
AKF-D52, a Synthetic Phenoxypyrimidine-Urea Derivative, Triggers Extrinsic/Intrinsic Apoptosis and Cytoprotective Autophagy in Human Non-Small Cell Lung Cancer Cells


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Culture
2.3. MTT Assay
2.4. DAPI Staining
2.5. Annexin V/PI Staining for Apoptosis Analysis
2.6. Nuclear Extraction
2.7. Western Blot Analysis
2.8. Analysis of Mitochondrial Membrane Potential (ΔΨm)
2.9. Preparation of Cytosolic and Mitochondrial Fractionation
2.10. Immunoprecipitation Assay
2.11. Acridine Orange Staining
2.12. Measurement of Reactive Oxygen Species (ROS)
2.13. Animals
2.14. In Vivo Tumor Xenograft Studies
2.15. Measurement of Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST), and Blood Urea Nitrogen (BUN) Levels
2.16. Statistical Analysis
3. Results
3.1. AKF-D52 Suppresses Cell Viability via Apoptotic Cell Death in A549 and NCI-H358 Cells
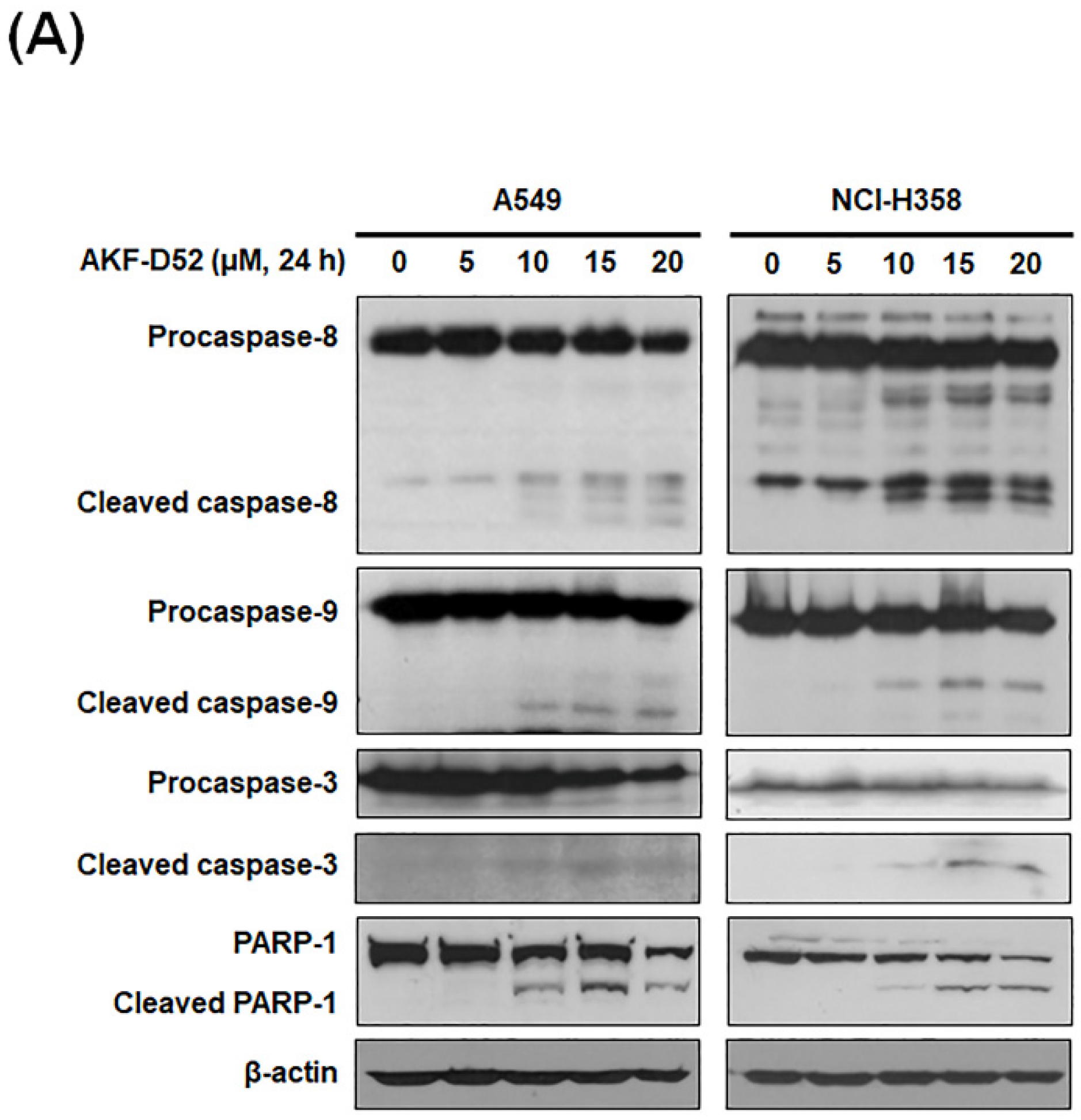
3.2. AKF-D52 Concurrently Induces Caspase-Dependent and -Independent Apoptosis in A549 and NCI-H358 Cells
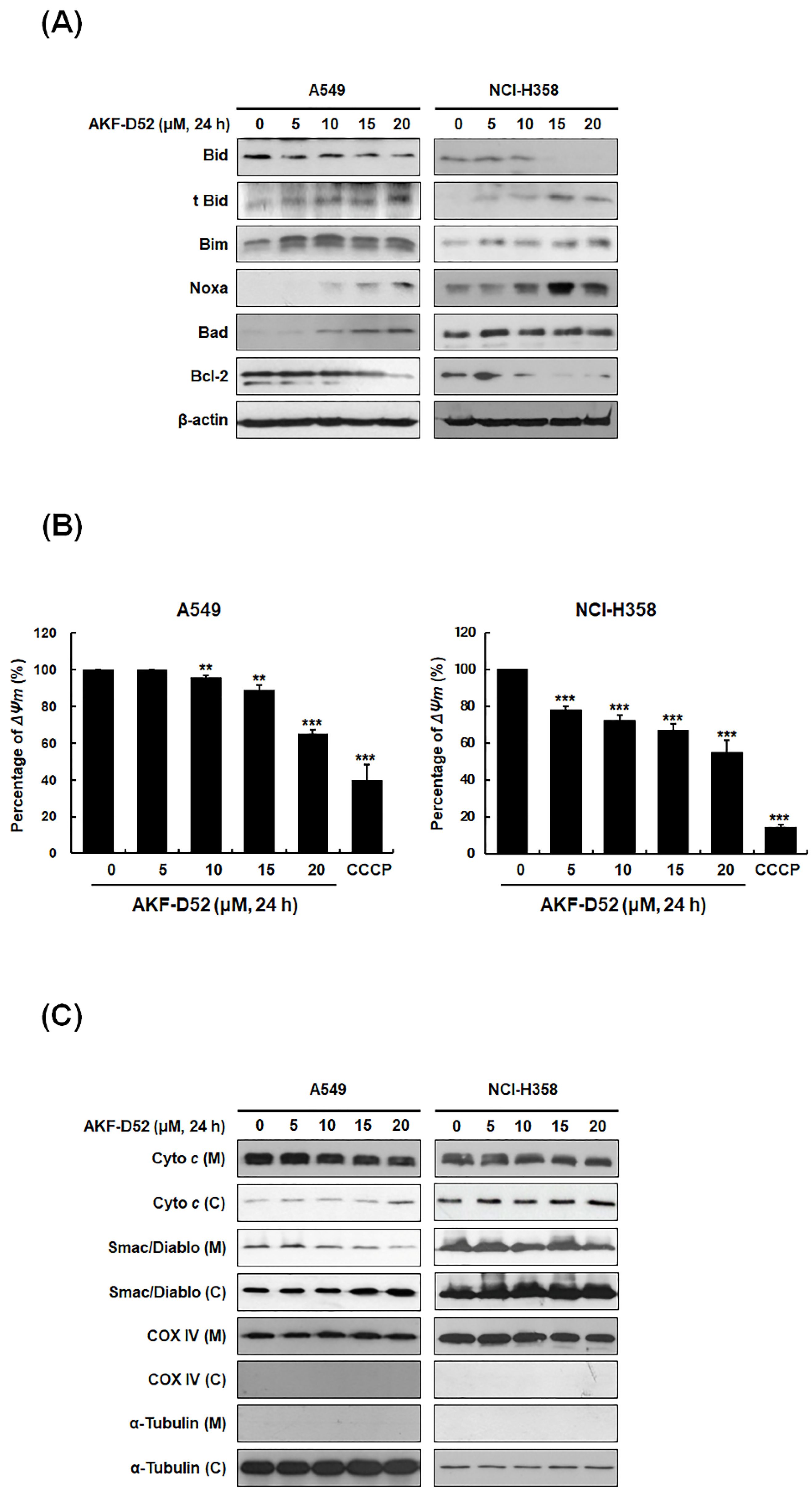
3.3. AKF-D52-Induced Apoptosis Is Dependent on Mitochondrial Dysfunction in A549 and NCI-H358 Cells
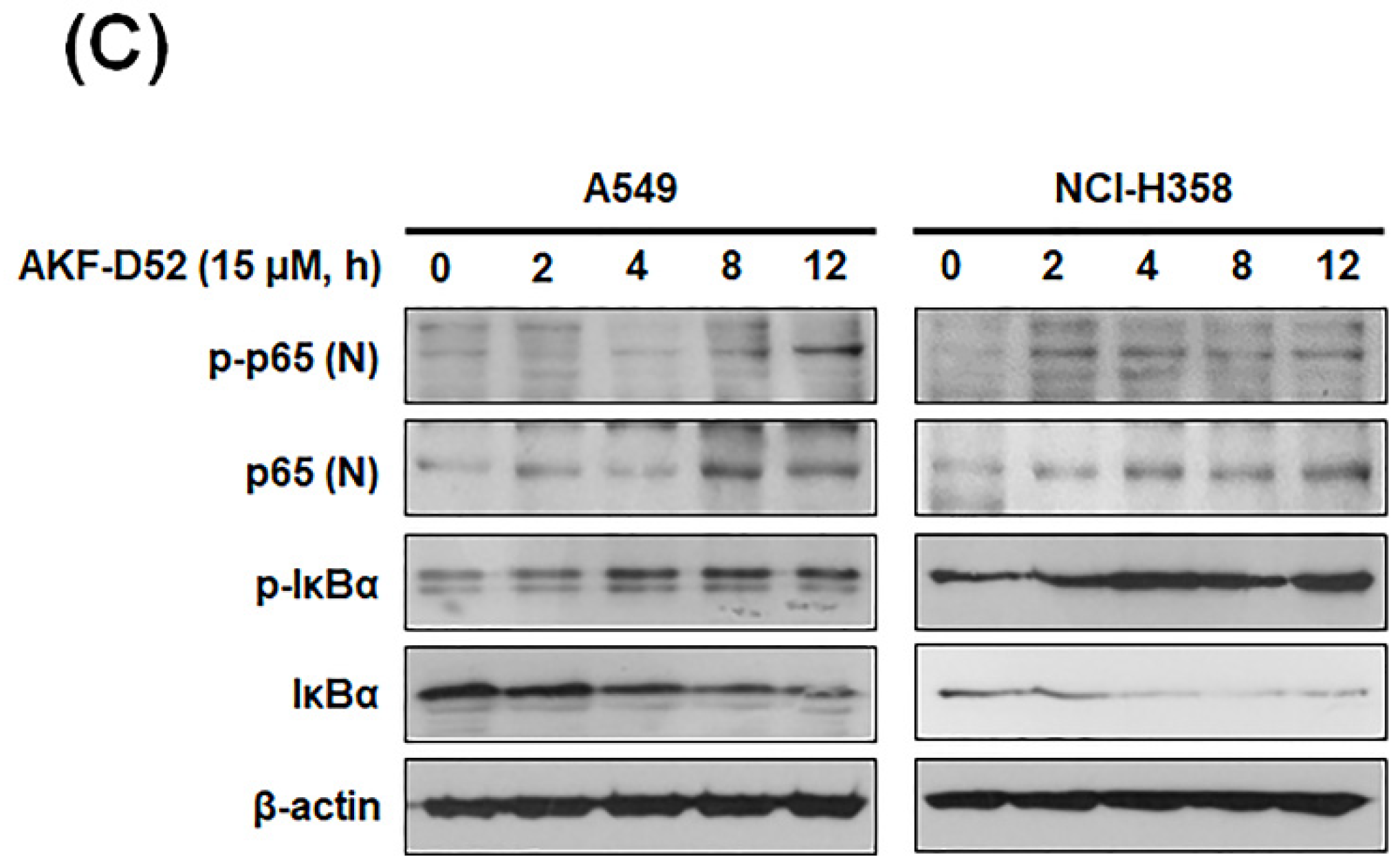
3.4. AKF-D52-Induced Apoptosis Requires Death-Inducing Signaling Complex Activation in A549 and NCI-H358 Cells
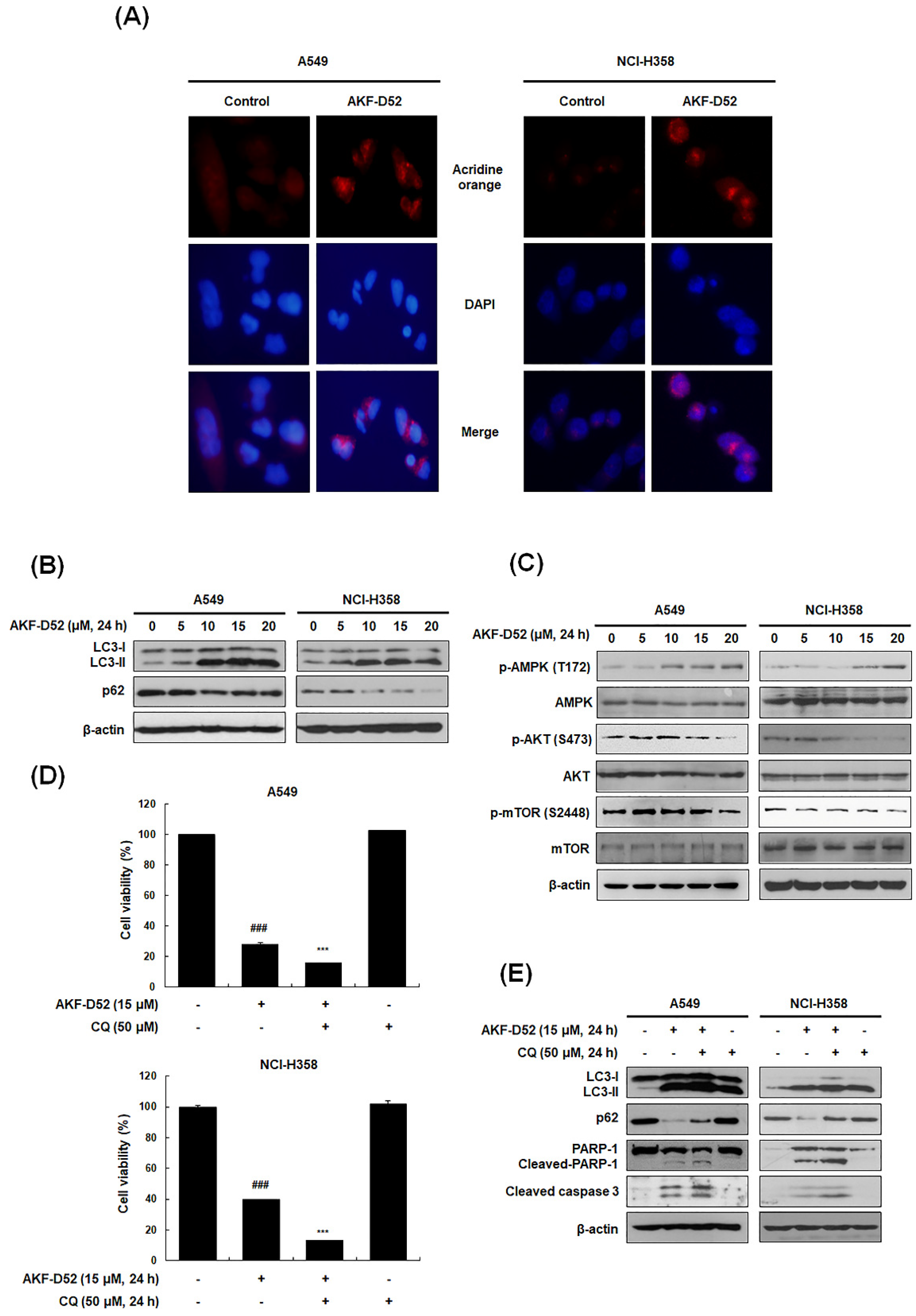
3.5. Inhibition of Cytoprotective Autophagy Enhances AKF-D52-Induced Apoptosis in A549 and NCI-H358 Cells
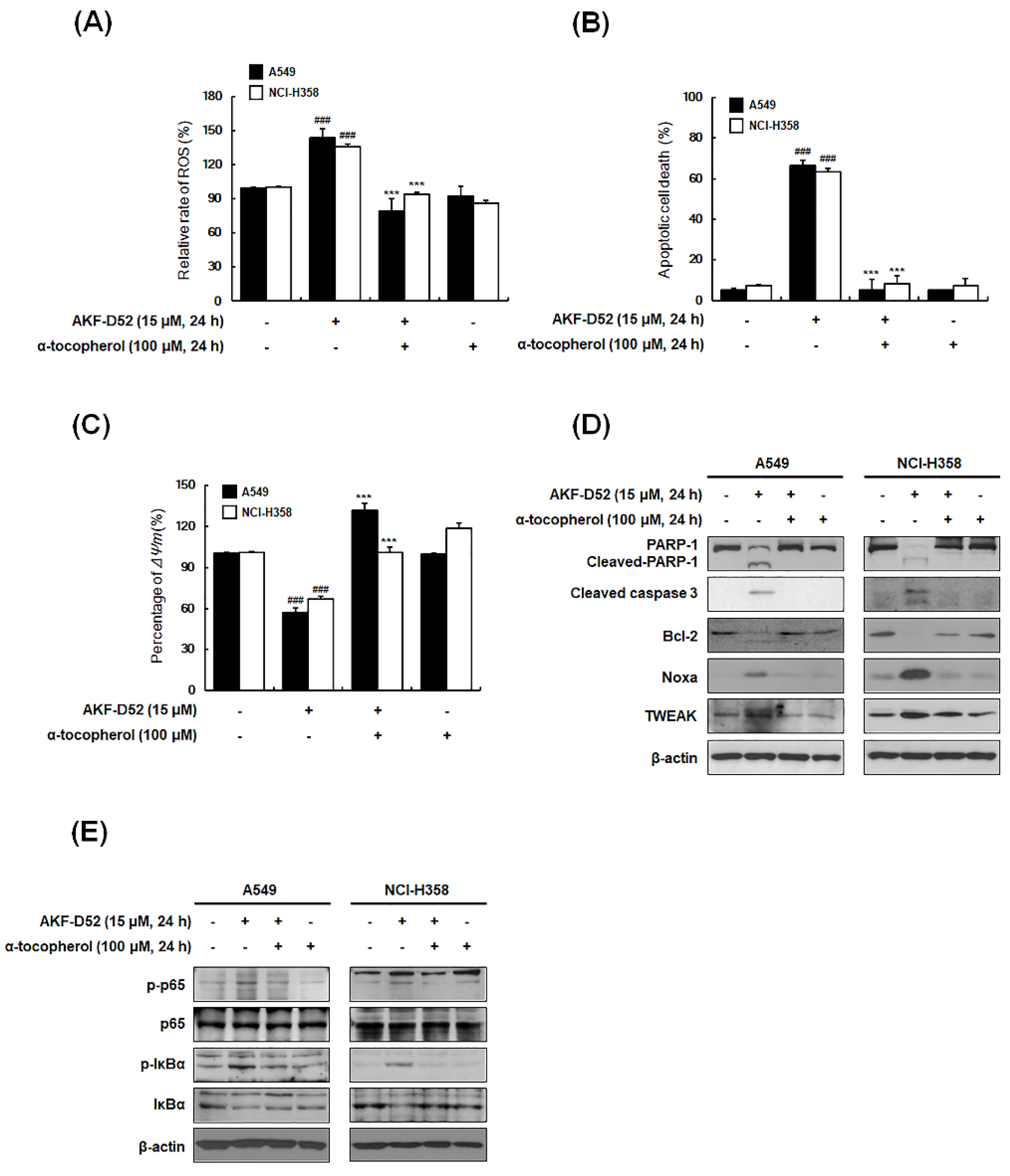
3.6. ROS Plays an Important Role in AKF-D52-Induced Apoptosis in A549 and NCI-H358 Cells
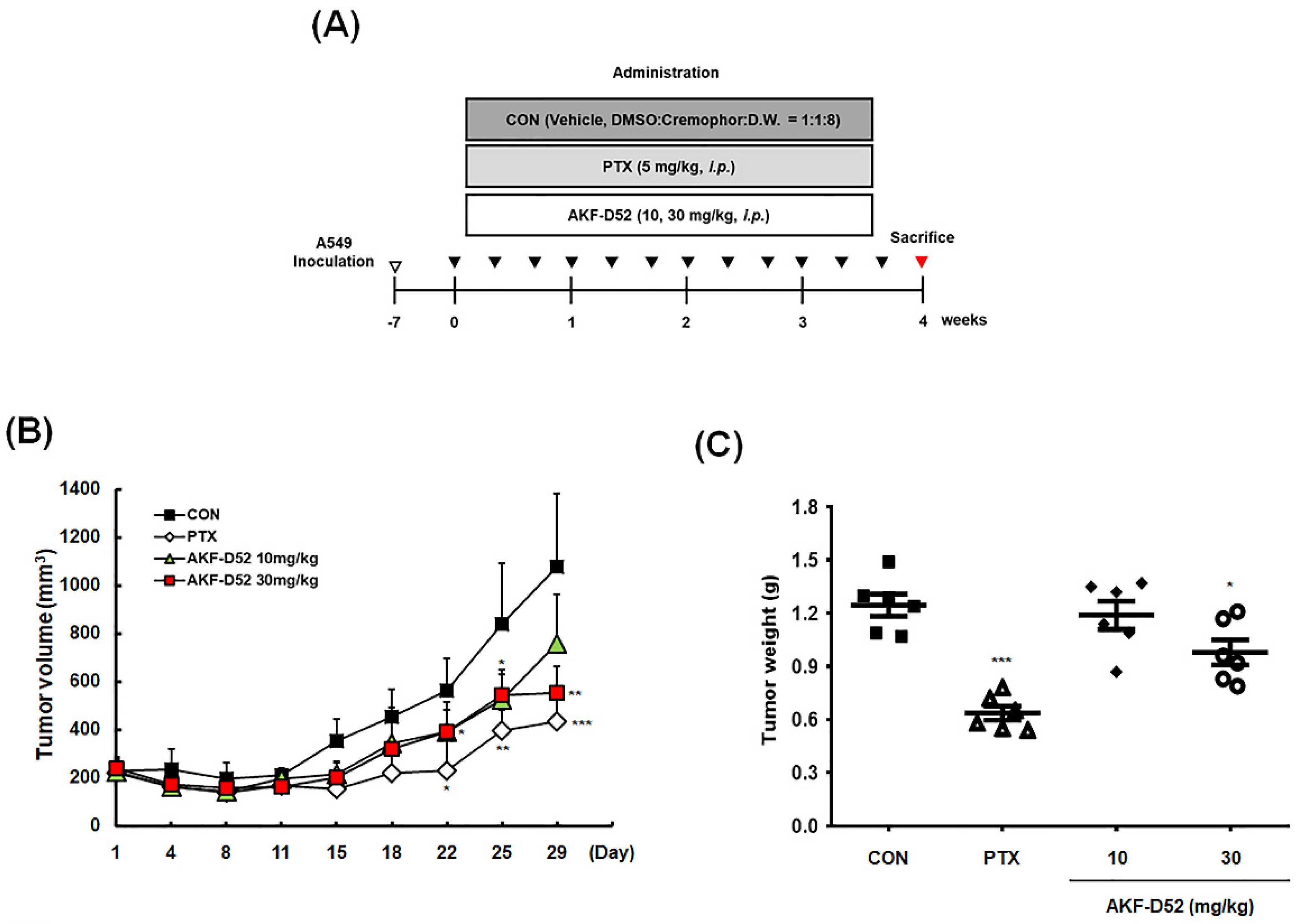
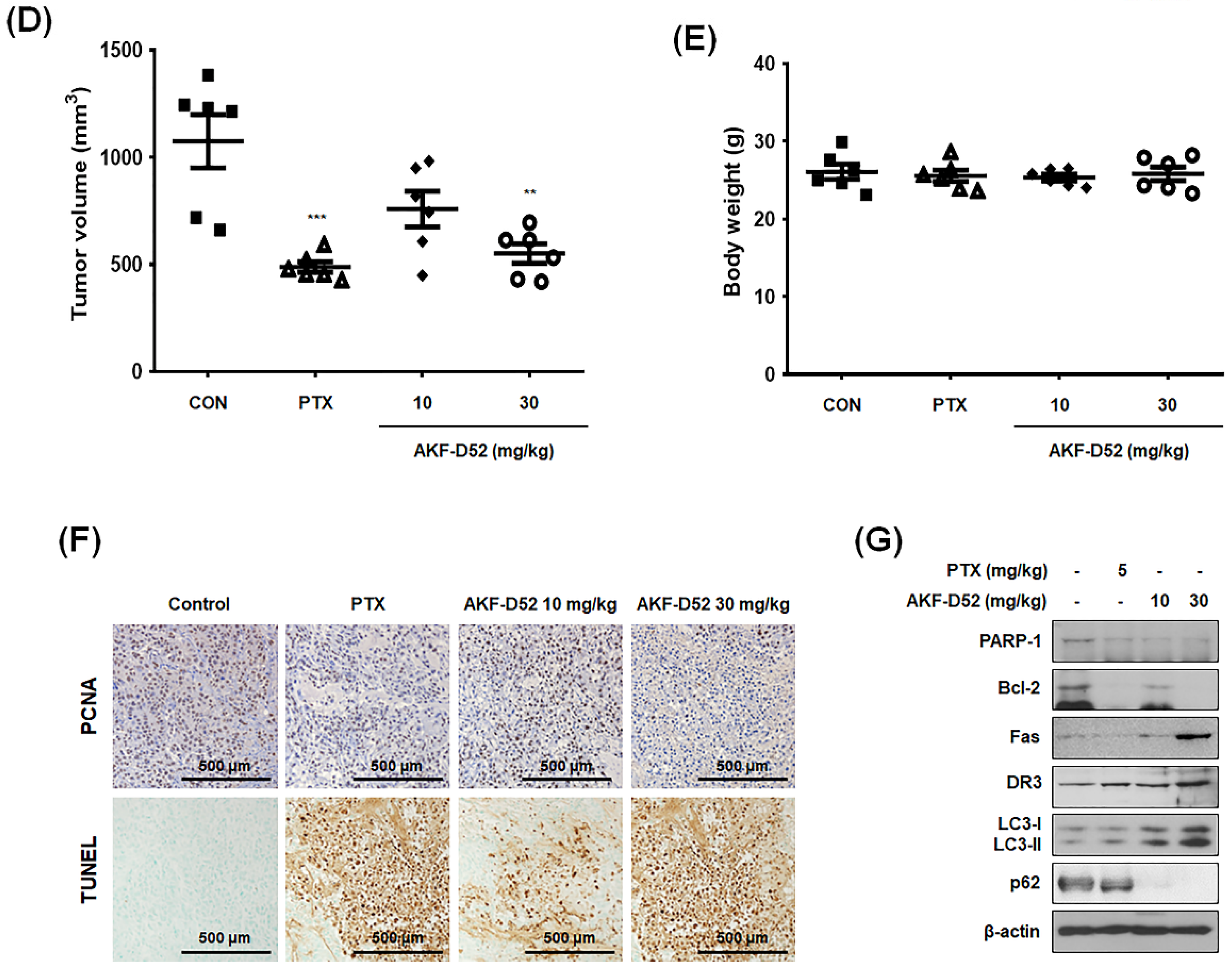
