
An Overview of Selected Rare B-Cell Lymphoproliferative Disorders: Imaging, Histopathologic, and Clinical Features

Abstract
:Simple Summary
Abstract
1. Introduction
2. Changes Contained in the Updated 2016 WHO Classification of B-Cell Lymphoproliferative Disorders (B-Cell LPD)
3. Uncommon B-Cell Lymphoproliferative Disorders
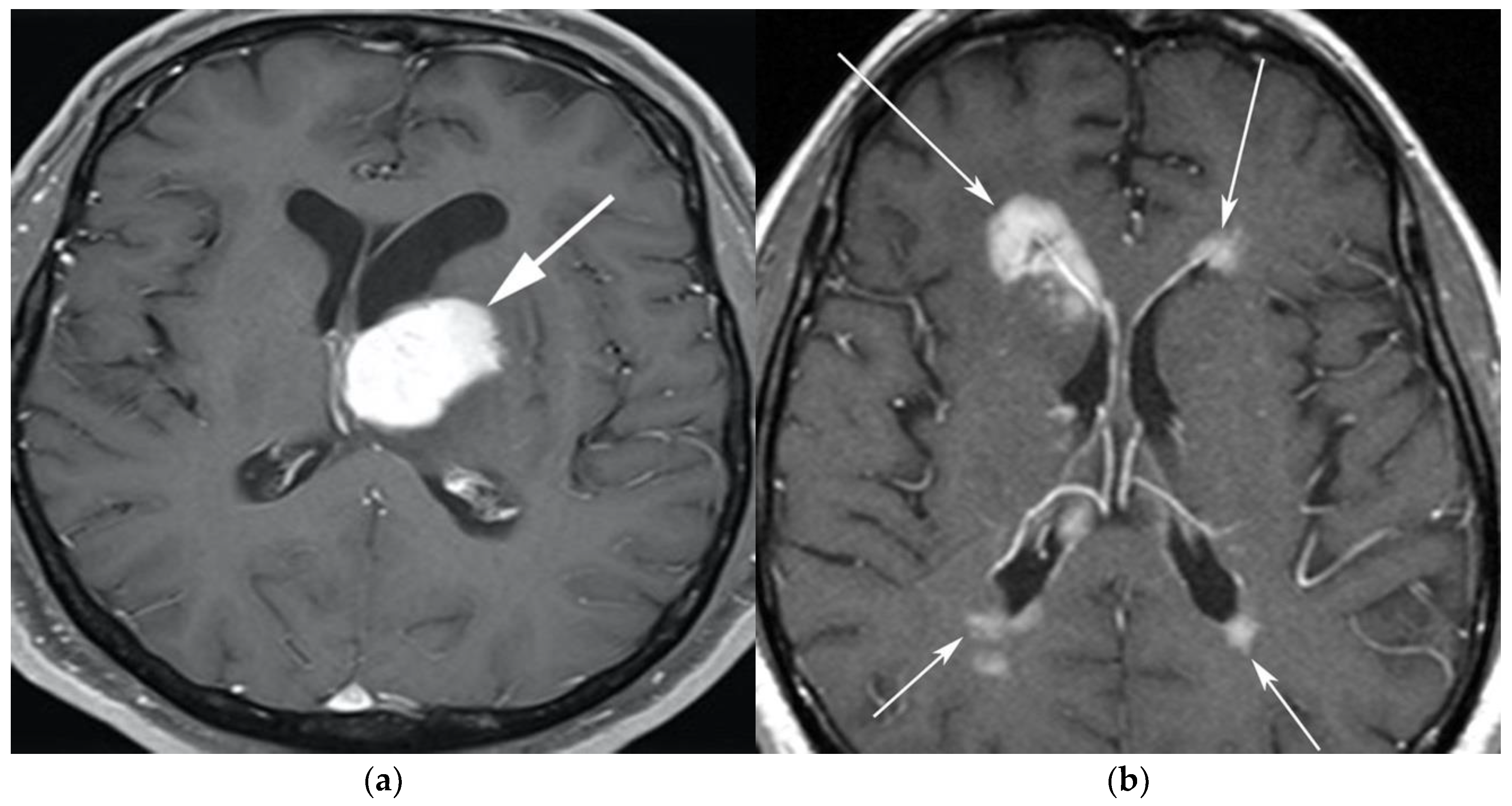
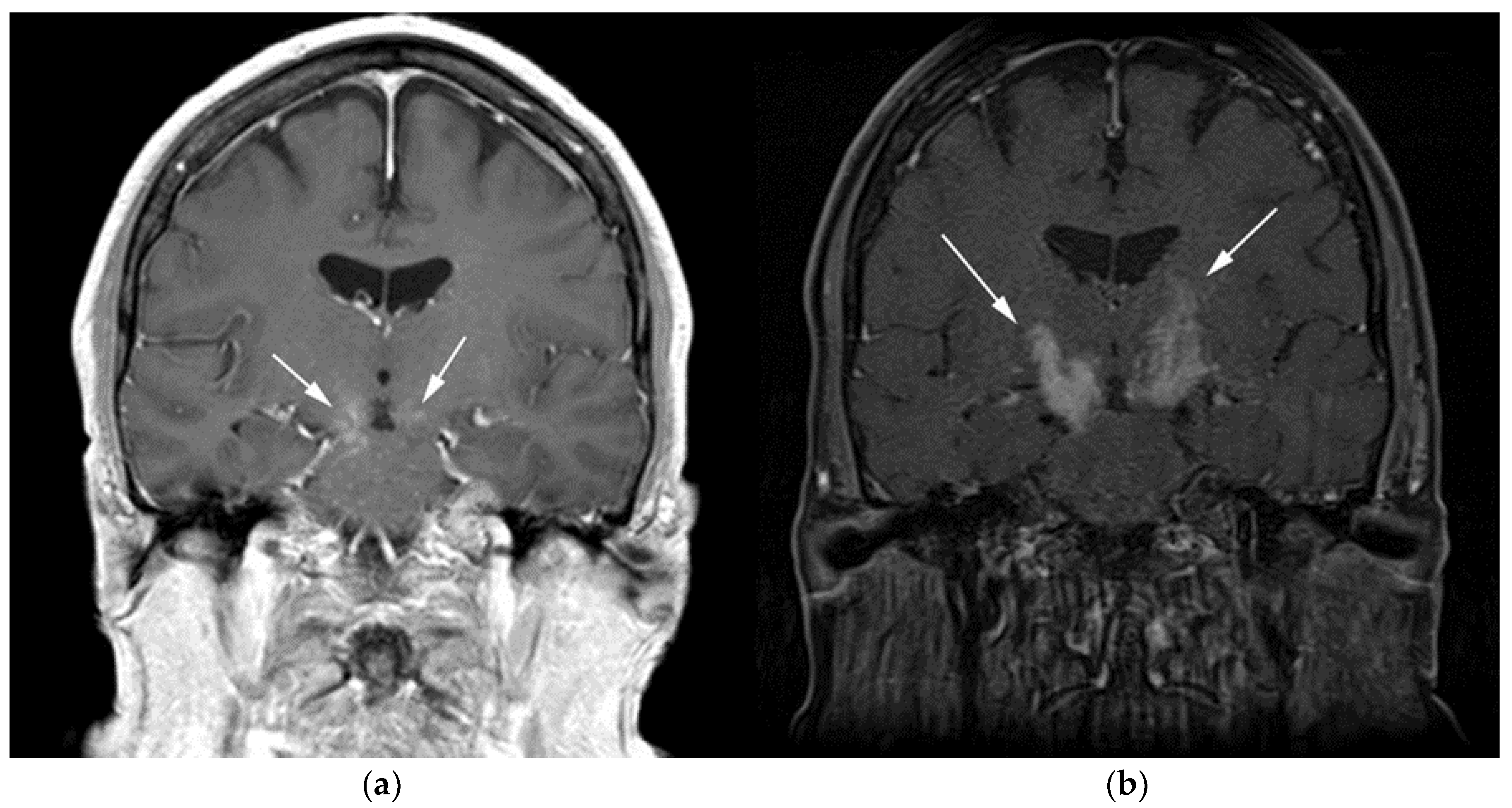
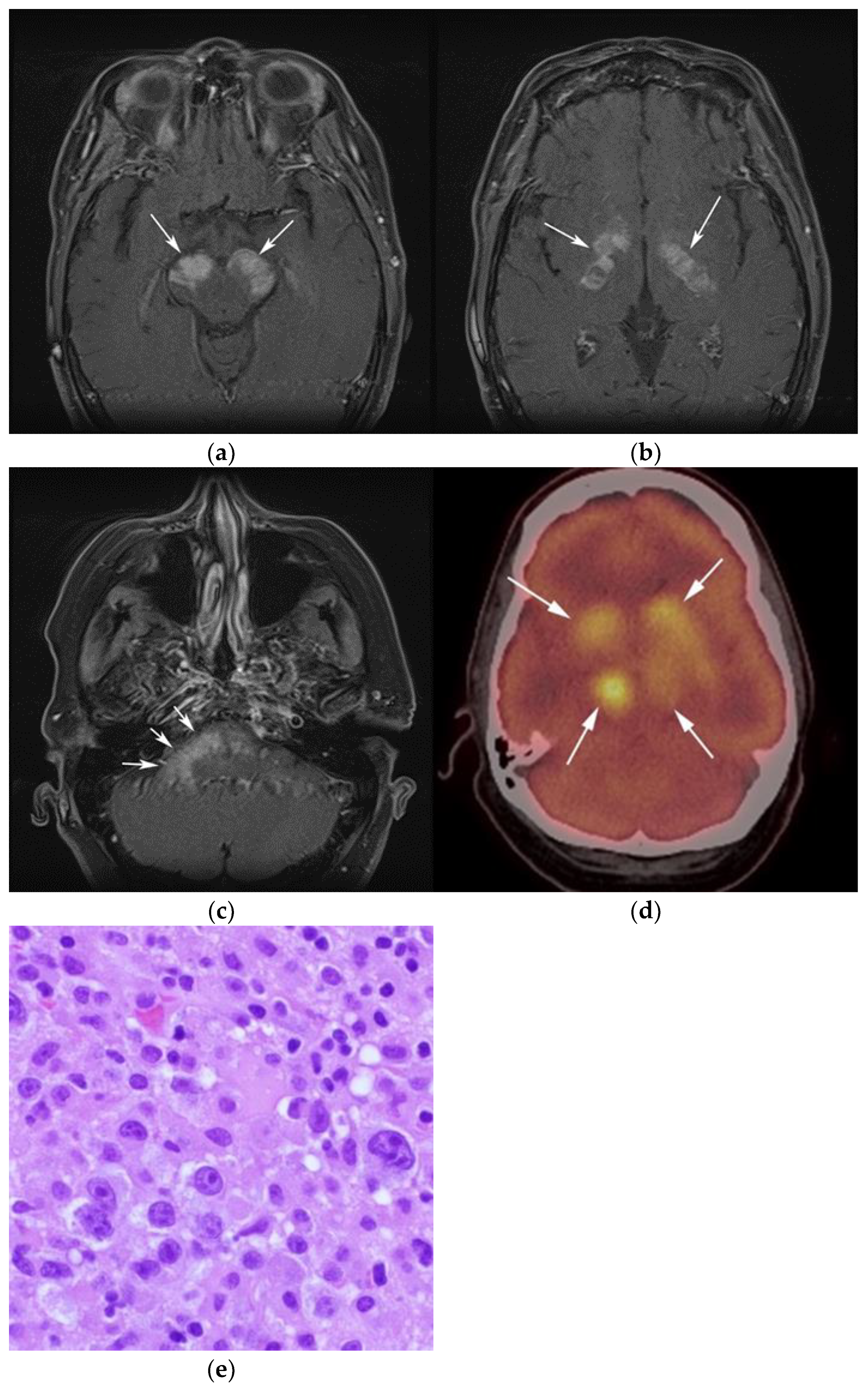
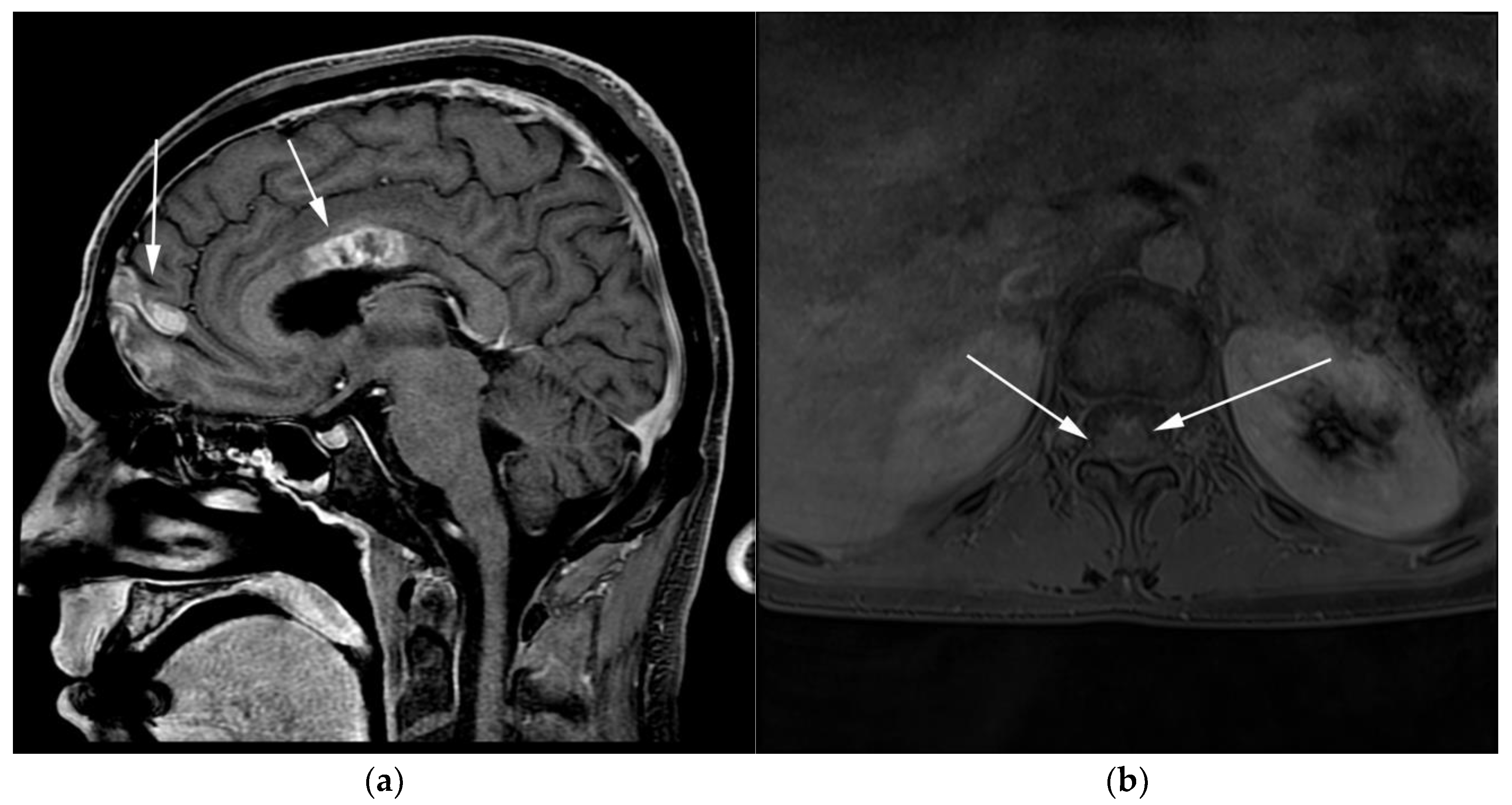
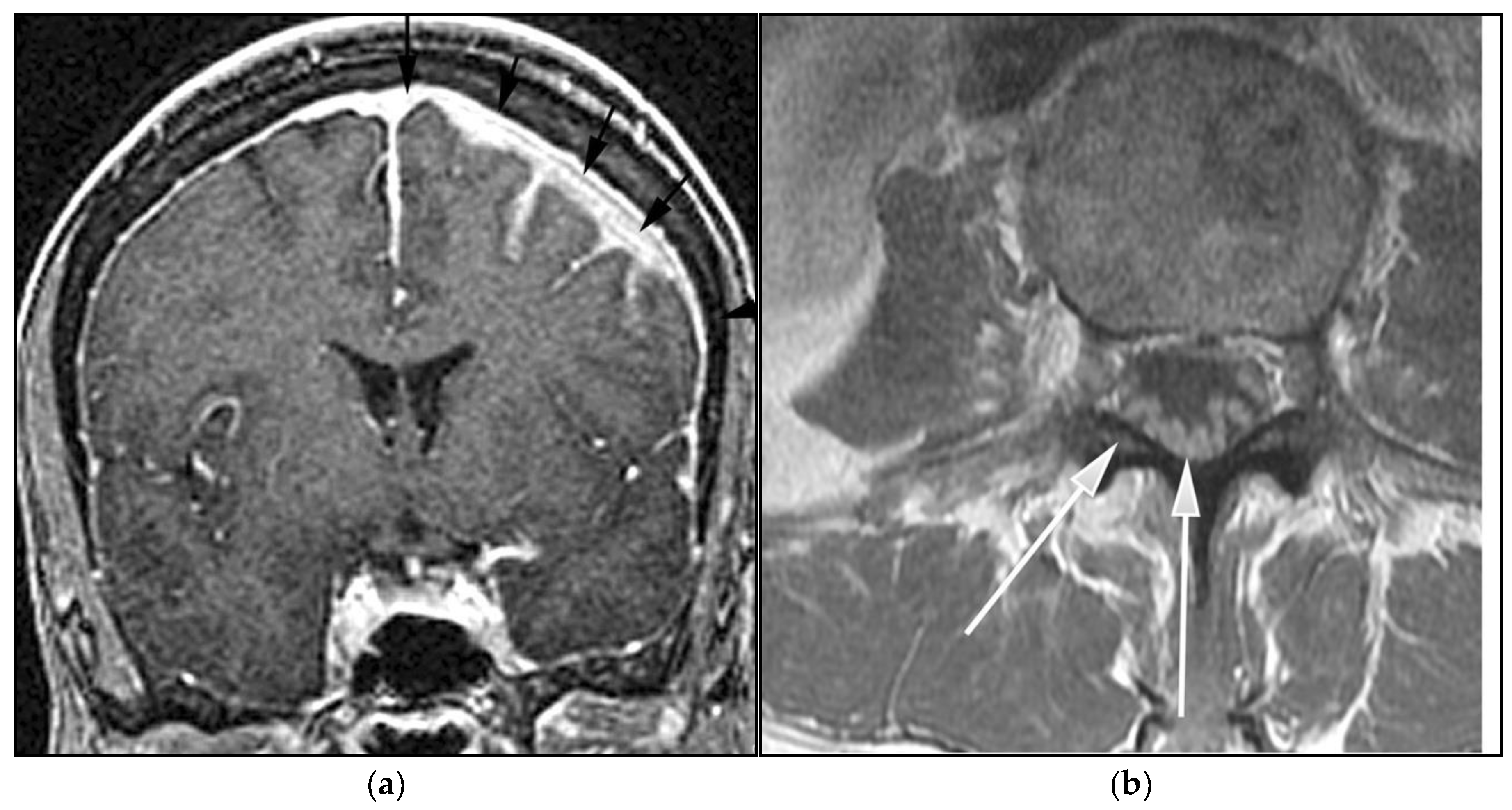
3.1. Primary Central Nervous System Lymphoma with Attention to Lymphomatosis Cerebri: An Extremely Rare Diffuse B-Cell Lymphoma Variant
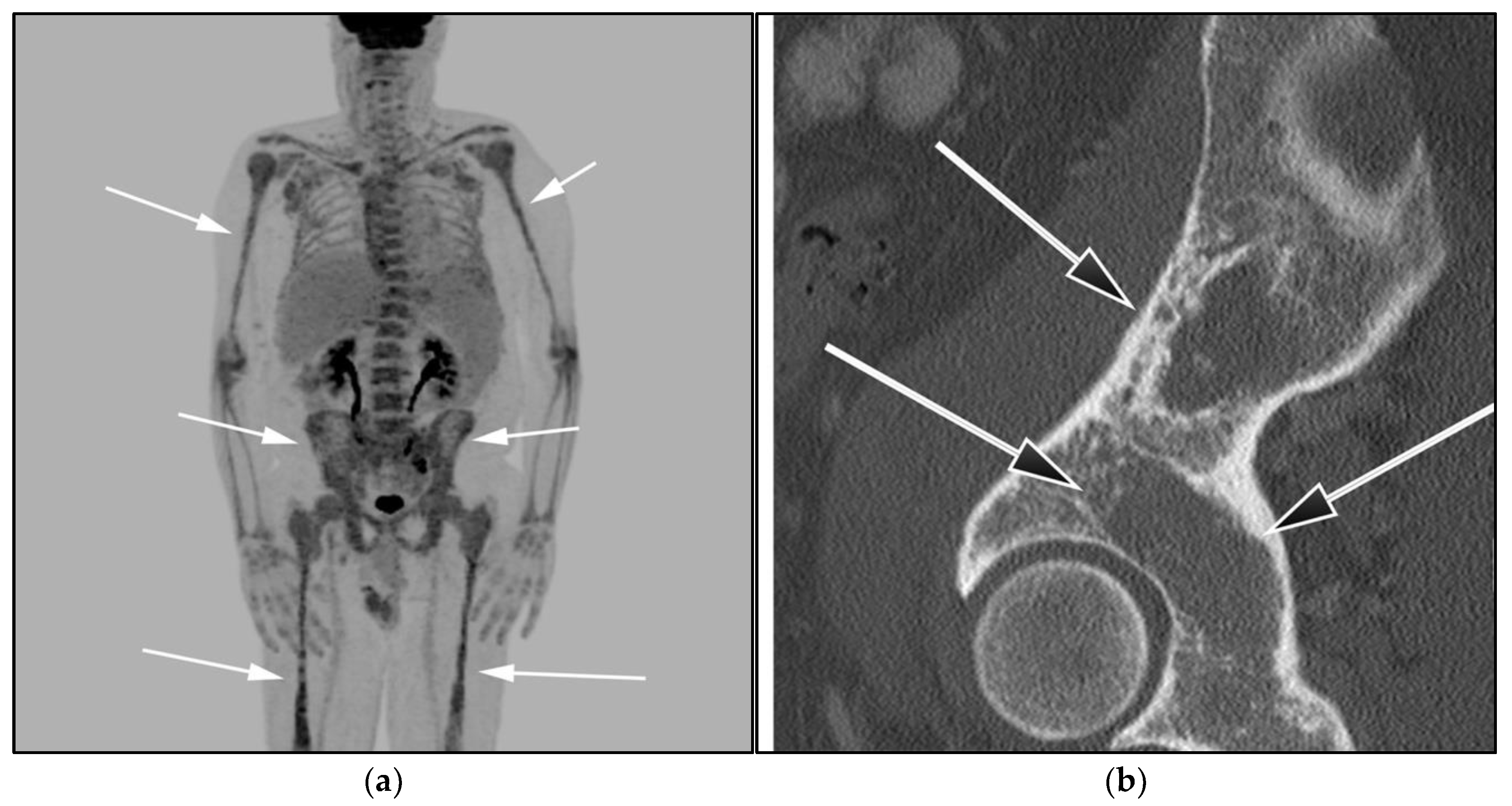
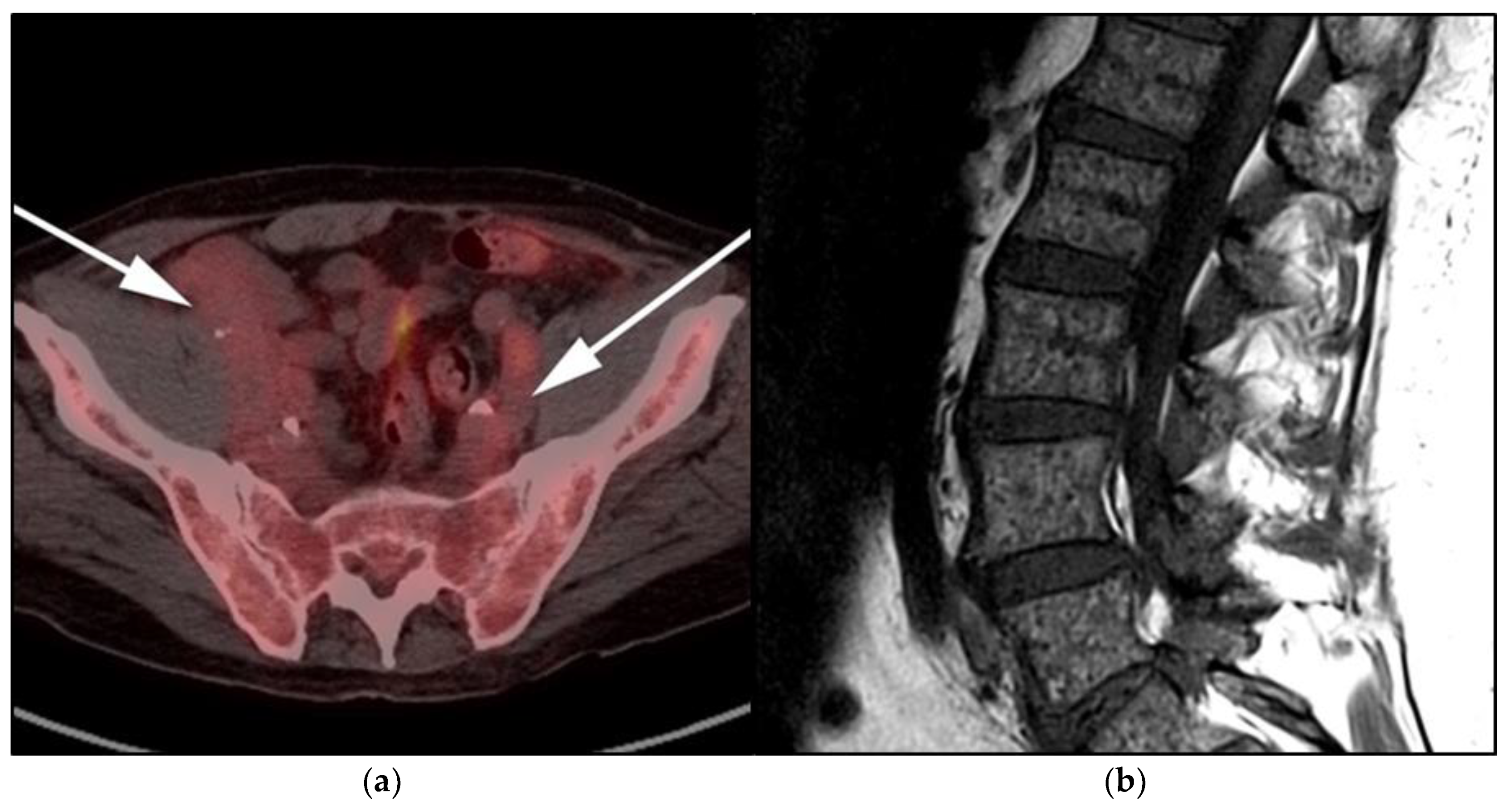
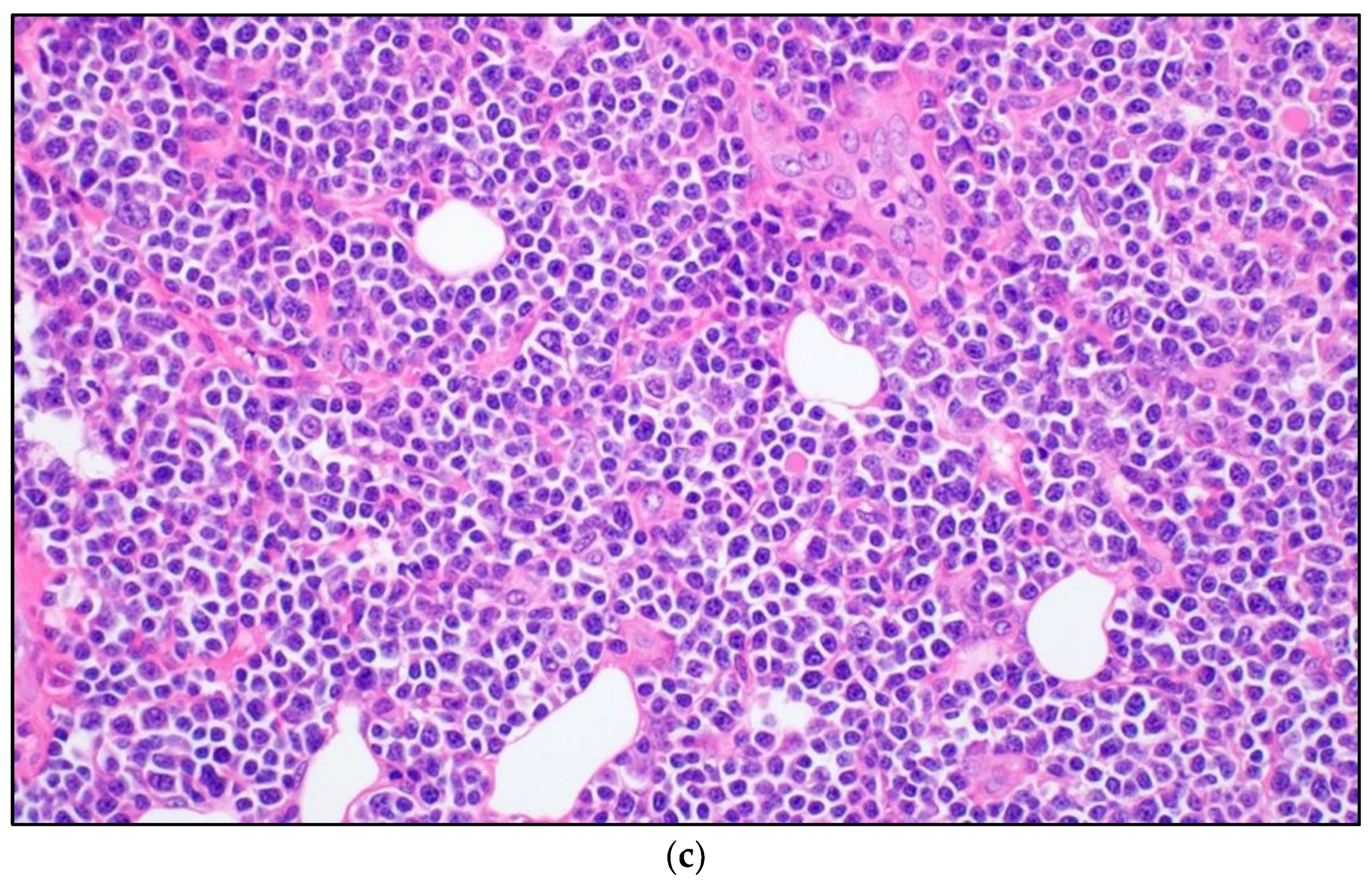
3.2. Autoimmune Lymphoproliferative Syndrome
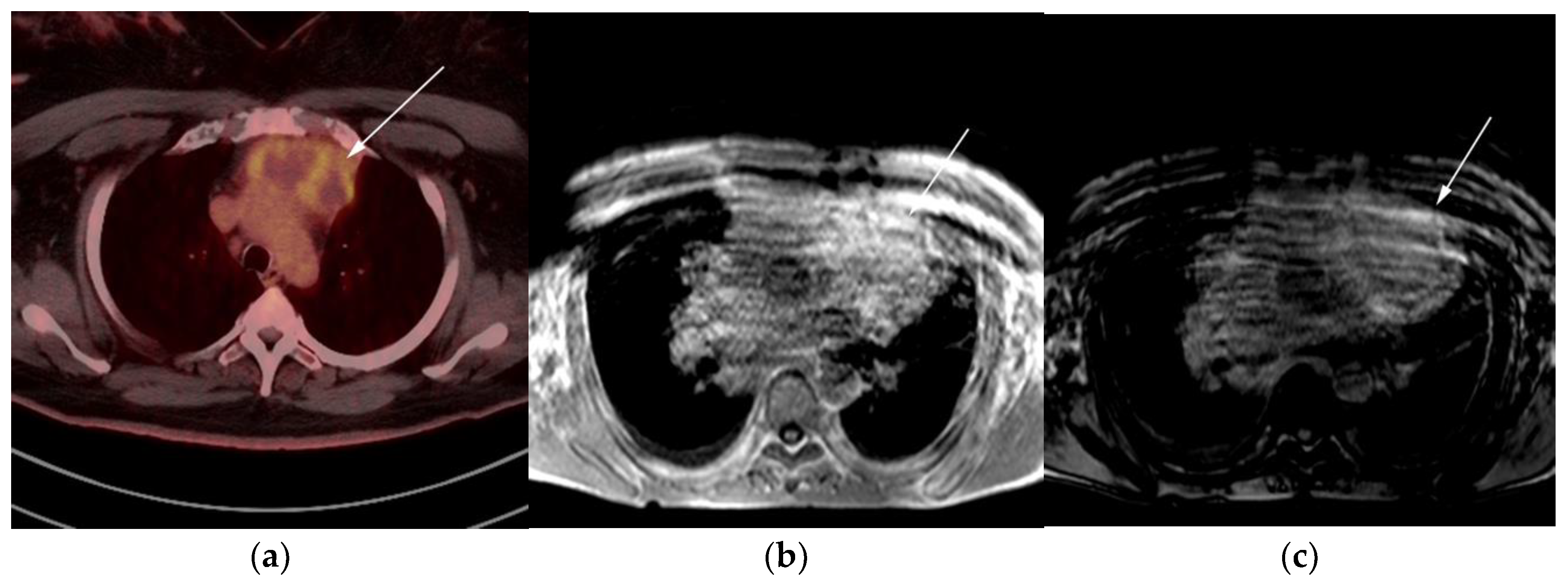
3.3. Primary Mediastinal Large B-Cell Lymphoma
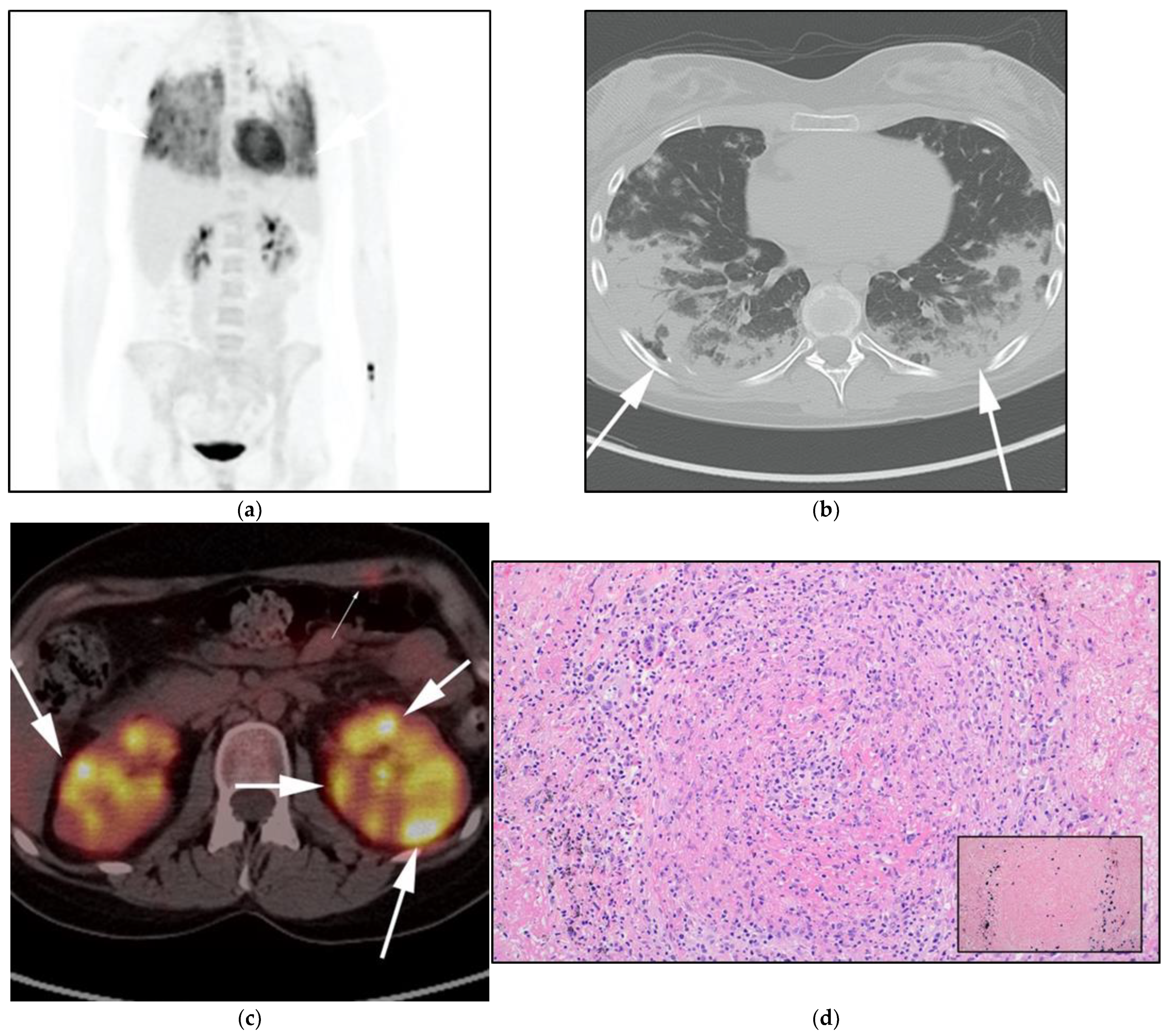
3.4. Lymphomatoid Granulomatosis
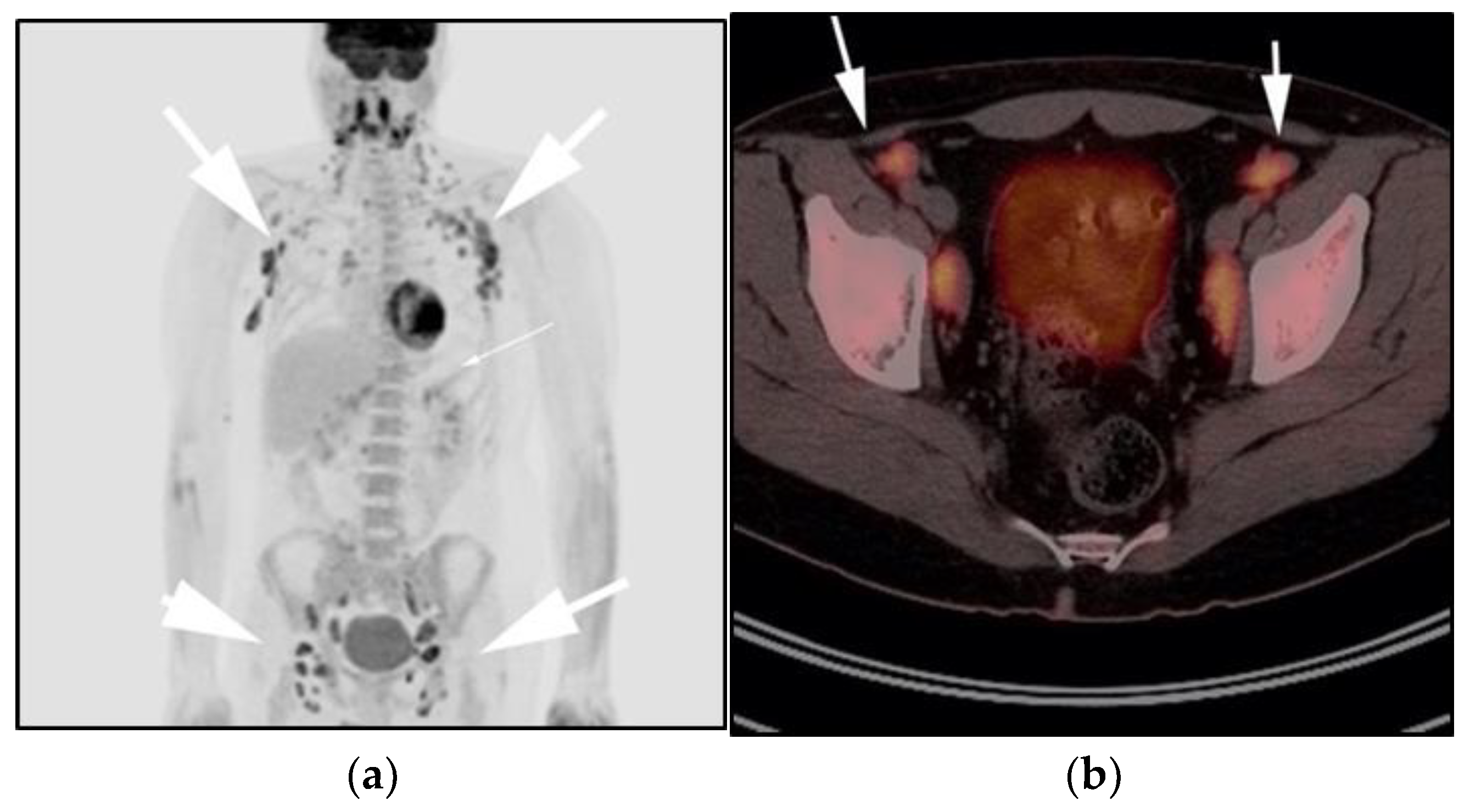
3.5. Lymphoplasmacytic Lymphoma, Waldenstrom Macroglobulinemia, and Bing–Neel Syndrome
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Justiz Vaillant, A.A.; Stang, C.M. Lymphoproliferative Disorders; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Toma, P.; Granata, C.; Rossi, A.; Garaventa, A. Multimodality imaging of Hodgkin disease and non-Hodgkin lymphomas in children. Radiographics 2007, 27, 1335–1354. [Google Scholar] [CrossRef]
- Ngeow, J.Y.Y.; Quek, R.H.H.; Ng, D.C.E.; Hee, S.W.; Tao, M.; Lim, L.C.; Tan, Y.H.; Lim, S.T. High SUV uptake on FDG-PET/CT predicts for an aggressive B-cell lymphoma in a prospective study of primary FDG-PET/CT staging in lymphoma. Ann. Oncol. 2009, 20, 1543–1547. [Google Scholar] [CrossRef]
