
MiRNA Deregulation Distinguishes Anaplastic Thyroid Carcinoma (ATC) and Supports Upregulation of Oncogene Expression

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Small RNA Library Construction, High-Throughput Sequencing, and Differential Expression Analysis
2.3. Unsupervised Expression Analysis
2.4. Prediction of microRNA-Target Interactions and Binding Site Positions
2.5. Pan-Cancer Loss of Function Studies
2.6. TCGA Based Survival Analysis
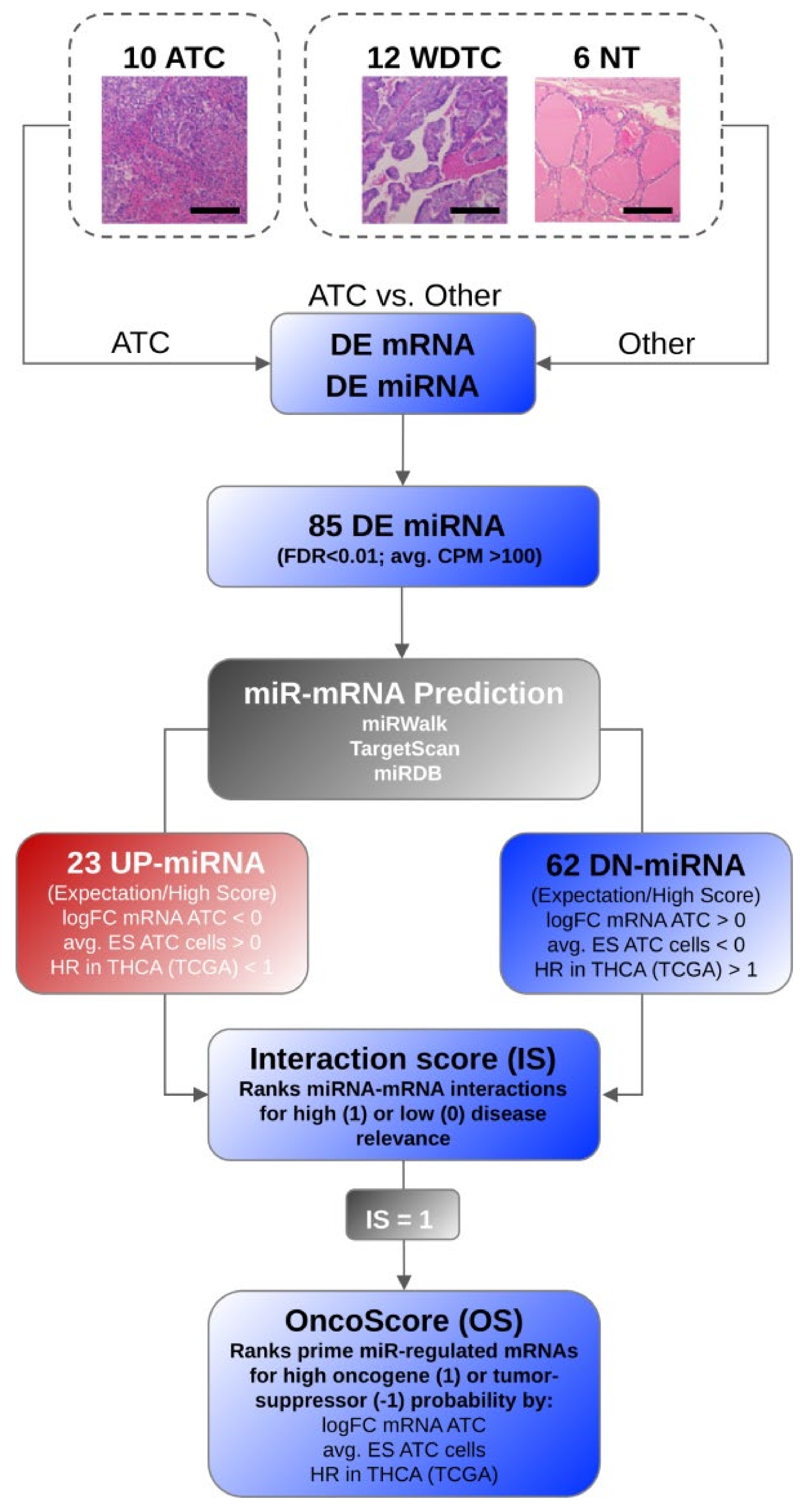
2.7. MicroRNA-Target Selection, Interaction Score, and OncoScore Calculation
- (1)
- Initially candidate miRNA–mRNA interactions were determined via miRWalk. Only interactions with a binding score > 0.95 were considered further and scored with the respective binding probability value determined by miRWalk (0.95–1). In addition, the binding predicted by miRDB and TargetScan were scored with 1 or 0 (no binding predicted), respectively. The sum of the binding scores was divided by three to yield a maximum score of 1 for the most likely (predicted with high probability by all three databases) candidate miRNA–mRNA interactions.
