | ↑ NLRP3, ↑ caspase-1, ↑ IL-1β mRNA levels | ↓ after AC-YVAD-CMK treatment | |||||
| / | ↑ NLRP3, ↑ cleaved caspase-1, ↑ cleaved IL-1β, ↑ cleaved IL-18 protein expression | ↓ after AC-YVAD-CMK treatment | |||||
| / | ↑ GSDMD protein expression | ||||||
| Berberine | Huang Lian Chinese herb (Coptis chinesis) and others plant spp. | HepG2 | 50 and 100 μM | / | Cell swelling | [204] | |
| 50 μM | 24, 48 and 72 h | ↓ Cell viability | ↑ after AC-YVAD-CMK treatment | ||||
| 48 h | ↓ Cell migration | ↑ after AC-YVAD-CMK treatment | |||||
| 48 h | ↓ Cell invasion | ↑ after AC-YVAD-CMK treatment | |||||
| 25, 50 and 100 μM | / | ↑ Caspase-1 mRNA levels | ↓ after AC-YVAD-CMK treatment | ||||
| ↑ Caspase-1 protein expression | ↓ after AC-YVAD-CMK treatment | ||||||
| Casticin | Vitex spp. | 5–8F | / | 24 h | ↑ LDH release | ↓ after SP60012 treatment | [205] |
| ↓ after JSH-23 treatment | |||||||
| ↓ after PKR knockdown | |||||||
| 3, 6 and 9 μM | ↑ Cleaved caspase-1, ↑ cleaved GSDMD, ↑ PKR, ↑ IL-1β, ↑ p-NF-κB, ↑ p-JNK protein expression | ↓ after SP600125 treatment | |||||
| ↓ after JSH-23 treatment | |||||||
| ↓ after PKR knockdown | |||||||
| 6 μM | ↑ IL-6, ↑ IL-1β, ↑ TLR4, ↑ ASC mRNA levels | ||||||
| Dioscin | Polygonatum zanlanscianense Pamp., Dioscorea nipponica Makino, and Dioscorea zingiberensis C. H. Wright | MNNG/HOS | / | 24 h | Bubbles at plasma membrane | [206] | |
| 2.5 and 5 μM | ↑ LDH release | ||||||
| ↑ Cleaved GSDME protein expression | |||||||
| MG63 | / | Bubbles at plasma membrane | |||||
| 2 and 4 μM | ↑ LDH release | ||||||
| ↑ Cleaved GSDME protein expression | |||||||
| U20S | / | Bubbles at plasma membrane | |||||
| 2 and 4 μM | ↑ LDH release | ↓ after Z-DEVD-FMK treatment | |||||
| ↑ Cleaved GSDME protein expression | ↓ after Z-DEVD-FMK treatment | ||||||
| ↓ after GSDME knockdown | |||||||
| 4 μM | ↓ Cell viability | ↑ after Z-DEVD-FMK treatment | |||||
| ↑ after SPSP600125 treatment | |||||||
| Galangin | Alpinia officinarum Hance, Alnus pendula Matsum, Plantago major L, and Scutellaria galericulata L. | U251, U87MG | 150 μM | 48 h | Bubbles at plasma membrane | [207] | |
| / | 12, 24, 48 and 72 h | ↑ LDH release | ↓ after GSDME knockdown | ||||
| 150 μM | 48 h | ↑ Cleaved GSDME protein expression | ↑ after 3-MA treatment | ||||
| Huaier extract | Trametes robiniophila Murr (Huaier) | H520, H358 | 5 and 10 mg/mL | 24 and 48 h | ↓ Cell viability | ↑ after NLRP3 knockdown | [208] |
| ↑ after MCC950 treatment | |||||||
| ↑ LDH release | ↓ after NLRP3 knockdown | ||||||
| ↓ after MCC950 treatment | |||||||
| ↑ NLRP3, ↑ caspase-1, ↑ IL-1β, ↑ IL-18 mRNA levels | |||||||
| ↑ NLRP3, ↑ cleaved caspase-1, ↑ cleaved IL-1β, ↑ cleaved IL-18 protein expression | ↓ after NLRP3 knockdown | ||||||
| ↓ after MCC950 treatment | |||||||
| L50377 (piperlongumine analogue) | Piper Longum L. | A549 | 10 μM | / | Cell swelling and bubbles at plasma membrane | ↓ after NAC treatment | [209] |
| ↓ after IKKβ overexpression | |||||||
| 0.74, 2.22, 6.67 and 20 μM | ↓ Cell viability | ↓ after NAC treatment | |||||
| ↓ after IKKβ overexpression | |||||||
| 5, 10 and 20 μM | 2 h | ↓ IKKα, ↓ IKKβ phosphorylation | |||||
| 2.5 μM | 8 h | ↑ ROS | ↓ after NAC treatment | ||||
| 5 and 10 μM | / | ↑ Cleaved GSDME protein expression | |||||
| Nobiletin | Citrus fruits | A2780, OVCAR-3 | 10, 20, 30, 40 and 50 μM | 24 h | ↓ Cell viability | [210] | |
| 50 μM | 12 h | ↑ ROS | ↓ after NAC treatment | ||||
| 10, 30 and 50 μM | ↓ ΔΨm | ||||||