- Valls, L.; Badve, C.; Avril, S.; Herrmann, K.; Faulhaber, P.; O’Donnell, J.; Avril, N. FDG-PET imaging in hematological malignancies. Blood Rev. 2016, 30, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood J. Am. Soc. Hematol. 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, E.S.; Barr, P.M.; Smith, S.M. Understanding the New WHO Classification of Lymphoid Malignancies: Why It’s Important and How It Will Affect Practice. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 535–546. [Google Scholar] [CrossRef]
- Das, J.; Ray, S.; Sen, S.; Chandy, M. Extranodal involvement in lymphoma-A Pictorial Essay and Retrospective Analysis of 281 PET/CT studies. Asia Ocean. J. Nucl. Med. Biol. 2014, 2, 42–56. [Google Scholar]
- Li, L.; Rong, J.H.; Feng, J. Neuroradiological features of lymphomatosis cerebri: A systematic review of the English literature with a new case report. Oncol. Lett. 2018, 16, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Diamond, C.; Taylor, T.H.; Aboumrad, T.; Anton-Culver, H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: Incidence, presentation, treatment, and survival. Cancer 2006, 106, 128–135. [Google Scholar] [CrossRef]
- Olson, J.E.; Janney, C.A.; Rao, R.D.; Cerhan, J.R.; Kurtin, P.J.; Schiff, D.; Kaplan, R.S.; O’Neill, B.P. The continuing increase in the incidence of primary central nervous system non-Hodgkin lymphoma: A surveillance, epidemiology, and end results analysis. Cancer 2002, 95, 1504–1510. [Google Scholar] [CrossRef]
- Bashir, R.; McManus, B.; Cunningham, C.; Weisenburger, D.; Hochberg, F. Detection of Eber-1 RNA in primary brain lymphomas in immunocompetent and immunocompromised patients. J. Neurooncol. 1994, 20, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, C.; Velasco, R.; Vidal, N.; Sánchez, J.J.; Argyriou, A.A.; Besora, S.; Graus, F.; Bruna, J. Lymphomatosis cerebri: A rare form of primary central nervous system lymphoma. Analysis of 7 cases and systematic review of the literature. Neuro-Oncol. 2016, 18, 707–715. [Google Scholar] [CrossRef] [Green Version]