- (2)
- The scaled fold change (logFC) of the target mRNA expression in ATC was considered with maximum upregulation = 1 and downregulation = −1, respectively.
- (3)
- In addition, the fold change of mRNA was further scored with 1 if (a) the fold change of target mRNA expression showed inverse association with miRNA deregulation, and (b) mRNA abundance was significantly changed (FDR < 0.05).
- (4)
- The average essentiality score (ES) determined for genes in ATC-derived cell lines—available via DepMap (cf. 2.5)—was scaled from −1 to 0 (ES < 0) or 0 to 1 (ES > 0) and considered as outlined in the formulas below. This scaling settled on the assumptions that the upregulation of target mRNAs by DN-miRNAs promotes the abundance of factors with a negative essentially score (ES), indicative of a rather pro-oncogenic role. The opposite, downregulation of target mRNAs by UP-miRNAs was expected for factors with a positive ES, indicative of a rather tumor-suppressive role.
- (5)
- The hazardous ratio (HR) of candidate target mRNA expression (cf. 2.6) was determined in the THCA cohort provided by the TCGA and considered as outlined below. HR values were scaled from −1 to 0 (HR < 1) or 0 to 1 (HR > 1).
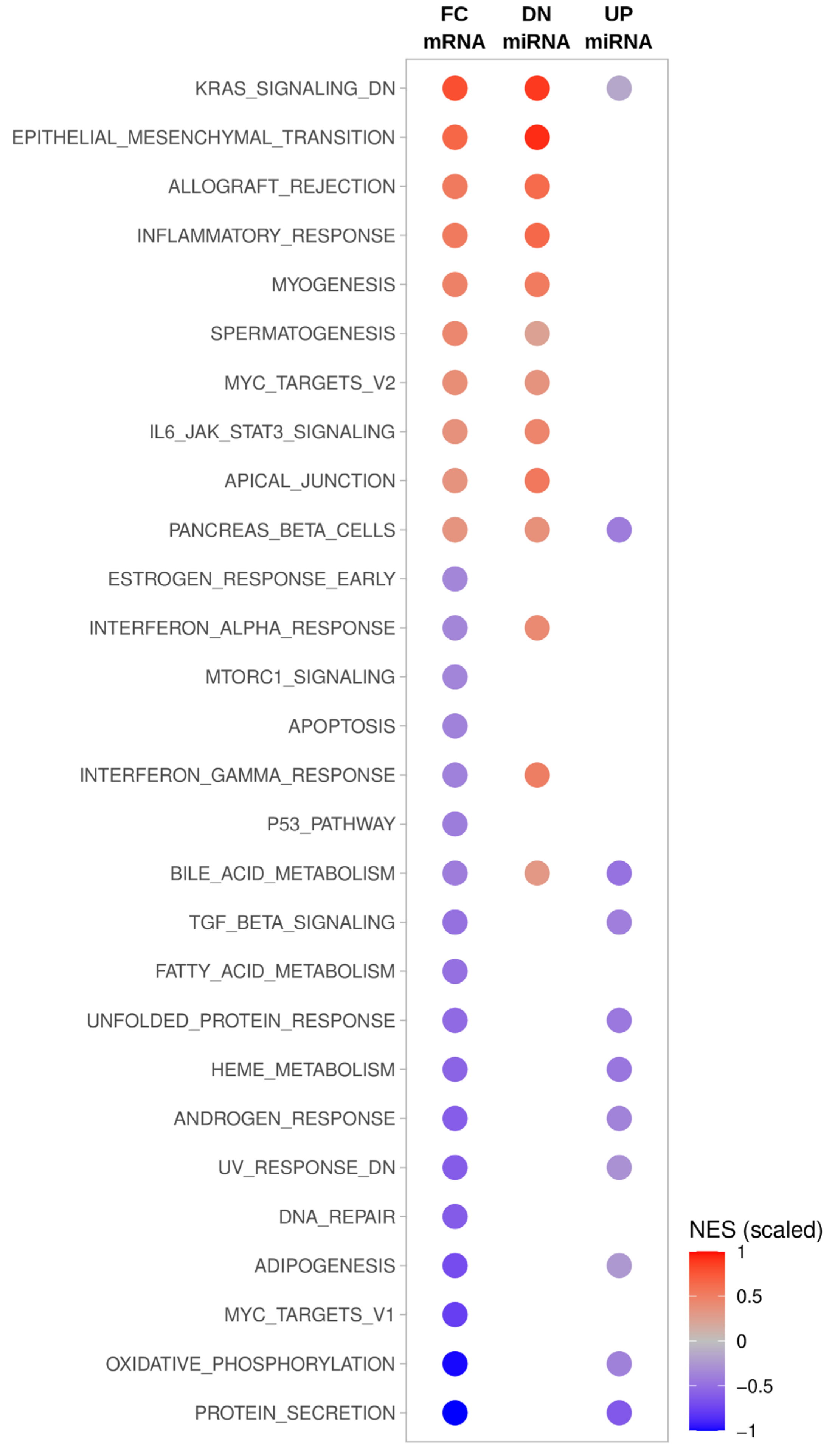
2.8. Gene Set Enrichment Analysis of miRNA Target mRNAs
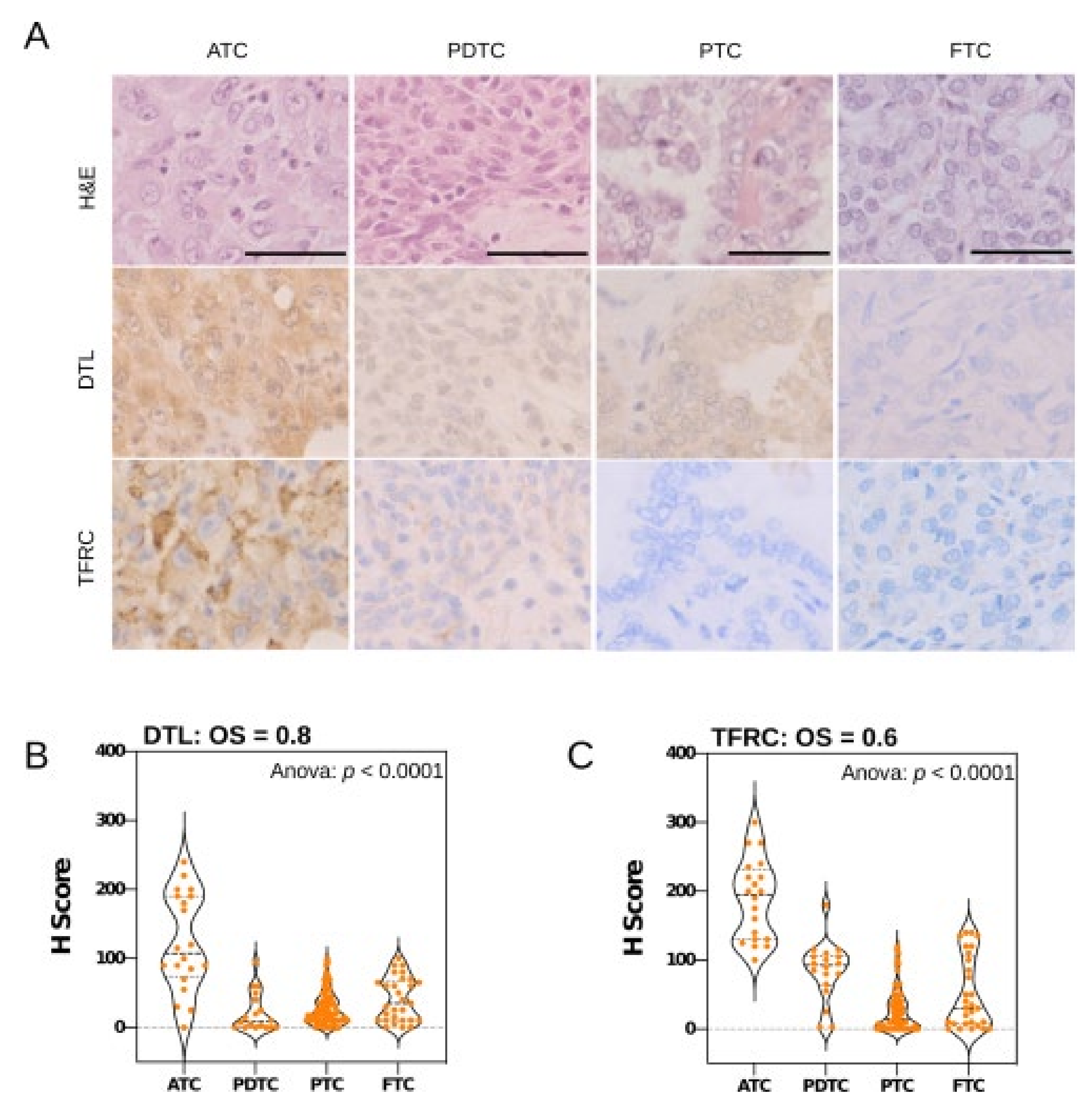
2.9. Tissue Microarray and Immunohistochemistry
3. Results
3.1. SmallRNA Sequencing Unravels miRNA Signatures Distinguishing ATC
3.2. MiRNA Deregulation in ATC Guides Gene Expression Shifts Distinguishing ATC
3.3. Key Effectors of miRNA Deregulation in ATC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Viola, D.; Valerio, L.; Molinaro, E.; Agate, L.; Bottici, V.; Biagini, A.; Lorusso, L.; Cappagli, V.; Pieruzzi, L.; Giani, C.; et al. Treatment of advanced thyroid cancer with targeted therapies: Ten years of experience. Endocr. Relat. Cancer 2016, 23, R185–R205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedje, V.; Stuschke, M.; Weber, F.; Dralle, H.; Moss, L.; Fuhrer, D. Anaplastic thyroid carcinoma: Review of treatment protocols. Endocr. Relat. Cancer 2018, 25, R153–R161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallridge, R.C.; Ain, K.B.; Asa, S.L.; Bible, K.C.; Brierley, J.D.; Burman, K.D.; Kebebew, E.; Lee, N.Y.; Nikiforov, Y.E.; Rosenthal, M.S.; et al. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid 2012, 22, 1104–1139. [Google Scholar] [CrossRef]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Invest. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Abe, I.; Lam, A.K. Anaplastic Thyroid Carcinoma: Current Issues in Genomics and Therapeutics. Curr. Oncol. Rep. 2021, 23, 31. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.; Misiak, D.; Bauer, M.; Pazaitis, N.; Braun, J.; Potschke, R.; Mensch, A.; Bell, J.L.; Dralle, H.; Siebolts, U.; et al. IGF2BP1 is the first positive marker for anaplastic thyroid carcinoma diagnosis. Mod. Pathol. 2021, 34, 32–41. [Google Scholar] [CrossRef]
- Sasanakietkul, T.; Murtha, T.D.; Javid, M.; Korah, R.; Carling, T. Epigenetic modifications in poorly differentiated and anaplastic thyroid cancer. Mol. Cell Endocrinol. 2018, 469, 23–37. [Google Scholar] [CrossRef]
- Bell, J.L.; Wachter, K.; Muhleck, B.; Pazaitis, N.; Kohn, M.; Lederer, M.; Huttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [Green Version]
- Busch, B.; Bley, N.; Muller, S.; Glass, M.; Misiak, D.; Lederer, M.; Vetter, M.; Strauss, H.G.; Thomssen, C.; Huttelmaier, S. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016, 44, 3845–3864. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Bley, N.; Busch, B.; Glass, M.; Lederer, M.; Misiak, C.; Fuchs, T.; Wedler, A.; Haase, J.; Bertoldo, J.B.; et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020, 48, 8576–8590. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Bley, N.; Glass, M.; Busch, B.; Rousseau, V.; Misiak, D.; Fuchs, T.; Lederer, M.; Huttelmaier, S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018, 46, 6285–6303. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Huttelmaier, S. Pathogenic mechanisms of deregulated microRNA expression in thyroid carcinomas of follicular origin. Thyroid Res. 2011, 4 Suppl 1, S1. [Google Scholar] [CrossRef] [Green Version]