3.7. AKF-D52 Inhibits Tumor Growth in an A549 Xenograft Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Testa, U.; Castelli, G.; Pelosi, E. Lung Cancers: Molecular Characterization, Clonal Heterogeneity and Evolution, and Cancer Stem Cells. Cancers 2018, 10, 248. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Chung, L.Y.; Tang, S.J.; Sun, G.H.; Chou, T.Y.; Yeh, T.S.; Yu, S.L.; Sun, K.H. Galectin-1 promotes lung cancer progression and chemoresistance by upregulating p38 MAPK, ERK, and cyclooxygenase-2. Clin. Cancer Res. 2012, 18, 4037–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Li, J.; Yang, Y.; Zhao, X.; Liu, Y.; Jiang, Y.; Zhou, L.; Feng, Y.; Yu, Y.; Cheng, Y. Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53dependent pathway: Integrated bioinformatics analysis and experimental validation. Int. J. Oncol. 2020, 57, 925–938. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, Y.; Cheng, X.; Fan, Y.; He, S.; Li, S.; Ye, H.; Xie, C.; Wu, W.; Li, C.; et al. Isogambogenic acid induces apoptosis-independent autophagic cell death in human non-small-cell lung carcinoma cells. Sci. Rep. 2015, 5, 7697. [Google Scholar] [CrossRef]
- Sui, Y.; Yao, H.; Li, S.; Jin, L.; Shi, P.; Li, Z.; Wang, G.; Lin, S.; Wu, Y.; Li, Y.; et al. Delicaflavone induces autophagic cell death in lung cancer via Akt/mTOR/p70S6K signaling pathway. J. Mol. Med. 2017, 95, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, X.; Hu, Y.; Lei, T.; Liu, T. Sotetsuflavone Induces Autophagy in Non-Small Cell Lung Cancer Through Blocking PI3K/Akt/mTOR Signaling Pathway in Vivo and in Vitro. Front. Pharmacol. 2019, 10, 1460. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wang, Z.; Lu, D.; Huang, J.; Liu, J.; Hong, L. Paeonol induces cytoprotective autophagy via blocking the Akt/mTOR pathway in ovarian cancer cells. Cell Death Dis. 2019, 10, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.Q.; Feng, Y.Y.; Luo, Y.H.; Zhai, Y.Q.; Ju, X.Y.; Feng, Y.C.; Sheng, Y.N.; Wang, J.R.; Yu, C.Q.; Jin, C.H. Quinalizarin induces ROSmediated apoptosis via the MAPK, STAT3 and NFkappaB signaling pathways in human