
Germline and Somatic Whole-Exome Sequencing Identifies New Candidate Genes Involved in Familial Predisposition to Serrated Polyposis Syndrome

 , , , , , , ,
, , , , , , ,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
- (1)
- at least 5 serrated polyps proximal to the sigmoid colon with two or more of these being >10 mm,
- (2)
- any number of serrated polyps proximal to the sigmoid colon in an individual who has a first-degree relative with serrated polyposis,
- (3)
- >20 serrated polyps of any size, but distributed throughout the colon.
2. Results
2.1. Patients
2.2. Whole Exome Sequencing Analysis
2.3. Germline-Somatic Paired Analysis
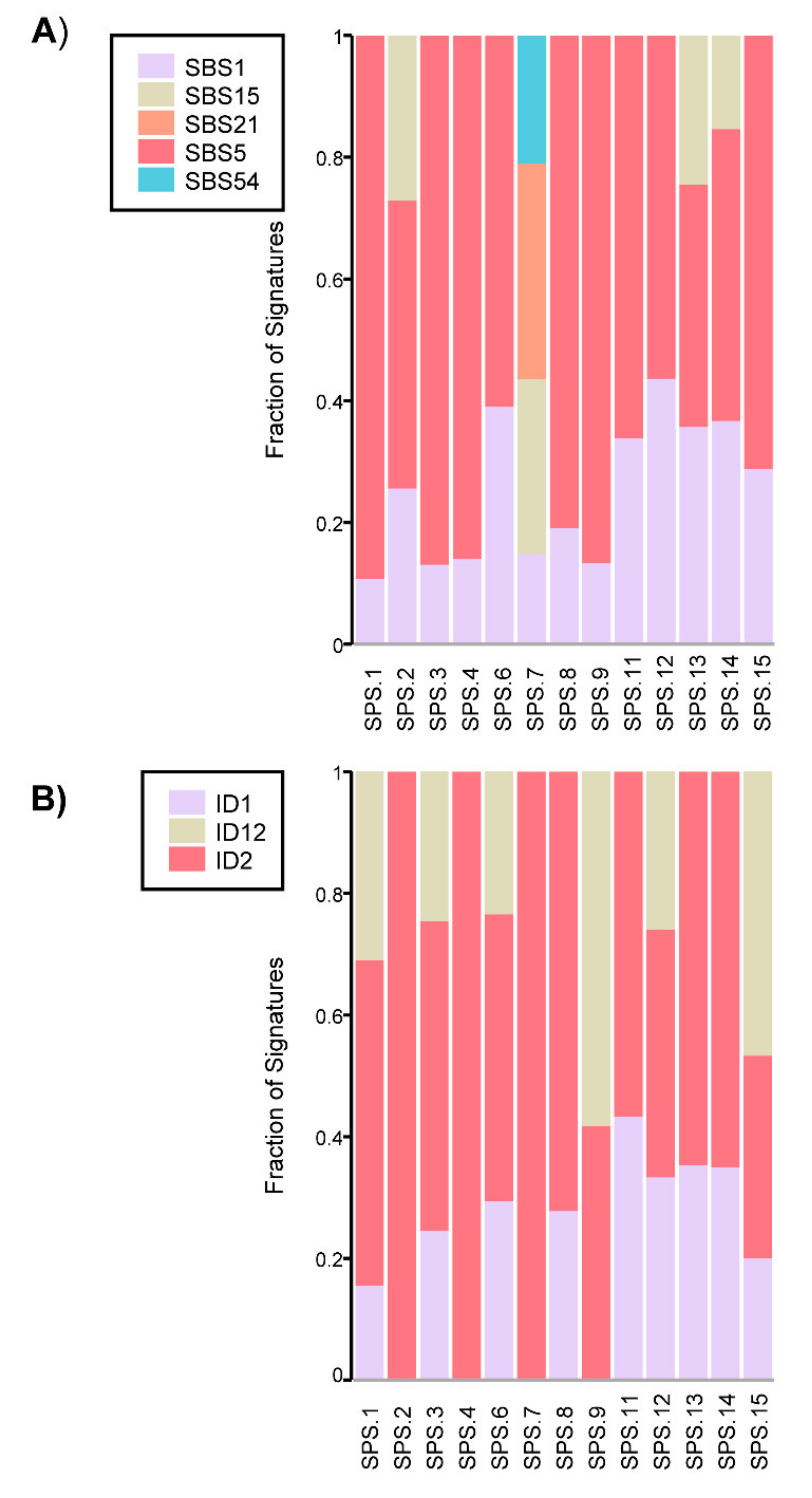
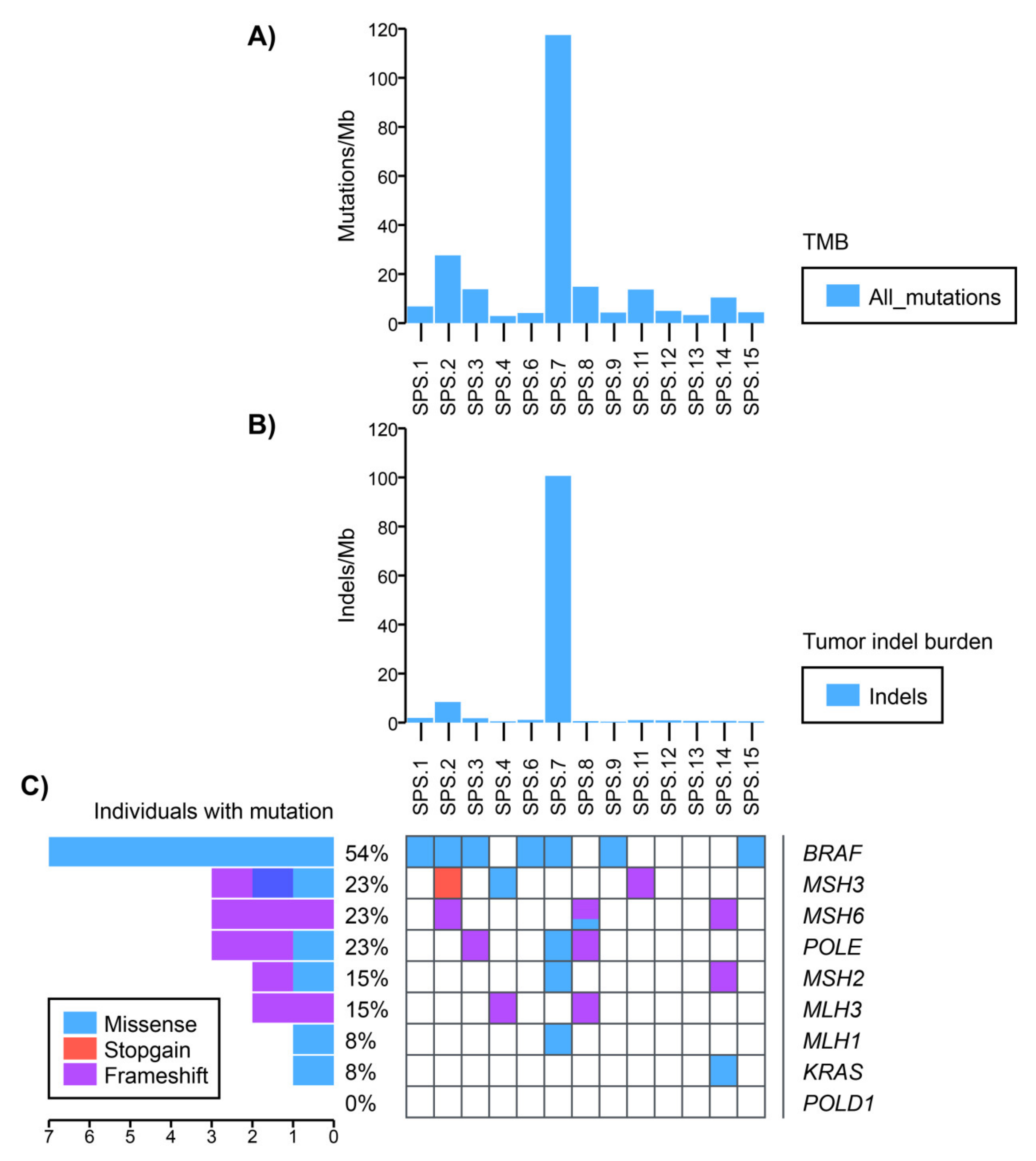
2.4. Somatic Mutational Profile
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole Exome Sequencing
4.3. Sequencing Quality Control and Alignment
4.4. Variant Calling
4.5. Copy number Variation Analysis
4.6. Variant Annotation
4.7. Variant Prioritization
4.8. Variant Validation
4.9. Somatic Data Analysis: LOH Analysis, Mutational Profiling and Mutational Signature Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the Global Cancer Incidence and Mortality in 2018: GLOBOCAN Sources and Methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Morson, B.C. The Evolution of Colorectal Carcinoma. Clin. Radiol. 1984, 35, 425–431. [Google Scholar] [CrossRef]
- Mankaney, G.; Rouphael, C.; Burke, C.A. Serrated Polyposis Syndrome. Clin. Gastroenterol. Hepatol. 2020, 18, 777–779. [Google Scholar] [CrossRef] [PubMed]
- IJspeert, J.E.G.; Vermeulen, L.; Meijer, G.A.; Dekker, E. Serrated Neoplasia—Role in Colorectal Carcinogenesis and Clinical Implications. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.; Pellisé, M.; Carballal, S.; Bessa, X.; Ocaña, T.; Serradesanferm, A.; Grau, J.; Macià, F.; Andreu, M.; Castells, A.; et al. High Prevalence of Serrated Polyposis Syndrome in Fit-Based Colorectal Cancer Screening Programmes. Gut 2013, 62, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Snover, D.; Ahnen, D.; Burt, R.; Odze, R.D. Serrated Polyps of the Colon and Rectum and Serrated Polyposis. In WHO Classification of Tumours of the Digestive System, 4th ed.; IARC: Lyon, France, 2010. [Google Scholar]
