
Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma

 , , , , , add
Show full author list
, , , , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
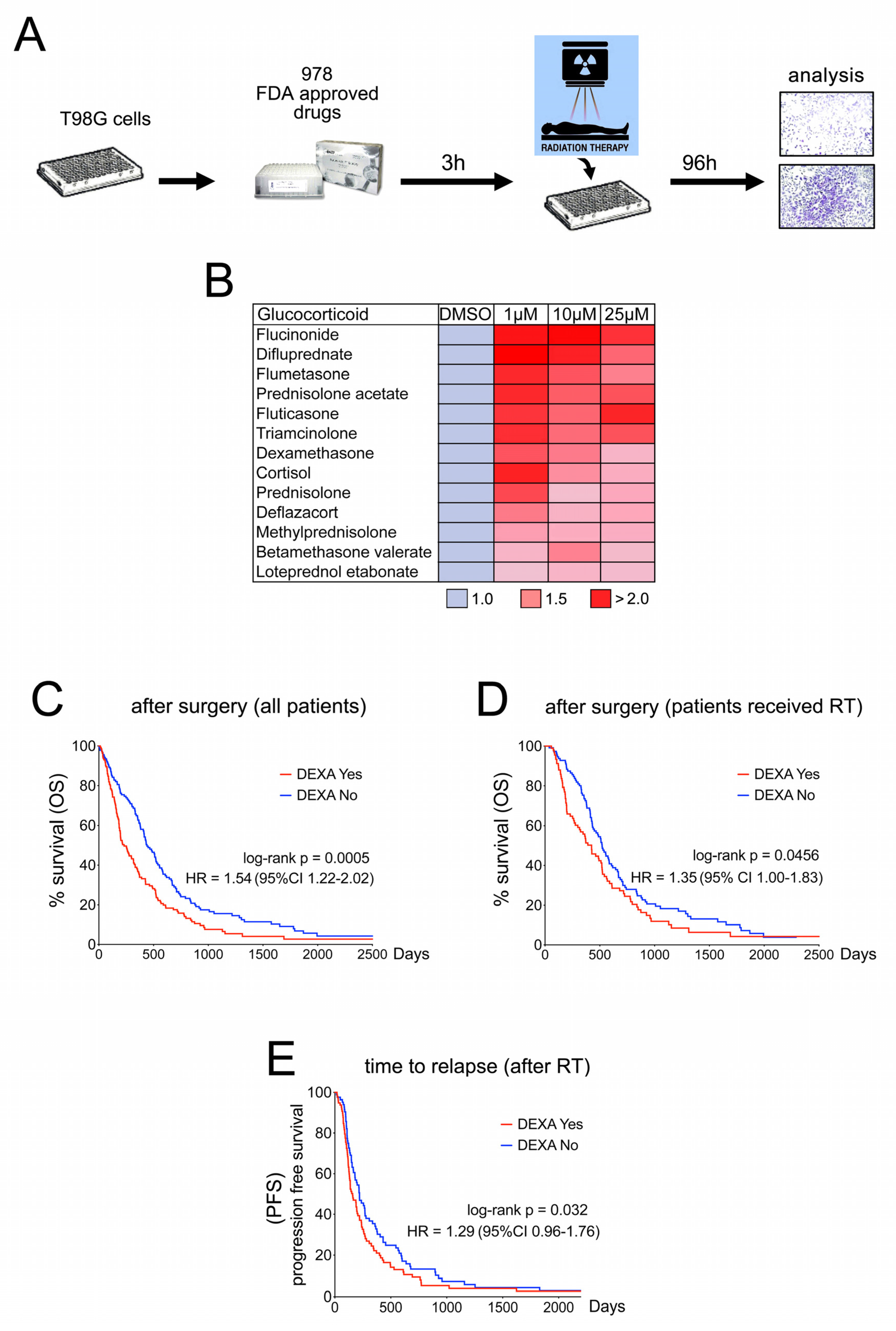
2.1. Dexamethasone Protects from Radiotherapy and Reduces Survival in GBM Patients
2.2. Dexamethasone Suppresses Genes Required for Accurate Mitosis Control
2.3. Dexamethasone Drives Proliferation by Overriding Cell Cycle Checkpoints
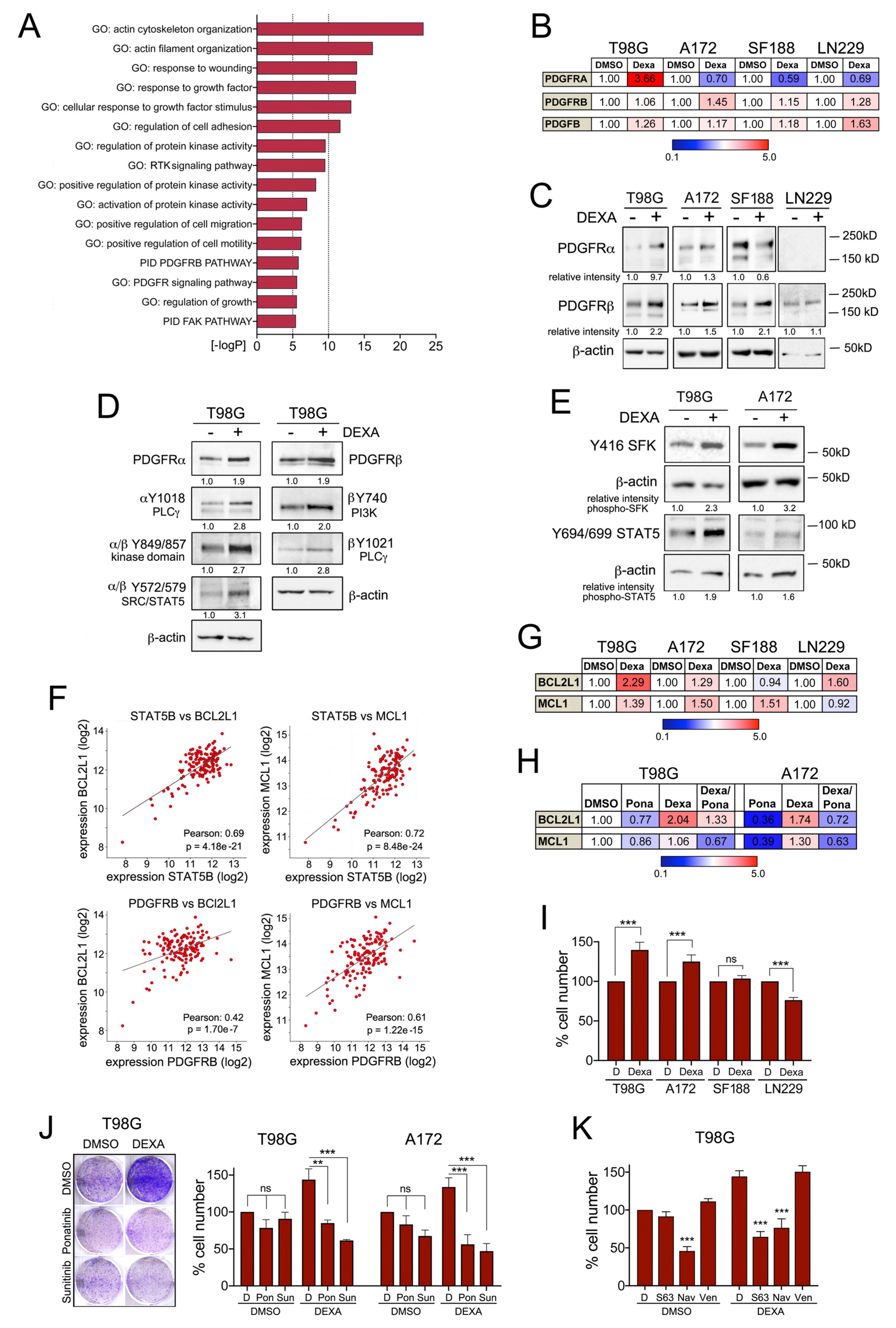
2.4. Dexamethasone Up-Egulates PDGFR Mediated Survival Signalling
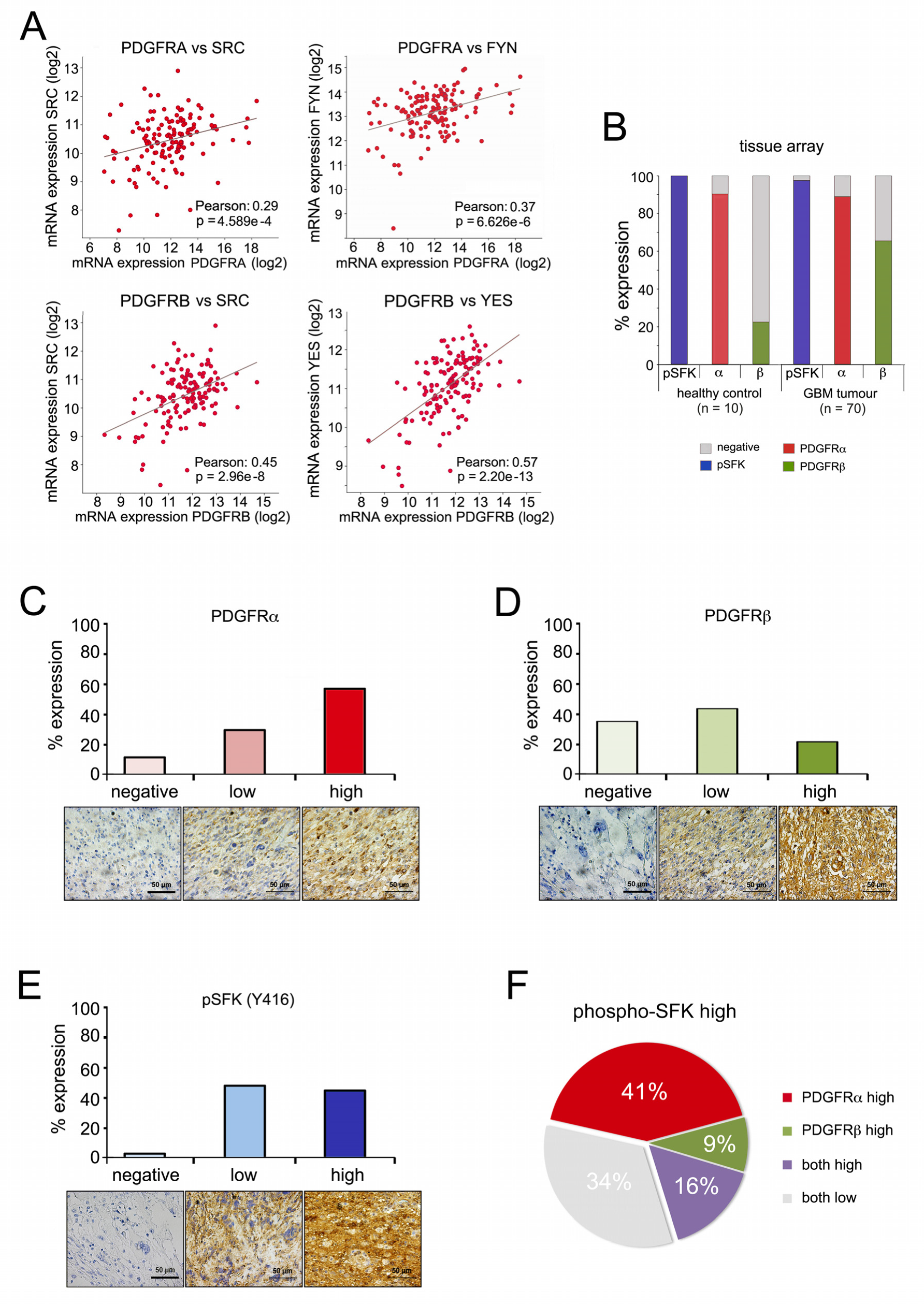
2.5. PDGFR Expression and SRC Kinase Activation Correlate in High-Grade Gliomas
2.6. Tyrosine Kinase Inhibition Overcomes Dexamethasone-Mediated Radioprotection
2.7. Dexamethasone Sensitizes Glioblastoma Cells to Sunitinib
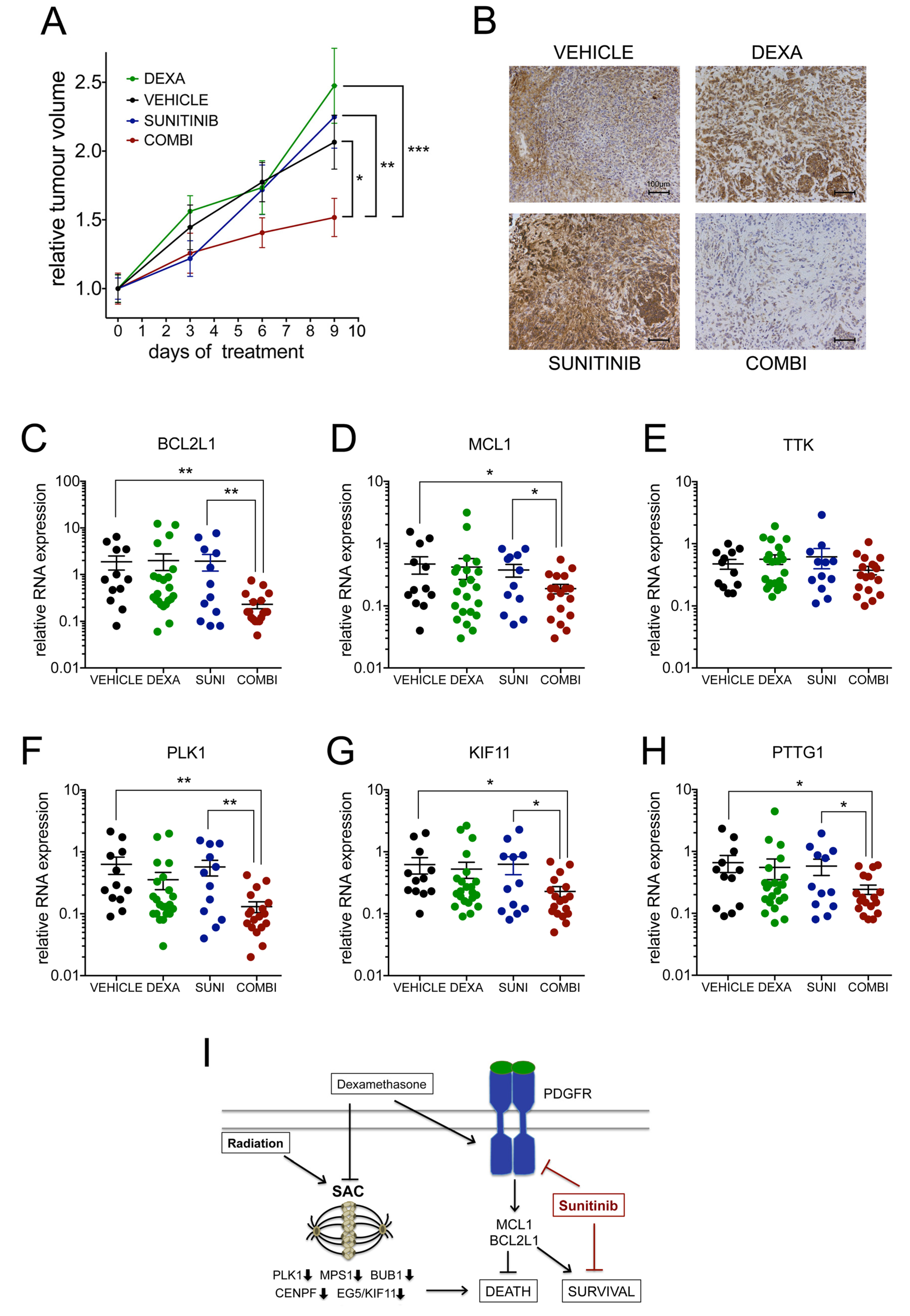
2.8. Dexamethasone Induces a Therapeutic Vulnerability for Sunitinib In Vivo
3. Discussion
4. Material and Methods
4.1. Patients
4.2. Cell Culture and Reagents
4.3. FDA-Approved Drug Screen
4.4. RNA Analysis
4.5. Colony Formation Assay
4.6. Neurosphere Formation Assays
4.7. Analysis of Mitotic Errors, SAC Override and Kinetochore Localisation
4.8. Tissue Microarray Analysis
4.9. Cell Lysis and Western Blot Analysis
4.10. In Vivo Drug Treatment
4.11. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for glioblastoma: Is it working? Drug Discov. Today 2019, 24, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Schiff, D.; Lee, E.Q.; Nayak, L.; Norden, A.D.; Reardon, D.A.; Wen, P.Y. Medical management of brain tumors and the sequelae of treatment. Neuro Oncol. 2015, 17, 488–504. [Google Scholar] [CrossRef] [Green Version]