- Das, P.K.; Asha, S.Y.; Abe, I.; Islam, F.; Lam, A.K. Roles of Non-Coding RNAs on Anaplastic Thyroid Carcinomas. Cancers 2020, 12, 3159. [Google Scholar] [CrossRef]
- Pishkari, S.; Paryan, M.; Hashemi, M.; Baldini, E.; Mohammadi-Yeganeh, S. The role of microRNAs in different types of thyroid carcinoma: A comprehensive analysis to find new miRNA supplementary therapies. J. Endocrinol. Invest. 2018, 41, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Hoang-Vu, C.; Dralle, H.; Huttelmaier, S. Downregulation of microRNAs directs the EMT and invasive potential of anaplastic thyroid carcinomas. Oncogene 2010, 29, 4237–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Paulsson, J.O.; Rafati, N.; DiLorenzo, S.; Chen, Y.; Haglund, F.; Zedenius, J.; Juhlin, C.C. Whole-genome Sequencing of Follicular Thyroid Carcinomas Reveal Recurrent Mutations in MicroRNA Processing Subunit DGCR8. J. Clin. Endocrinol. Metab 2021, 106, 3265–3282. [Google Scholar] [CrossRef]
- Frezzetti, D.; Reale, C.; Cali, G.; Nitsch, L.; Fagman, H.; Nilsson, O.; Scarfo, M.; De Vita, G.; Di Lauro, R. The microRNA-processing enzyme Dicer is essential for thyroid function. PLoS ONE 2011, 6, e27648. [Google Scholar] [CrossRef]
- Ramirez-Moya, J.; Wert-Lamas, L.; Riesco-Eizaguirre, G.; Santisteban, P. Impaired microRNA processing by DICER1 downregulation endows thyroid cancer with increased aggressiveness. Oncogene 2019, 38, 5486–5499. [Google Scholar] [CrossRef]
- Stokowy, T.; Eszlinger, M.; Swierniak, M.; Fujarewicz, K.; Jarzab, B.; Paschke, R.; Krohn, K. Analysis options for high-throughput sequencing in miRNA expression profiling. BMC Res. Notes 2014, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [Green Version]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366. [Google Scholar] [CrossRef] [PubMed]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef]