- Rosty, C.; Brosens, L.; Dekker, E.; Nagtegaal, I. Serrated Polyposis. In WHO Classification of Tumours of the Digestive System, 5th ed.; IARC: Lyon, France, 2019. [Google Scholar]
- Crockett, S.D.; Nagtegaal, I.D. Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology 2019, 157, 949–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnau-Collell, C.; De Lima, Y.S.; Diáz-Gay, M.; Munõz, J.; Carballal, S.; Bonjoch, L.; Moreira, L.; Lozano, J.J.; Ocanã, T.; Cautrecasas, M.; et al. Colorectal Cancer Genetic Variants Are Also Associated with Serrated Polyposis Syndrome Susceptibility. J. Med. Genet. 2020, 57, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Gala, M.K.; Mizukami, Y.; Le, L.P.; Moriichi, K.; Austin, T.; Yamamoto, M.; Lauwers, G.Y.; Bardeesy, N.; Chung, D.C. Germline Mutations in Oncogene-Induced Senescence Pathways Are Associated with Multiple Sessile Serrated Adenomas. Gastroenterology 2014, 146, 520–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, D.D.; Clendenning, M.; Zhuoer, L.; Stewart, J.R.; Joseland, S.; Arnold, J.; Macrae, F.A.; Holter, S.; Gallinger, S.; Winship, I.M.; et al. Lack of Evidence for Germline RNF43 Mutations in Patients with Serrated Polyposis Syndrome from a Large Multinational Study. Gut 2016, 66, 1170–1172. [Google Scholar] [CrossRef]
- Quintana, I.; Mejías-Luque, R.; Terradas, M.; Navarro, M.; Piñol, V.; Belhadj, S.; Capellá, G.; Darder, E.; Mur, P.; Solanes, A.; et al. Evidence Suggests That Germline RNF43 Mutations Are a Rare Cause of Serrated Polyposis. Gut 2018, 67, 2230–2232. [Google Scholar] [CrossRef]
- Clendenning, M.; Young, J.P.; Walsh, M.D.; Woodall, S.; Arnold, J.; Jenkins, M.; Ko Win, A.; Hopper, J.L.; Sweet, K.; Gallinger, S.; et al. Germline Mutations in the Polyposis-Associated Genes BMPR1A, SMAD4, PTEN, MUTYH and GREM1 Are Not Common in Individuals with Serrated Polyposis Syndrome. PLoS ONE 2013, 8, e66705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome Sequencing Identifies the Cause of a Mendelian Disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Valle, L.; de Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on Genetic Predisposition to Colorectal Cancer and Polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Franch-Expósito, S.; Arnau-Collell, C.; Park, S.; Supek, F.; Muñoz, J.; Bonjoch, L.; Gratacós-Mulleras, A.; Sánchez-Rojas, P.; Esteban-Jurado, C.; et al. Integrated Analysis of Germline and Tumor DNA Identifies New Candidate Genes Involved in Familial Colorectal Cancer. Cancers 2019, 11, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolleman, J.E.; Díaz-Gay, M.; Franch-Expósito, S.; Castellví-Bel, S.; de Voer, R.M. Somatic Mutational Signatures in Polyposis and Colorectal Cancer. Mol. Asp. Med. 2019, 69, 62–72. [Google Scholar] [CrossRef]
- De Palma, F.; D’Argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.; Salvatore, F. The Molecular Hallmarks of the Serrated Pathway in Colorectal Cancer. Cancers 2019, 11, 1017. [Google Scholar] [CrossRef] [Green Version]
- Briggs, S.; Tomlinson, I. Germline and Somatic Polymerase ϵ and δ Mutations Define a New Class of Hypermutated Colorectal and Endometrial Cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Min, J.N.; Tian, Y.; Xiao, Y.; Wu, L.; Li, L.; Chang, S. The MINO80 Chromatin Remodeling Complex Is Required for Efficient Telomere Replication and Maintenance of Genome Stability. Cell Res. 2013, 23, 1396–1413. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Huang, A.; Zheng, X.; Liu, T.; Lin, Z.; Zhang, S.; Yang, Q.; Zhang, T.; Ma, H. 53BP1 Loss Induces Chemoresistance of Colorectal Cancer Cells to 5-Fluorouracil by Inhibiting the ATM–CHK2–P53 Pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Sinha, D.; Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Asaithamby, A. Werner Syndrome Protein and Dna Replication. Int. J. Mol. Sci. 2018, 19, 3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Choi, M.; Kim, J.E. The Histone Methyltransferase Dot1/DOT1L as a Critical Regulator of the Cell Cycle. Cell Cycle 2014, 13, 726–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kari, V.; Raul, S.K.; Henck, J.M.; Kitz, J.; Kramer, F.; Kosinsky, R.L.; Übelmesser, N.; Mansour, W.Y.; Eggert, J.; Spitzner, M.; et al. The Histone Methyltransferase DOT1L Is Required for Proper DNA Damage Response, DNA Repair, and Modulates Chemotherapy Responsiveness. Clin. Epigenet. 