- Hohwieler Schloss, M.; Freidberg, S.R.; Heatley, G.J.; Lo, T.C. Glucocorticoid dependency as a prognostic factor in radiotherapy for cerebral gliomas. Acta Oncol. 1989, 28, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Michaelsen, S.R.; Christensen, I.J.; Grunnet, K.; Stockhausen, M.T.; Broholm, H.; Kosteljanetz, M.; Poulsen, H.S. Clinical variables serve as prognostic factors in a model for survival from glioblastoma multiforme: An observational study of a cohort of consecutive non-selected patients from a single institution. BMC Cancer 2013, 13, 402. [Google Scholar] [CrossRef] [Green Version]
- Shields, L.B.; Shelton, B.J.; Shearer, A.J.; Chen, L.; Sun, D.A.; Parsons, S.; Bourne, T.D.; LaRocca, R.; Spalding, A.C. Dexamethasone administration during definitive radiation and temozolomide renders a poor prognosis in a retrospective analysis of newly diagnosed glioblastoma patients. Radiat. Oncol. 2015, 10, 222. [Google Scholar] [CrossRef] [Green Version]
- Watne, K.; Hannisdal, E.; Nome, O.; Hager, B.; Hirschberg, H. Prognostic factors in malignant gliomas with special reference to intra-arterial chemotherapy. Acta Oncol. 1993, 32, 307–310. [Google Scholar] [CrossRef]
- Wong, E.T.; Lok, E.; Gautam, S.; Swanson, K.D. Dexamethasone exerts profound immunologic interference on treatment efficacy for recurrent glioblastoma. Br. J. Cancer 2015, 113, 1642. [Google Scholar] [CrossRef] [PubMed]
- Cenciarini, M.; Valentino, M.; Belia, S.; Sforna, L.; Rosa, P.; Ronchetti, S.; D’Adamo, M.C.; Pessia, M. Dexamethasone in Glioblastoma Multiforme Therapy: Mechanisms and Controversies. Front. Mol. Neurosci. 2019, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Celestino, J.C.; Okada, Y.; Louis, D.N.; Fuller, G.N.; Holland, E.C. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001, 15, 1913–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Amankulor, N.M.; Helmy, K.Y.; Becher, O.J.; Holland, E.C. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl. Oncol. 2009, 2, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, E.C.; Hively, W.P.; DePinho, R.A.; Varmus, H.E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998, 12, 3675–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, C.Y.; Rudra, S.; Ma, S.; Campian, J.L.; Huang, J. Impact of overall corticosteroid exposure during chemoradiotherapy on lymphopenia and survival of glioblastoma patients. J. Neurooncol. 2019, 143, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [CrossRef] [Green Version]
- Ruan, W.; Lim, H.H.; Surana, U. Mapping Mitotic Death: Functional Integration of Mitochondria, Spindle Assembly Checkpoint and Apoptosis. Front. Cell Dev. Biol. 2018, 6, 177. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, A. Different cell fates after mitotic slippage: From aneuploidy to polyploidy. Mol. Cell Oncol. 2016, 3, e1088503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topham, C.; Tighe, A.; Ly, P.; Bennett, A.; Sloss, O.; Nelson, L.; Ridgway, R.A.; Huels, D.; Littler, S.; Schandl, C.; et al. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell 2015, 28, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santaguida, S.; Tighe, A.; D’Alise, A.M.; Taylor, S.S.; Musacchio, A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 2010, 190, 73–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amabile, G.; D’Alise, A.M.; Iovino, M.; Jones, P.; Santaguida, S.; Musacchio, A.; Taylor, S.; Cortese, R. The Aurora B kinase activity is required for the maintenance of the differentiated state of murine myoblasts. Cell Death Differ. 2009, 16, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Lennartsson, J. Structural and functional properties of platelet-derived growth factor and stem cell factor receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar]
