
Genomic Sub-Classification of Ovarian Clear Cell Carcinoma Revealed by Distinct Mutational Signatures


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Biological Samples
2.2. DNA Extraction
2.3. Microsatellite Instability Assay
2.4. DNA Next-Generation Sequencing
2.5. Data Analysis
2.6. Statistical Analysis
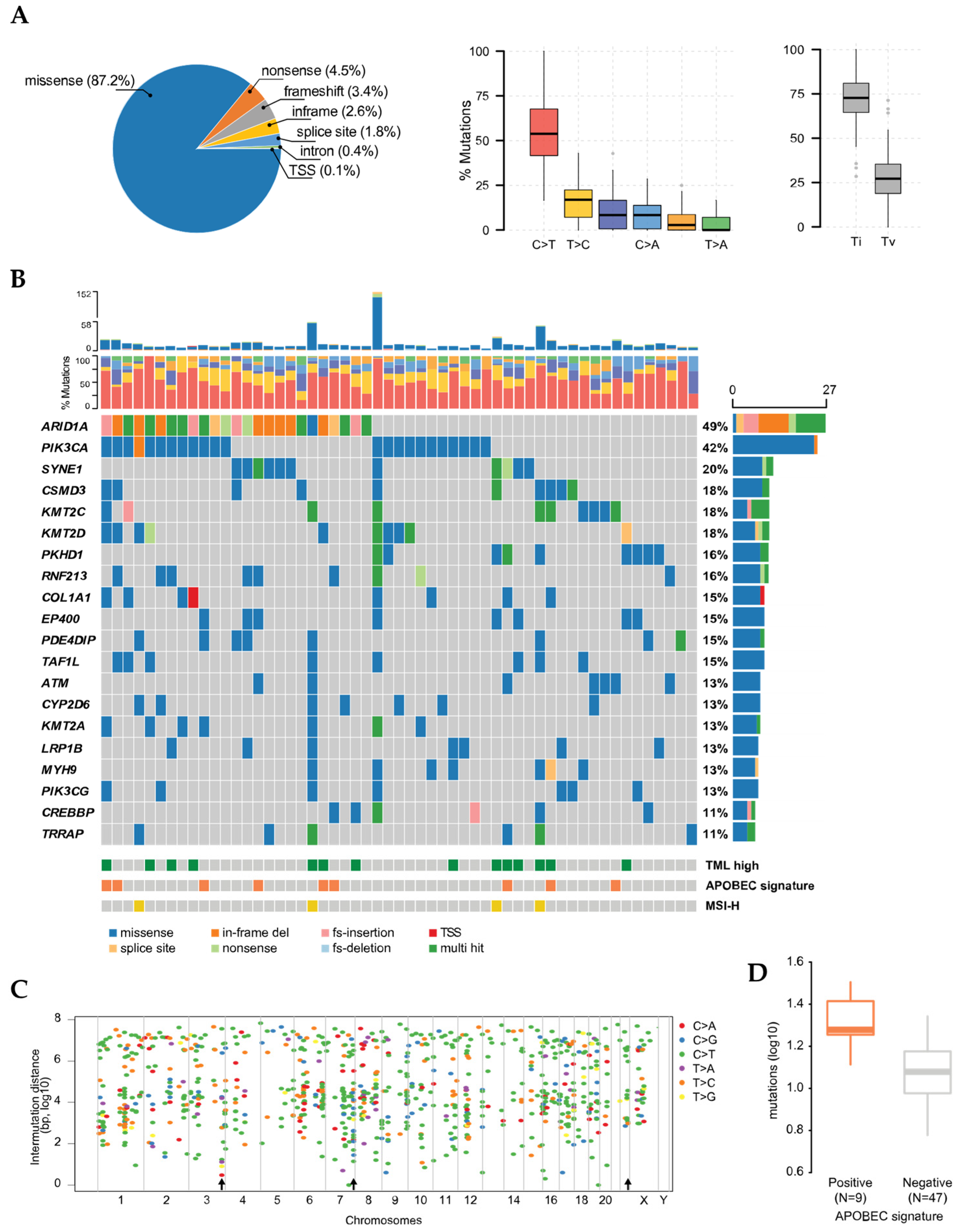
3. Results
3.1. Molecular Stratification and Localized Hypermutation
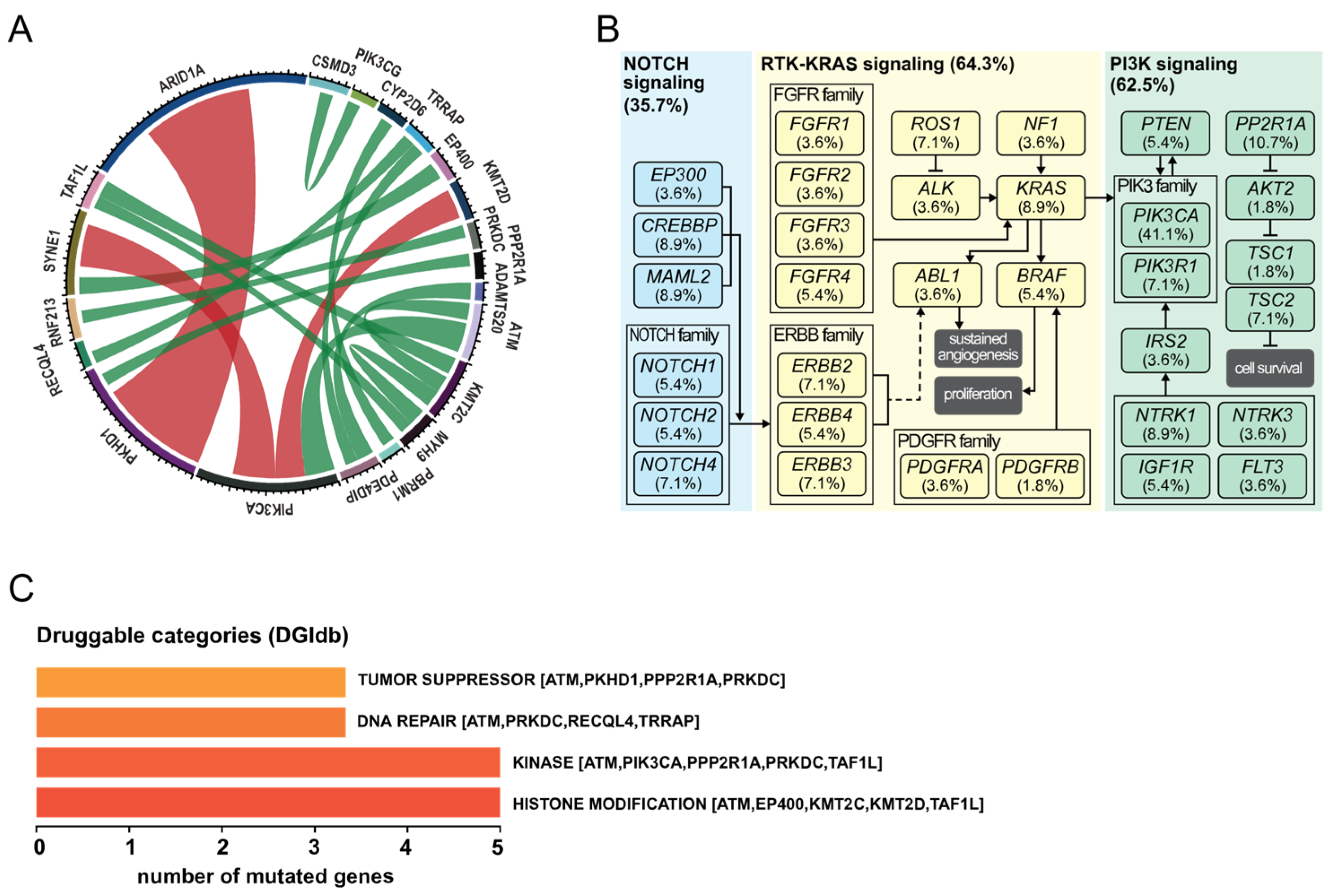
3.2. Co-Occurrence of Mutations and Metabolic Pathways
3.3. Mutational Signatures in OCCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.M.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef]
- Takahashi, K.; Takenaka, M.; Kawabata, A.; Yanaihara, N.; Okamoto, A. Rethinking of treatment strategies and clinical management in ovarian clear cell carcinoma. Int. J. Clin. Oncol. 2020, 25, 425–431. [Google Scholar] [CrossRef]
- Schnack, T.H.; Hogdall, E.; Nedergaard, L.; Hogdall, C. Demographic Clinical and Prognostic Factors of Primary Ovarian Adenocarcinomas of Serous and Clear Cell Histology—A Comparative Study. Int. J. Gynecol. Cancer 2016, 26, 82–90. [Google Scholar] [CrossRef]
- Zhu, C.; Zhu, J.; Qian, L.; Liu, H.; Shen, Z.; Wu, D.; Zhao, W.; Xiao, W.; Zhou, Y. Clinical characteristics and prognosis of ovarian clear cell carcinoma: A 10-year retrospective study. BMC Cancer 2021, 21, 322. [Google Scholar] [CrossRef]
- Danish Gynecologic Cancer Database. Landsdækkende Klinisk Database for Gynækologisk Kræft—National Årsrapport 2017/2018 og 2018/2019; Danish Gynecologic Cancer Database: Denmark, 2019; p. 77. Available online: https://www.sundhed.dk/content/cms/80/4680_aarsrapport_dansk_gynaekologisk_cancer_database_2017-2019_offentlig_annonymiseret.pdf (accessed on 20 August 2021).
- Sorensen, S.M.; Bjorn, S.F.; Jochumsen, K.M.; Jensen, P.T.; Thranov, I.R.; Hare-Bruun, H.; Seibaek, L.; Hogdall, C. Danish Gynecological Cancer Database. Clin. Epidemiol. 2016, 8, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Schnack, T.H.; Hogdall, E.; Thomsen, L.N.; Hogdall, C. Demographic, Clinical, and Prognostic Factors of Ovarian Clear Cell Adenocarcinomas According to Endometriosis Status. Int. J. Gynecol. Cancer 2017, 27, 1804–1812. [Google Scholar] [CrossRef]