- Haldorsen, I.S.; Espeland, A.; Larsson, E.M. Central nervous system lymphoma: Characteristic findings on traditional and advanced imaging. Am. J. Neuroradiol. 2011, 32, 984–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathla, G.; Hegde, A. Lymphomatous involvement of the central nervous system. Clin. Radiol. 2016, 71, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, A.; Upchurch, K.; Batchelor, T. Clinical features and diagnosis of primary central nervous system lymphoma. Hematol. Oncol. Clin. 2005, 19, 689–703. [Google Scholar] [CrossRef]
- Eichler, A.F.; Batchelor, T.T. Primary central nervous system lymphoma: Presentation, diagnosis and staging. Neurosurg. Focus 2006, 21, E15. [Google Scholar] [CrossRef]
- Koeller, K.K.; Smirniotopoulos, J.G.; Jones, R.V. Primary central nervous system lymphoma: Radiologic-pathologic correlation. Radiographics 1997, 17, 1497–1526. [Google Scholar] [CrossRef] [PubMed]
- Go, J.L.; Lee, S.C.; Kim, P.E. Imaging of primary central nervous system lymphoma. Neurosurg. Focus 2006, 21, E4. [Google Scholar] [CrossRef]
- Kitai, R.; Hashimoto, N.; Yamate, K.; Ikawa, M.; Yoneda, M.; Nakajima, T.; Arishima, H.; Takeuchi, H.; Sato, K.; Kikuta, K.I. Lymphomatosis cerebri: Clinical characteristics, neuroimaging, and pathological findings. Brain Tumor Pathol. 2012, 29, 47–53. [Google Scholar] [CrossRef]
- Lewerenz, J.; Ding, X.Q.; Matschke, J.; Schnabel, C.; Emami, P.; von Borczyskowski, D.; Buchert, R.; Krieger, T.; de Wit, M.; Münchau, A. Dementia and leukoencephalopathy due to lymphomatosis cerebri. BMJ Case Rep. 2009, 2009, 777. [Google Scholar] [CrossRef]
- Lee, I.H.; Kim, S.T.; Kim, H.J.; Kim, K.H.; Jeon, P.; Byun, H.S. Analysis of perfusion weighted image of CNS lymphoma. Eur. J. Radiol. 2010, 76, 48–51. [Google Scholar] [CrossRef]
- Bettinardi, A.; Brugnoni, D.; Quiros-Roldan, E.; Malagoli, A.; La Grutta, S.; Correra, A.; Notarangelo, L.D. Missense mutations in the Fas gene resulting in autoimmune lymphoproliferative syndrome: A molecular and immunological analysis. Blood J. Am. Soc. Hematol. 1997, 89, 902–909. [Google Scholar] [CrossRef]