- Kim, L.C.; Song, L.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef]
- Latha, K.; Li, M.; Chumbalkar, V.; Gururaj, A.; Hwang, Y.; Dakeng, S.; Sawaya, R.; Aldape, K.; Cavenee, W.K.; Bogler, O.; et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. Int. J. Cancer 2013, 132, 509–520. [Google Scholar] [CrossRef]
- Gressot, L.V.; Doucette, T.A.; Yang, Y.; Fuller, G.N.; Heimberger, A.B.; Bogler, O.; Rao, A.; Latha, K.; Rao, G. Signal transducer and activator of transcription 5b drives malignant progression in a PDGFB-dependent proneural glioma model by suppressing apoptosis. Int. J. Cancer 2015, 136, 2047–2054. [Google Scholar] [CrossRef] [Green Version]
- Vieira de Castro, J.; Goncalves, C.S.; Hormigo, A.; Costa, B.M. Exploiting the Complexities of Glioblastoma Stem Cells: Insights for Cancer Initiation and Therapeutic Targeting. Int. J. Mol. Sci. 2020, 21, 5278. [Google Scholar] [CrossRef]
- Moroz, M.A.; Huang, R.; Kochetkov, T.; Shi, W.; Thaler, H.; de Stanchina, E.; Gamez, I.; Ryan, R.P.; Blasberg, R.G. Comparison of corticotropin-releasing factor, dexamethasone, and temozolomide: Treatment efficacy and toxicity in U87 and C6 intracranial gliomas. Clin. Cancer Res. 2011, 17, 3282–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, J.; Galarneau, H.; Beaudet, M.J.; Tremblay, P.; Chernomoretz, A.; Vallieres, L. Reduced glioma growth following dexamethasone or anti-angiopoietin 2 treatment. Brain Pathol. 2008, 18, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Garcia, J.; Bott, M.; Klingeman Plati, S.; Dinh, C.T.; Bracho, O.R.; Yan, D.; Zou, B.; Mittal, R.; Telischi, F.F.; et al. Ponatinib promotes a G1 cell-cycle arrest of merlin/NF2-deficient human schwann cells. Oncotarget 2017, 8, 31666–31681. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Yozgat, Y.; de Stanchina, E.; Veach, D.; Clarkson, B.; Manova, K.; Giancotti, F.G.; Antonescu, C.R.; Besmer, P. Imatinib upregulates compensatory integrin signaling in a mouse model of gastrointestinal stromal tumor and is more effective when combined with dasatinib. Mol. Cancer Res. 2010, 8, 1271–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitadze, G.; Fluh, C.; Quabius, E.S.; Freitag-Wolf, S.; Peters, C.; Lettau, M.; Bhat, J.; Wesch, D.; Oberg, H.H.; Luecke, S.; et al. In-depth immunophenotyping of patients with glioblastoma multiforme: Impact of steroid treatment. Oncoimmunology 2017, 6, e1358839. [Google Scholar] [CrossRef]
- Dubinski, D.; Won, S.Y.; Gessler, F.; Quick-Weller, J.; Behmanesh, B.; Bernatz, S.; Forster, M.T.; Franz, K.; Plate, K.H.; Seifert, V.; et al. Dexamethasone-induced leukocytosis is associated with poor survival in newly diagnosed glioblastoma. J. Neurooncol. 2018, 137, 503–510. [Google Scholar] [CrossRef]
- Luedi, M.M.; Singh, S.K.; Mosley, J.C.; Hassan, I.S.A.; Hatami, M.; Gumin, J.; Andereggen, L.; Sulman, E.P.; Lang, F.F.; Stueber, F.; et al. Dexamethasone-mediated oncogenicity in vitro and in an animal model of glioblastoma. J. Neurosurg. 2018, 129, 1446–1455. [Google Scholar] [CrossRef] [Green Version]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; Martinez-Lacaci, I.; Mayor-Lopez, L.; Tristante, E.; Carballo-Santana, M.; Garcia-Morales, P.; Ventero Martin, M.P.; Fuentes-Baile, M.; Rodriguez-Lescure, A.; Saceda, M. PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells 2018, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.J.; Appleman, V.A.; Wu, H.J.; Rose, C.M.; Pineda, J.J.; Yeo, A.T.; Delcuze, B.; Lee, C.; Gyuris, A.; Zhu, H.; et al. A PDGFRalpha-driven mouse model of glioblastoma reveals a stathmin1-mediated mechanism of sensitivity to vinblastine. Nat. Commun. 2018, 9, 3116. [Google Scholar] [CrossRef]