- Chan, J.K.; Teoh, D.; Hu, J.M.; Shin, J.Y.; Osann, K.; Kapp, D.S. Do clear cell ovarian carcinomas have poorer prognosis compared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecol. Oncol. 2008, 109, 370–376. [Google Scholar] [CrossRef]
- McCabe, M.J.; Gauthier, M.A.; Chan, C.L.; Thompson, T.J.; De Sousa, S.M.C.; Puttick, C.; Grady, J.P.; Gayevskiy, V.; Tao, J.; Ying, K.; et al. Development and validation of a targeted gene sequencing panel for application to disparate cancers. Sci. Rep. 2019, 9, 17052. [Google Scholar] [CrossRef]
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.M.; Patterson, N.E.; Zechmeister, J.M.; Bejerano-Sagie, M.; Delio, M.; Patel, K.; Ravi, N.; Quispe-Tintaya, W.; Maslov, A.; Simmons, N.; et al. Development and validation of a targeted next generation DNA sequencing panel outperforming whole exome sequencing for the identification of clinically relevant genetic variants. Oncotarget 2017, 8, 102033–102045. [Google Scholar] [CrossRef]
- Castellanos, E.; Gel, B.; Rosas, I.; Tornero, E.; Santin, S.; Pluvinet, R.; Velasco, J.; Sumoy, L.; Del Valle, J.; Perucho, M.; et al. A comprehensive custom panel design for routine hereditary cancer testing: Preserving control, improving diagnostics and revealing a complex variation landscape. Sci. Rep. 2017, 7, 39348. [Google Scholar] [CrossRef]
- Maggi, E.; Montagna, C. AACR precision medicine series: Highlights of the integrating clinical genomics and cancer therapy meeting. Mutat. Res. 2015, 782, 44–51. [Google Scholar] [CrossRef]
- Itamochi, H.; Oishi, T.; Oumi, N.; Takeuchi, S.; Yoshihara, K.; Mikami, M.; Yaegashi, N.; Terao, Y.; Takehara, K.; Ushijima, K.; et al. Whole-genome sequencing revealed novel prognostic biomarkers and promising targets for therapy of ovarian clear cell carcinoma. Br. J. Cancer 2017, 117, 717–724. [Google Scholar] [CrossRef]
- Kim, S.I.; Lee, J.W.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; Seo, J.S. Genomic landscape of ovarian clear cell carcinoma via whole exome sequencing. Gynecol. Oncol. 2018, 148, 375–382. [Google Scholar] [CrossRef]