- Spergel, A.R.; Walkovich, K.; Price, S.; Niemela, J.E.; Wright, D.; Fleisher, T.A.; Rao, V.K. Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis. Pediatrics 2013, 132, e1440–e1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.S.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.T.; et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood J. Am. Soc. Hematol. 2010, 116, e35–e40. [Google Scholar] [CrossRef] [PubMed]
- Avila, N.A.; Dwyer, A.J.; Dale, J.K.; Lopatin, U.A.; Sneller, M.C.; Jaffe, E.S.; Puck, J.M.; Straus, S.E. Autoimmune lymphoproliferative syndrome: A syndrome associated with inherited genetic defects that impair lymphocytic apoptosis—CT and US features. Radiology 1999, 212, 257–263. [Google Scholar] [CrossRef]
- Sneller, M.C.; Straus, S.E.; Jaffe, E.S.; Jaffe, J.S.; Fleisher, T.A.; Stetler-Stevenson, M.; Strober, W. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J. Clin. Investig. 1992, 90, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Martelli, M.; Ferreri, A.; Di Rocco, A.; Ansuinelli, M.; Johnson, P.W. Primary mediastinal large B-cell lymphoma. Crit. Rev. Oncol. Hematol. 2017, 113, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, K.E.; O’Malley, D.P. Aggressive B cell lymphomas in the 2017 revised WHO classification of tumors of hematopoietic and lymphoid tissues. Ann. Diagn. Pathol. 2019, 38, 6–10. [Google Scholar] [CrossRef]
- Ceriani, L.; Milan, L.; Martelli, M.; Ferreri, A.J.; Cascione, L.; Zinzani, P.L.; Di Rocco, A.; Conconi, A.; Stathis, A.; Cavalli, F.; et al. Metabolic heterogeneity on baseline 18FDG-PET/CT scan is a predictor of outcome in primary mediastinal B-cell lymphoma. Blood J. Am. Soc. Hematol. 2018, 132, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, J.O.; Aisenberg, A.C.; Lamarre, L.; Willett, C.G.; Linggood, R.M.; Miketic, L.M.; Harris, N.L. Mediastinal large cell lymphoma. An uncommon subset of adult lymphoma curable with combined modality therapy. Cancer 1988, 62, 1893–1898. [Google Scholar] [CrossRef]
- Giulino-Roth, L. How I treat primary mediastinal B-cell lymphoma. Blood J. Am. Soc. Hematol. 2018, 132, 782–790. [Google Scholar] [CrossRef]