- Balana, C.; Gil, M.J.; Perez, P.; Reynes, G.; Gallego, O.; Ribalta, T.; Capellades, J.; Gonzalez, S.; Verger, E. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: Results of a phase II study. Target. Oncol. 2014, 9, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, S.; Ferrari, V.D.; Buglione, M.; Agazzi, G.M.; Liserre, R.; Poliani, L.; Buttolo, L.; Gipponi, S.; Pedersini, R.; Consoli, F.; et al. Second line treatment of recurrent glioblastoma with sunitinib: Results of a phase II study and systematic review of literature. J. Neurosurg. Sci. 2019, 63, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Nowosielski, M.; Haybaeck, J.; Embacher, S.; Stockhammer, F.; Gotwald, T.; Holzner, B.; Capper, D.; Preusser, M.; Marosi, C.; et al. A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (SURGE 01-07). Neuro Oncol. 2014, 16, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisl, T.N.; Smith, P.; Sul, J.; Salgado, C.; Iwamoto, F.M.; Shih, J.H.; Fine, H.A. Continuous daily sunitinib for recurrent glioblastoma. J. Neurooncol. 2013, 111, 41–48. [Google Scholar] [CrossRef]
- Pan, E.; Yu, D.; Yue, B.; Potthast, L.; Chowdhary, S.; Smith, P.; Chamberlain, M. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. J. Neurooncol. 2012, 110, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Gore, M.E.; Hariharan, S.; Porta, C.; Bracarda, S.; Hawkins, R.; Bjarnason, G.A.; Oudard, S.; Lee, S.H.; Carteni, G.; Nieto, A.; et al. Sunitinib in metastatic renal cell carcinoma patients with brain metastases. Cancer 2011, 117, 501–509. [Google Scholar] [CrossRef]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Pariente, A.; Perez-Sala, A.; Ochoa, R.; Pelaez, R.; Larrayoz, I.M. Genome-Wide Transcriptomic Analysis Identifies Pathways Regulated by Sterculic Acid in Retinal Pigmented Epithelium Cells. Cells 2020, 9, 1187. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Ferguson, J.; Smith, M.; Zudaire, I.; Wellbrock, C.; Arozarena, I. Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 2017, 8, 32946–32959. [Google Scholar] [CrossRef] [Green Version]
- Erice, O.; Smith, M.P.; White, R.; Goicoechea, I.; Barriuso, J.; Jones, C.; Margison, G.P.; Acosta, J.C.; Wellbrock, C.; Arozarena, I. MGMT Expression Predicts PARP-Mediated Resistance to Temozolomide. Mol. Cancer Ther. 2015, 14, 1236–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.L.; Scott, M.I.; Holt, S.V.; Hussein, D.; Taylor, S.S. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 2004, 117, 1577–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldaz, P.; Auzmendi-Iriarte, J.; Durántez, M.; Lasheras-Otero, I.; Carrasco-Garcia, E.; Zelaya, M.V.; Bragado, L.; Olías-Arjona, A.; Egaña, L.; Samprón, N.; et al. Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma. Cancers 2021, 13, 361. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020361
Aldaz P, Auzmendi-Iriarte J, Durántez M, Lasheras-Otero I, Carrasco-Garcia E, Zelaya MV, Bragado L, Olías-Arjona A, Egaña L, Samprón N, et al. Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma. Cancers. 2021; 13(2):361. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020361
Chicago/Turabian StyleAldaz, Paula, Jaione Auzmendi-Iriarte, Maika Durántez, Irene Lasheras-Otero, Estefania Carrasco-Garcia, M. Victoria Zelaya, Laura Bragado, Ana Olías-Arjona, Larraitz Egaña, Nicolás Samprón, and et al. 2021. "Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma" Cancers 13, no. 2: 361. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020361