- Rainer, J.; Gatto, L.; Weichenberger, C.X. ensembldb: An R package to create and use Ensembl-based annotation resources. Bioinformatics 2019, 35, 3151–3153. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (N Y) 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hebrant, A.; Floor, S.; Saiselet, M.; Antoniou, A.; Desbuleux, A.; Snyers, B.; La, C.; de Saint Aubain, N.; Leteurtre, E.; Andry, G.; et al. miRNA expression in anaplastic thyroid carcinomas. PLoS ONE 2014, 9, e103871. [Google Scholar] [CrossRef] [PubMed]
- Visone, R.; Pallante, P.; Vecchione, A.; Cirombella, R.; Ferracin, M.; Ferraro, A.; Volinia, S.; Coluzzi, S.; Leone, V.; Borbone, E.; et al. Specific microRNAs are downregulated in human thyroid anaplastic carcinomas. Oncogene 2007, 26, 7590–7595. [Google Scholar] [CrossRef]
- Boyerinas, B.; Park, S.M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of let-7-regulated oncofetal genes. Cancer Res. 2008, 68, 2587–2591. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Hung, S.W.; Krentz, M.; Patel, D.; Lovin, D.; Manoharan, R.; Thomson, J.M.; Govindarajan, R. Differential processing of let-7a precursors influences RRM2 expression and chemosensitivity in pancreatic cancer: Role of LIN-28 and SET oncoprotein. PLoS ONE 2013, 8, e53436. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses ce.ell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Aljahdali, I.; Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study? J. Exp. Clin. Cancer Res. 2019, 38, 368. [Google Scholar] [CrossRef] [Green Version]
- Haase, D.; Meister, M.; Muley, T.; Hess, J.; Teurich, S.; Schnabel, P.; Hartenstein, B.; Angel, P. FRMD3, a novel putative tumour suppressor in NSCLC. Oncogene 2007, 26, 4464–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Noncoding RNA 2017, 3, 0022. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A Current Overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar] [CrossRef] [Green Version]
- Ohmura, S.; Marchetto, A.; Orth, M.F.; Li, J.; Jabar, S.; Ranft, A.; Vinca, E.; Ceranski, K.; Carreno-Gonzalez, M.J.; Romero-Perez, L.; et al. Translational evidence for RRM2 as a prognostic biomarker and therapeutic target in Ewing sarcoma. Mol. Cancer 2021, 20, 97. [Google Scholar] [CrossRef] [PubMed]