breast cancer cells. Mol. Med. Rep. 2019, 20, 4576–4586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.Y.; Park, K.I.; Kim, S.H.; Yu, S.N.; Lee, D.; Kim, Y.W.; Noh, K.T.; Ma, J.Y.; Seo, Y.K.; Ahn, S.C. Salinomycin Induces Reactive Oxygen Species and Apoptosis in Aggressive Breast Cancer Cells as Mediated with Regulation of Autophagy. Anticancer Res. 2017, 37, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Wei, S.; Wang, J.; Liu, X. Isoorientin induces apoptosis and autophagy simultaneously by reactive oxygen species (ROS)-related p53, PI3K/Akt, JNK, and p38 signaling pathways in HepG2 cancer cells. J. Agric. Food Chem. 2014, 62, 5390–5400. [Google Scholar] [CrossRef]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G.; Ferraro, M. Diaryl Urea: A Privileged Structure in Anticancer Agents. Curr. Med. Chem. 2016, 23, 1528–1548. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.T.; Sun, C.C.; Wu, C.H.; Lee, Y.J.; Chiang, C.H.; Wang, W.S. Regorafenib Induces Apoptosis and Inhibits Metastatic Potential of Human Bladder Carcinoma Cells. Anticancer Res. 2017, 37, 4919–4926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.N.; Wang, X.F.; Li, T.; Wu, D.W.; Fu, X.B.; Zhang, G.J.; Shen, X.C.; Wang, H.S. Design, synthesis, and biological evaluation of novel quinazolinyl-diaryl urea derivatives as potential anticancer agents. Eur. J. Med. Chem. 2016, 107, 12–25. [Google Scholar] [CrossRef]
- Azimian, F.; Hamzeh-Mivehroud, M.; Shahbazi Mojarrad, J.; Hemmati, S.; Dastmalchi, S. Synthesis and biological evaluation of diaryl urea derivatives designed as potential anticarcinoma agents using de novo structure-based lead optimization approach. Eur. J. Med. Chem. 2020, 201, 112461. [Google Scholar] [CrossRef]
- Farag, A.K.; Hassan, A.H.E.; Chung, K.S.; Lee, J.H.; Gil, H.S.; Lee, K.T.; Roh, E.J. Diarylurea derivatives comprising 2,4-diarylpyrimidines: Discovery of novel potential anticancer agents via combined failed-ligands repurposing and molecular hybridization approaches. Bioorg. Chem. 2020, 103, 104121. [Google Scholar] [CrossRef]