- Lee, K.S.; Im, J.G.; Han, C.H.; Han, M.C.; Kim, C.W.; Kim, W.S. Malignant primary germ cell tumors of the mediastinum: CT features. Am. J. Roentgenol. 1989, 153, 947–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, T.T.; Kamaludin, Z.; Husin, A. Primary mediastinal large B-cell lymphoma and its mimickers: A rare case report with literature review. Malays. J. Pathol. 2016, 38, 153–157. [Google Scholar] [PubMed]
- Savage, K.J.; Al-Rajhi, N.; Voss, N.; Paltiel, C.; Klasa, R.; Gascoyne, R.D.; Connors, J.M. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: The British Columbia experience. Ann. Oncol. 2006, 17, 123–130. [Google Scholar] [CrossRef]
- Aoki, T.; Shimada, K.; Suzuki, R.; Izutsu, K.; Tomita, A.; Maeda, Y.; Takizawa, J.; Mitani, K.; Igarashi, T.; Sakai, K.; et al. High-dose chemotherapy followed by autologous stem cell transplantation for relapsed/refractory primary mediastinal large B-cell lymphoma. Blood Cancer J. 2015, 5, e372. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, C.C.; Dabaja, B.; Ahmed, M.A.; Chuang, H.H.; Costelloe, C.; Wogan, C.F.; Reed, V.; Romaguera, J.E.; Neelapu, S.; Oki, Y.; et al. Single-institution experience in the treatment of primary mediastinal B cell lymphoma treated with immunochemotherapy in the setting of response assessment by 18fluorodeoxyglucose positron emission tomography. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, M.; Ceriani, L.; Zucca, E.; Zinzani, P.L.; Ferreri, A.J.; Vitolo, U.; Stelitano, C.; Brusamolino, E.; Cabras, M.G.; Rigacci, L.; et al. [18F]Fluorodeoxyglucose Positron Emission Tomography Predicts Survival After Chemoimmunotherapy for Primary Mediastinal Large B-Cell Lymphoma: Results of the International Extranodal Lymphoma Study Group IELSG-26 Study. J. Clin. Oncol. 2014, 32, 1769–1775. [Google Scholar] [CrossRef] [Green Version]
- Liebow, A.A.; Carrington, C.R.; Friedman, P.J. Lymphomatoid granulomatosis. Hum. Pathol. 1972, 3, 457–558. [Google Scholar] [CrossRef] [Green Version]