- R.C. Team. A Language and Environment for Statistical Computing; 3.4.1 (Single Candle); R.C. Team: Vienna, Austria, 2017. [Google Scholar]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Bi, R.; Ma, P.; Zhang, S.; Zhang, Y.; Sun, Y.; Zhang, Y.; Jing, Y.; Yu, M.; Wang, W.; et al. Multiregion whole-genome sequencing depicts intratumour heterogeneity and punctuated evolution in ovarian clear cell carcinoma. J. Med. Genet. 2020, 57, 605–609. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Gehring, J.S.; Fischer, B.; Lawrence, M.; Huber, W. SomaticSignatures: Inferring mutational signatures from single-nucleotide variants. Bioinformatics 2015, 31, 3673–3675. [Google Scholar] [CrossRef] [Green Version]
- Huebschmann, D.; Jopp-Saile, L.; Andresen, C.; Gu, Z.; Schlesner, M. YAPSA: Yet Another Package for Signature Analysis, version 1.18.0. 2021. Available online: http://bioconductor.org/packages/release/bioc/html/YAPSA.html (accessed on 20 August 2021).
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef]
- Okamoto, A.; Glasspool, R.M.; Mabuchi, S.; Matsumura, N.; Nomura, H.; Itamochi, H.; Takano, M.; Takano, T.; Susumu, N.; Aoki, D.; et al. Gynecologic Cancer InterGroup (GCIG) consensus review for clear cell carcinoma of the ovary. Int. J. Gynecol. Cancer 2014, 24, S20–S25. [Google Scholar] [CrossRef]
- Mabuchi, S.; Sugiyama, T.; Kimura, T. Clear cell carcinoma of the ovary: Molecular insights and future therapeutic perspectives. J. Gynecol. Oncol. 2016, 27, e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Tokunaga, H.; Saito, S.; Shimokawa, K.; Katsuoka, F.; Bin, L.; Kojima, K.; Nagasaki, M.; Yamamoto, M.; Yaegashi, N.; et al. Identification of somatic genetic alterations in ovarian clear cell carcinoma with next generation sequencing. Genes Chromosomes Cancer 2018, 57, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Iizuka, T.; Endo, S.; Horie, K.; Kanda, H.; Niki, T. Ovarian clear cell carcinoma with an immature teratoma component showing ARID1A deficiency and an identical PIK3CA mutation. J. Obstet. Gynaecol. Res. 2021, 47, 3401–3407. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.; Fournier, L.A.; Chang, E.Y.; Wells, J.P.; Minaker, S.W.; Zhu, Y.D.; Wang, A.Y.; Wang, Y.; Huntsman, D.G.; Stirling, P.C. ARID1A regulates R-loop associated DNA replication stress. PLoS Genet. 2021, 17, e1009238. [Google Scholar] [CrossRef]
- Maeda, D.; Mao, T.L.; Fukayama, M.; Nakagawa, S.; Yano, T.; Taketani, Y.; Shih Ie, M. Clinicopathological significance of loss of ARID1A immunoreactivity in ovarian clear cell carcinoma. Int. J. Mol. Sci. 2010, 11, 5120–5128. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Gao, M.; Wu, C.H.; Wang, T.L.; Shih Ie, M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 2012, 14, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Hong, D.S.; Stepanek, V.M.; Fu, S.; Piha-Paul, S.A.; Lee, J.J.; Luthra, R.; et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013, 73, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Chandler, R.L.; Damrauer, J.S.; Raab, J.R.; Schisler, J.C.; Wilkerson, M.D.; Didion, J.P.; Starmer, J.; Serber, D.; Yee, D.; Xiong, J.; et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun. 2015, 6, 6118. [Google Scholar] [CrossRef] [Green Version]
- Samartzis, E.P.; Gutsche, K.; Dedes, K.J.; Fink, D.; Stucki, M.; Imesch, P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget 2014, 5, 5295–5303. [Google Scholar] [CrossRef]
- Wu, Y.H.; Huang, Y.F.; Chen, C.C.; Huang, C.Y.; Chou, C.Y. Comparing PI3K/Akt Inhibitors Used in Ovarian Cancer Treatment. Front. Pharmacol. 2020, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Browning, E.A.; Hong, N.; DeBolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.; Girio, A.; Cebola, I.; Santos, C.I.; Antunes, F.; Barata, J.T. Intracellular reactive oxygen species are essential for PI3K/Akt/mTOR-dependent IL-7-mediated viability of T-cell acute lymphoblastic leukemia cells. Leukemia 2011, 25, 960–967. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.A.; Morales-Oyarvide, V.; Campbell, S.; Longacre, T.A.; Oliva, E. Mismatch Repair Protein Expression in Clear Cell Carcinoma of the Ovary: Incidence and Morphologic Associations in 109 Cases. Am. J. Surg. Pathol. 2016, 40, 656–663. [Google Scholar] [CrossRef]