- Hong, J.Y.; Chung, K.S.; Shin, J.S.; Lee, J.H.; Gil, H.S.; Lee, H.H.; Choi, E.; Choi, J.H.; Hassan, A.H.E.; Lee, Y.S.; et al. The Anti-Proliferative Activity of the Hybrid TMS-TMF-4f Compound Against Human Cervical Cancer Involves Apoptosis Mediated by STAT3 Inactivation. Cancers 2019, 11, 1927. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.W.; Chung, K.S.; Seo, J.H.; Yim, S.V.; Park, H.J.; Choi, J.H.; Lee, K.T. Sulfuretin from heartwood of Rhus verniciflua triggers apoptosis through activation of Fas, Caspase-8, and the mitochondrial death pathway in HL-60 human leukemia cells. J. Cell Biochem. 2012, 113, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.A.; Prewitt, J.M.; Mendelsohn, M.L. Analysis of tumor growth curves. J. Natl. Cancer Inst. 1968, 40, 389–405. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Gaume, B.; Karbowski, M.; Sharpe, J.C.; Cecconi, F.; Youle, R.J. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003, 22, 4385–4399. [Google Scholar] [CrossRef]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef] [Green Version]
- Billen, L.P.; Shamas-Din, A.; Andrews, D.W. Bid: A Bax-like BH3 protein. Oncogene 2008, 27 (Suppl. S1), S93–S104. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death--a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [Green Version]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, P.; Gao, Z.; Zhao, X.; Qi, G. Surfactin inducing mitochondria-dependent ROS to activate MAPKs, NF-kappaB and inflammasomes in macrophages for adjuvant activity. Sci. Rep. 2016, 6, 39303. [Google Scholar] [CrossRef] [PubMed]
- Andon, F.T.; Fadeel, B. Programmed cell death: Molecular mechanisms and implications for safety assessment of nanomaterials. Acc. Chem. Res. 2013, 46, 733–742. [Google Scholar] [CrossRef]