- Song, J.Y.; Pittaluga, S.; Dunleavy, K.; Grant, N.; White, T.; Jiang, L.; Davies-Hill, T.; Raffeld, M.; Wilson, W.H.; Jaffe, E.S. Lymphomatoid granulomatosis--a single institute experience: Pathologic findings and clinical correlations. Am. J. Surg. Pathol. 2015, 39, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Guinee Jr, D.; Jaffe, E.; Kingma, D.; Fishback, N.; Wallberg, K.; Krishnan, J.; Frizzera, G.; Travis, W.; Koss, M. Pulmonary lymphomatoid granulomatosis. Evidence for a proliferation of Epstein-Barr virus infected B-lymphocytes with a prominent T-cell component and vasculitis. Am. J. Surg. Pathol. 1994, 18, 753–764. [Google Scholar] [CrossRef]
- Sheehy, N.; Bird, B.; O’Briain, D.S.; Daly, P.; Wilson, G. Synchronous regression and progression of pulmonary nodules on chest CT in untreated lymphomatoid granulomatosis. Clin. Radiol. 2004, 59, 451–454. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Yoshizawa, A.; Kudo, K.; Okuwaki, H.; Niino, H.; Morita, T. A case of lymphomatoid granulomatosis with multiple thin-walled cavities. Nihon Kokyuki Gakkai Zasshi 2002, 40, 292–298. [Google Scholar]
- Chung, J.H.; Wu, C.C.; Gilman, M.D.; Palmer, E.L.; Hasserjian, R.P.; Shepard, J.A.O. Lymphomatoid granulomatosis: CT and FDG-PET findings. Korean J. Radiol. 2011, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Katzenstein, A.L.; Doxtader, E.; Narendra, S. Lymphomatoid granulomatosis: Insights gained over 4 decades. Am. J. Surg. Pathol. 2010, 34, e35–e48. [Google Scholar] [CrossRef]
- Janz, S. Waldenstrom macroglobulinemia: Clinical and immunological aspects, natural history, cell of origin, and emerging mouse models. Int. Sch. Res. Not. 2013, 2013, 815325. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Li, F.; Delasalle, K.; Wang, J.; Alexanian, R.; Kwak, L.; Rustveld, L.; Du, X.L.; Wang, M. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: A large population-based study. Cancer 2012, 118, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Johnson, S.A.; Morgan, G.J. Waldenstrom’s macroglobulinaemia: Laboratory diagnosis and treatment. Hematol. Oncol. 2000, 18, 41–49. [Google Scholar]