- Warrier, N.M.; Agarwal, P.; Kumar, P. Emerging Importance of Survivin in Stem Cells and Cancer: The Development of New Cancer Therapeutics. Stem Cell Rev. Rep. 2020, 16, 828–852. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Wang, Q.; Lei, Z.; Feng, M.; Zhao, Z.; Wang, Y.; Wei, G. DTL promotes cancer progression by PDCD4 ubiquitin-dependent degradation. J. Exp. Clin. Cancer Res. 2019, 38, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Falvo, E.; Damiani, V.; Conti, G.; Boschi, F.; Messana, K.; Giacomini, P.; Milella, M.; De Laurenzi, V.; Morea, V.; Sala, G.; et al. High activity and low toxicity of a novel CD71-targeting nanotherapeutic named The-0504 on preclinical models of several human aggressive tumors. J. Exp. Clin. Cancer Res. 2021, 40, 63. [Google Scholar] [CrossRef]
- Elcheva, I.; Goswami, S.; Noubissi, F.K.; Spiegelman, V.S. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 2009, 35, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misiak, D.; Bauer, M.; Lange, J.; Haase, J.; Braun, J.; Lorenz, K.; Wickenhauser, C.; Hüttelmaier, S. MiRNA Deregulation Distinguishes Anaplastic Thyroid Carcinoma (ATC) and Supports Upregulation of Oncogene Expression. Cancers 2021, 13, 5913. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13235913
Misiak D, Bauer M, Lange J, Haase J, Braun J, Lorenz K, Wickenhauser C, Hüttelmaier S. MiRNA Deregulation Distinguishes Anaplastic Thyroid Carcinoma (ATC) and Supports Upregulation of Oncogene Expression. Cancers. 2021; 13(23):5913. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13235913
Chicago/Turabian StyleMisiak, Danny, Marcus Bauer, Jana Lange, Jacob Haase, Juliane Braun, Kerstin Lorenz, Claudia Wickenhauser, and Stefan Hüttelmaier. 2021. "MiRNA Deregulation Distinguishes Anaplastic Thyroid Carcinoma (ATC) and Supports Upregulation of Oncogene Expression" Cancers 13, no. 23: 5913. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13235913