| 50 μM | 24 h | Cell swelling and bubbles at plasma membrane | |||||
| 10, 30 and 50 μM | ↑ Cleaved GSDMD protein expression | ↓ after NAC treatment | |||||
| ↓ after 3-MA treatment | |||||||
| ↑ Cleaved GSDME protein expression | ↓ after NAC treatment | ||||||
| ↓ after 3-MA treatment | |||||||
| ↓ after NSA treatment | |||||||
| 50 μM | ↑ IL-1β, ↑ ASC mRNA levels | ||||||
| Osthole | Cnidium monnieri L. Cusson | A2780, OVCAR-3 | 10, 20, 30, 40, 50, 60, 70 and 80 μM | 24 h | ↓ Cell viability | [211] | |
| 20, 40 and 80 μM | Cell swelling and bubbles at plasma membrane | ||||||
| ↑ Cleaved GSDME protein expression | |||||||
| Paclitaxel | Pacific yew | A549 | 20 and 60 μM | 24 and 48 h | ↑ Lytic cell death | ↓ after AC-DEVD-CHO treatment | [212] |
| ↓ after GSDME knockdown | |||||||
| 60, 120, 180 and 240 μM | ↑ Cleaved caspase-3, ↑ cleaved caspase-7, ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ cleaved GSDME protein expression | ||||||
| Polyphyllin VI | Trillium tschonoskii Maxim | A549, H1299 | 3, 4, 5 and 6 μM | 24 h | ↑ Pyroptotic cells | ↓ after VX-765 treatment | [198] |
| ↓ after NSA treatment | |||||||
| 4 μM | ↑ Activated caspase-1 expression | ||||||
| ↑ PI positive cells | ↓ after VX-765 treatment | ||||||
| ↓ after BAY treatment | |||||||
| 3, 4, 5 and 6 μM | ↑ NLRP3, ↑ cleaved caspase-1, ↑ cleaved IL-1β, ↑ cleaved IL-18, ↑ cleaved GSDMD protein expression | ||||||
| ↑ IL-1β, ↑ IL-18 secretion | |||||||
| ↑ ROS | ↓ after NAC treatment | ||||||
| ↑ p65/NF-kB protein expression | ↓ after NAC treatment | ||||||
| Tanshinone IIA | Salvia miltiorrhiza Bunge (Danshen) | HeLa | 2, 4 and 8 μM | 24, 48 and 72 h | ↓ Cell proliferation | [213] | |
| 72 h | ↑ IL-1β, ↑ IL-18, ↑ GSDMD protein expression | ↓ after miR-145 knockdown |
Abbreviations: ↑: Increase; ↓: Decrease; 3-MA: 3-methyladenine; AC-DEVD-CHO: Caspase-3 inhibitor; AC-YVAD-CMK: Caspase-1 inhibitor; ASC: Apoptosis-associated speck-like protein containing a CARD (caspase activation and recruitment domain); ATG5: Autophagy related 5; BAY: Bay 11-7082, NF-kB inhibitor; GSDMD: Gasdermin D; GSDME: Gasdermin E; IKKα: Inhibitor of nuclear factor kappa-B kinase subunit alfa; IKKβ: Inhibitor of NF-kB kinase subunit beta; IL-6: Interleukin-6; IL-18: Interleukin-18; IL-1β: Interleukin 1 beta; JSH-23: NF-κB inhibitor; LDH: Lactate dehydrogenase; MCC950: NLRP3 inhibitor; NAC: N-acetylcysteine; NF-kB: Nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: NLR (nucleotide-binding oligomerization domain (NOD)-like receptor) family pyrin domain-containing 3; NSA: Necrosulfonamide; PKR: Protein kinase R; p-JNK1: Phospho-c-Jun N-terminal kinase; p65: Transcription factor p65; Spp.: species; SPSP600125: Inhibitor of c-Jun N-terminal kinase (JNK); VX-765: Caspase-1 inhibitor; Z-DEVD-FMK: Caspase-3 inhibitor.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Greco, G.; Catanzaro, E.; Fimognari, C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers 2021, 13, 304. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020304
AMA Style
Greco G, Catanzaro E, Fimognari C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers. 2021; 13(2):304. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020304
Chicago/Turabian StyleGreco, Giulia, Elena Catanzaro, and Carmela Fimognari. 2021. "Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer" Cancers 13, no. 2: 304. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020304
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.