- Bursch, W.; Ellinger, A.; Gerner, C.; Frohwein, U.; Schulte-Hermann, R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann. N. Y. Acad. Sci. 2000, 926, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Tompkins, K.D.; Thorburn, A. Regulation of Apoptosis by Autophagy to Enhance Cancer Therapy. Yale J. Biol. Med. 2019, 92, 707–718. [Google Scholar] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Caspase-independent cell death: Leaving the set without the final cut. Oncogene 2008, 27, 6452–6461. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M. Ways of dying: Multiple pathways to apoptosis. Genes Dev. 2003, 17, 2481–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.; Shi, Y. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Elumalai, P.; Gunadharini, D.N.; Senthilkumar, K.; Banudevi, S.; Arunkumar, R.; Benson, C.S.; Sharmila, G.; Arunakaran, J. Induction of apoptosis in human breast cancer cells by nimbolide through extrinsic and intrinsic pathway. Toxicol. Lett. 2012, 215, 131–142. [Google Scholar] [CrossRef]
- Kavurma, M.M.; Khachigian, L.M. Signaling and transcriptional control of Fas ligand gene expression. Cell Death Differ. 2003, 10, 36–44. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Kamalakaran, S. Pro-apoptotic role of NF-kappaB: Implications for cancer therapy. Biochim. Biophys. Acta 2006, 1766, 53–62. [Google Scholar] [CrossRef]
- Strozyk, E.; Poppelmann, B.; Schwarz, T.; Kulms, D. Differential effects of NF-kappaB on apoptosis induced by DNA-damaging agents: The type of DNA damage determines the final outcome. Oncogene 2006, 25, 6239–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M.; Haisa, M.; Uetsuka, H.; Takaoka, M.; Ohkawa, T.; Kawashima, R.; Yamatsuji, T.; Gunduz, M.; Kaneda, Y.; Tanaka, N.; et al. TNF combined with IFN-alpha accelerates NF-kappaB-mediated apoptosis through enhancement of Fas expression in colon cancer cells. Cell Death Differ. 