- Chui, M.H.; Gilks, C.B.; Cooper, K.; Clarke, B.A. Identifying Lynch syndrome in patients with ovarian carcinoma: The significance of tumor subtype. Adv. Anat. Pathol. 2013, 20, 378–386. [Google Scholar] [CrossRef]
- Helleman, J.; van Staveren, I.L.; Dinjens, W.N.; van Kuijk, P.F.; Ritstier, K.; Ewing, P.C.; van der Burg, M.E.; Stoter, G.; Berns, E.M. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer 2006, 6, 201. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.Q.; Albarracin, C.; Rosen, D.; Zhong, R.; Zheng, W.; Luthra, R.; Broaddus, R.; Liu, J. Microsatellite instability and alteration of the expression of hMLH1 and hMSH2 in ovarian clear cell carcinoma. Hum. Pathol. 2004, 35, 552–559. [Google Scholar] [CrossRef]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J., III; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, T.; Park, P.H.; Wu, S.; Fatkhutdinov, N.; Karakashev, S.; Nacarelli, T.; Kossenkov, A.V.; Speicher, D.W.; Jean, S.; Zhang, L.; et al. Repurposing Pan-HDAC Inhibitors for ARID1A-Mutated Ovarian Cancer. Cell Rep. 2018, 22, 3393–3400. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, D.V.N.P.; Schnack, T.H.; Poulsen, T.S.; Christiansen, A.P.; Høgdall, C.K.; Høgdall, E.V. Genomic Sub-Classification of Ovarian Clear Cell Carcinoma Revealed by Distinct Mutational Signatures. Cancers 2021, 13, 5242. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205242
Oliveira DVNP, Schnack TH, Poulsen TS, Christiansen AP, Høgdall CK, Høgdall EV. Genomic Sub-Classification of Ovarian Clear Cell Carcinoma Revealed by Distinct Mutational Signatures. Cancers. 2021; 13(20):5242. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205242
Chicago/Turabian StyleOliveira, Douglas V. N. P., Tine H. Schnack, Tim S. Poulsen, Anne P. Christiansen, Claus K. Høgdall, and Estrid V. Høgdall. 2021. "Genomic Sub-Classification of Ovarian Clear Cell Carcinoma Revealed by Distinct Mutational Signatures" Cancers 13, no. 20: 5242. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205242