- Dimopoulos, M.A.; Panayiotidis, P.; Moulopoulos, L.A.; Sfikakis, P.; Dalakas, M. Waldenstrom’s macroglobulinemia: Clinical features, complications, and management. J. Clin. Oncol. 2000, 18, 214–226. [Google Scholar] [CrossRef]
- Rossi, D. Role of MYD88 in lymphoplasmacytic lymphoma diagnosis and pathogenesis. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, C.; Alonso-Álvarez, S.; Alcoceba, M.; Ordóñez, G.R.; García-Álvarez, M.; Prieto-Conde, M.I.; Chillón, M.C.; Balanzategui, A.; Corral, R.; Marín, L.A.; et al. From Waldenstrom’s macroglobulinemia to aggressive diffuse large B-cell lymphoma: A whole-exome analysis of abnormalities leading to transformation. Blood Cancer J. 2017, 7, e591. [Google Scholar] [CrossRef] [Green Version]
- Royer, R.H.; Koshiol, J.; Giambarresi, T.R.; Vasquez, L.G.; Pfeiffer, R.M.; McMaster, M.L. Differential characteristics of Waldenstrom macroglobulinemia according to patterns of familial aggregation. Blood J. Am. Soc. Hematol. 2010, 115, 4464–4471. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Braschi-Amirfarzan, M.; Laferriere, S.L.; Jagannathan, J.P. Imaging of Waldenstrom Macroglobulinemia: A Comprehensive Review for the Radiologist in the Era of Personalized Medicine. Am. J. Roentgenol. 2019, 213, W248–W256. [Google Scholar] [CrossRef] [PubMed]
- Imhof, J.W.; Baars, H.; Verloop, M.C. Clinical and haematological aspects of macroglobulinaemia Waldenstrom. Acta Med. Scand. 1959, 163, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Merlini, G. The elusive pathogenesis of Schnitzler syndrome. Blood J. Am. Soc. Hematol. 2018, 131, 944–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders | Changes | New Diagnostic Implications |
|---|---|---|
| I-Premalignant conditions |
|
|
| II-Frank Lymphomas: A-Mature B-cell lymphoma (BCL) | ||
| Lymphoplasmacytic lymphoma (LPL) |
|
|
| Waldenstrom macroglobinema (WM) | ||
| IgM monoclonal gammopathy of undetermined significance (MGUS) | ||