2003, 10, 718–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiener, Z.; Ontsouka, E.C.; Jakob, S.; Torgler, R.; Falus, A.; Mueller, C.; Brunner, T. Synergistic induction of the Fas (CD95) ligand promoter by Max and NFkappaB in human non-small lung cancer cells. Exp. Cell Res. 2004, 299, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Abdalla, M.; Li, Y.; Bai, Y. NF-kappaB mediates the induction of Fas receptor and Fas ligand by microcystin-LR in HepG2 cells. Mol. Cell. Biochem. 2011, 352, 209–219. [Google Scholar] [CrossRef]
- Muniraj, N.; Siddharth, S.; Shriver, M.; Nagalingam, A.; Parida, S.; Woo, J.; Elsey, J.; Gabrielson, K.; Gabrielson, E.; Arbiser, J.L.; et al. Induction of STK11-dependent cytoprotective autophagy in breast cancer cells upon honokiol treatment. Cell Death Discov. 2020, 6, 81. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Yang, J.; Li, H.; Zhang, H.; Zheng, P. Curcumin induces apoptotic cell death and protective autophagy in human gastric cancer cells. Oncol. Rep. 2017, 37, 3459–3466. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, J.; Fan, M.; Shen, C.; Dai, W.; Bao, Y.; Liu, J.H.; Yu, B.Y. Novel dihydroartemisinin derivative DHA-37 induces autophagic cell death through upregulation of HMGB1 in A549 cells. Cell Death Dis. 2018, 9, 1048. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Feng, Y.; Wang, Y.; Ji, Q.; Cai, G.; Shi, L.; Wang, Y.; Huang, Y.; Zhang, J.; Li, Q. alpha-hederin induces autophagic cell death in colorectal cancer cells through reactive oxygen species dependent AMPK/mTOR signaling pathway activation. Int. J. Oncol. 2019, 54, 1601–1612. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Ouchida, A.T.; Li, Y.; Geng, J.; Najafov, A.; Ofengeim, D.; Sun, X.; Yu, Q.; Yuan, J. Synergistic effect of a novel autophagy inhibitor and Quizartinib enhances cancer cell death. Cell Death Dis. 2018, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.M.; Tetreault, M.P.; Hamilton, K.E.; Whelan, K.A. Autophagy as a cytoprotective mechanism in esophageal squamous cell carcinoma. Curr. Opin. Pharmacol. 2018, 41, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bazhin, A.V.; Philippov, P.P.; Karakhanova, S. Reactive Oxygen Species in Cancer Biology and Anticancer Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 4197815. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Essers, M.A.; de Vries-Smits, L.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.; Korswagen, H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef]
- Fan, J.; Ren, D.; Wang, J.; Liu, X.; Zhang, H.; Wu, M.; Yang, G. Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo. Cell Death Dis. 2020, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Sun, X.; Wu, Z.; Yuan, H.; Han, H.; Huang, H.; Shu, Y.; Xu, M.; Gao, R.; Li, S.; et al. A Novel Benzofuran Derivative Moracin N Induces Autophagy and Apoptosis Through ROS Generation in Lung Cancer. Front. Pharmacol. 2020, 11, 391. [Google Scholar] [CrossRef]
- Tang, Z.H.; Cao, W.X.; Su, M.X.; Chen, X.; Lu, J.J. Osimertinib induces autophagy and apoptosis via reactive oxygen species generation in non-small cell lung cancer cells. Toxicol. Appl. Pharmacol. 2017, 321, 18–26. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E: Protective role of a Janus molecule. FASEB J. 2001, 15, 2314–2325. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazdro, R.; Burgess, J.R. Differential effects of alpha-tocopherol and N-acetyl-cysteine on advanced glycation end product-induced oxidative damage and neurite degeneration in SH-SY5Y cells. Biochim. Biophys. Acta 2012, 1822, 550–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Ibrahim, P.N.; Zhang, J.; Burton, E.A.; Habets, G.; Zhang, Y.; Powell, B.; West, B.L.; Matusow, B.; Tsang, G.; et