| Plasma cell myeloma |
|
|
| Solitary plasmacytoma of the bone | ||
| Extraosseous plasmacytoma | ||
| Mantle cell lymphoma (MTL) |
|
|
| Follicular lymphoma (FL) subtypes |
|
|
| Lymphomatoid granulomatosis (LG) | No changes | No changes |
| Diffuse large B-cell lymphoma (DLBCL) |
|
|
| Primary diffuse large B-cell lymphoma of the CNS (PCNSL) |
|
|
| Primary mediastinal (thymic) B-cell lymphoma (PMLBCL) B-cell lymphoma unclassifiable, with features intermediate between DLBCL and classic Hodgkin lymphoma (CHL) |
|
|
| High-grade B-cell lymphoma with MYC and BCL2 and or BCL6 rearrangements |
|
|
| High-grade B-cell lymphoma, NOS |
|
|
| Burkitt-like lymphoma with 11q aberration |
|
|
| Entity | Pathologic Features | Clinical Features | Imaging Features | Differential Considerations |
|---|---|---|---|---|
| LC |
|
|
|
|
| PMLBCL |
|
|
|
|
| LG |
|
|
|
|
| LPL-WM |
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, A.E.; Zaki, Y.H.; El-Hussieny, G.; ElNoueam, K.I.; Shaaban, A.M.; Koppula, B.R.; Bustoros, M.; Salama, M.; Elsayes, K.M.; Morton, K.; et al. An Overview of Selected Rare B-Cell Lymphoproliferative Disorders: Imaging, Histopathologic, and Clinical Features. Cancers 2021, 13, 5853. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225853
Salem AE, Zaki YH, El-Hussieny G, ElNoueam KI, Shaaban AM, Koppula BR, Bustoros M, Salama M, Elsayes KM, Morton K, et al. An Overview of Selected Rare B-Cell Lymphoproliferative Disorders: Imaging, Histopathologic, and Clinical Features. Cancers. 2021; 13(22):5853. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225853
Chicago/Turabian StyleSalem, Ahmed Ebada, Yehia H. Zaki, Gamal El-Hussieny, Khaled I. ElNoueam, Akram M. Shaaban, Bhasker Rao Koppula, Mark Bustoros, Mohamed Salama, Khaled M. Elsayes, Kathryn Morton, and et al. 2021. "An Overview of Selected Rare B-Cell Lymphoproliferative Disorders: Imaging, Histopathologic, and Clinical Features" Cancers 13, no. 22: 5853. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225853