al. Design and pharmacology of a highly specific dual FMS and KIT kinase inhibitor. Proc. Natl. Acad. Sci. USA 2013, 110, 5689–5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hao, Y.; Chen, J.; Huang, L.; Ao, W.; Yang, J.; Li, L.; Heng, J.; Chen, Z.; Liang, W.; et al. Colony stimulating factor-1 receptor promotes proliferation, migration and invasion in the human nasopharyngeal carcinoma 6-10B cell line via the phosphoinositide 3-kinase/Akt pathway. Oncol. Lett. 2018, 16, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.J.; Lee, J.Y.; Shin, M.G.; Kim, H.J.; Choi, Y.J.; Rho, S.B.; Kim, B.R.; Jang, I.S.; Lee, S.H. TSC-22 inhibits CSF-1R function and induces apoptosis in cervical cancer. Oncotarget 2017, 8, 97990–98003. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil, H.-S.; Lee, J.-H.; Farag, A.K.; Hassan, A.H.E.; Chung, K.-S.; Choi, J.-H.; Roh, E.-J.; Lee, K.-T. AKF-D52, a Synthetic Phenoxypyrimidine-Urea Derivative, Triggers Extrinsic/Intrinsic Apoptosis and Cytoprotective Autophagy in Human Non-Small Cell Lung Cancer Cells. Cancers 2021, 13, 5849. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225849
Gil H-S, Lee J-H, Farag AK, Hassan AHE, Chung K-S, Choi J-H, Roh E-J, Lee K-T. AKF-D52, a Synthetic Phenoxypyrimidine-Urea Derivative, Triggers Extrinsic/Intrinsic Apoptosis and Cytoprotective Autophagy in Human Non-Small Cell Lung Cancer Cells. Cancers. 2021; 13(22):5849. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225849
Chicago/Turabian StyleGil, Hyo-Sun, Jeong-Hun Lee, Ahmed K. Farag, Ahmed H. E. Hassan, Kyung-Sook Chung, Jung-Hye Choi, Eun-Joo Roh, and Kyung-Tae Lee. 2021. "AKF-D52, a Synthetic Phenoxypyrimidine-Urea Derivative, Triggers Extrinsic/Intrinsic Apoptosis and Cytoprotective Autophagy in Human Non-Small Cell Lung Cancer Cells" Cancers 13, no. 22: 5849. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225849