2019, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Takei, Y.; Assenberg, M.; Tsujimoto, G.; Laskey, R. The MCM3 Acetylase MCM3AP Inhibits Initiation, but Not Elongation, of DNA Replication via Interaction with MCM3. J. Biol. Chem. 2002, 277, 43121–43125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Bekker-Jensen, I.H.; Christensen, J.; Rasmussen, K.D.; Sidoli, S.; Qi, Y.; Kong, Y.; Wang, X.; Cui, Y.; Xiao, Z.; et al. Tumor Suppressor ASXL1 Is Essential for the Activation of INK4B Expression in Response to Oncogene Activity and Anti-Proliferative Signals. Cell Res. 2015, 25, 1205–1218. [Google Scholar] [CrossRef] [Green Version]
- Scott, P.; Anderson, K.; Singhania, M.; Cormier, R. Cystic Fibrosis, CFTR, and Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Song, C.; Li, J.; Sun, Q. Cftr Functions as a Tumor Suppressor and Is Regulated by Dna Methylation in Colorectal Cancer. Cancer Manag. Res. 2020, 12, 4261–4270. [Google Scholar] [CrossRef]
- Wen, Y.C.; Wang, D.H.; RayWhay, C.Y.; Luo, J.; Gu, W.; Baylin, S.B. Tumor Suppressor HIC1 Directly Regulates SIRT1 to Modulate P53-Dependent DNA-Damage Responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef]
- Szczepny, A.; Carey, K.; McKenzie, L.; Jayasekara, W.S.N.; Rossello, F.; Gonzalez-Rajal, A.; McCaw, A.S.; Popovski, D.; Wang, D.; Sadler, A.J.; et al. The Tumor Suppressor Hic1 Maintains Chromosomal Stability Independent of Tp53. Oncogene 2018, 37, 1939–1948. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jiang, Z.; Zhang, Y.; Wang, X.; Liu, L.; Fan, Z. RNA Sequencing Analysis Reveals Protective Role of Kruppel-like Factor 3 in Colorectal Cancer. Oncotarget 2017, 8, 21984–21993. [Google Scholar] [CrossRef] [PubMed]
- Golubicki, M.; Bonjoch, L.; Acuña-Ochoa, J.G.; Díaz-Gay, M.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline Biallelic MCM8 Variants Are Associated with Early-Onset Lynch-like Syndrome. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Xiao, G.; Fu, J. NF-ΚB and Cancer: A Paradigm of Yin-Yang. Am. J. Cancer Res. 2011, 1, 192–221. [Google Scholar]
- Palles, C.; Cazier, J.-B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Israel Salguero, I.; et al. Germline Mutations Affecting the Proofreading Domains of POLE and POLD1 Predispose to Colorectal Adenomas and Carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Dehghanizadeh, S.; Khoddami, V.; Mosbruger, T.L.; Hammoud, S.S.; Edes, K.; Berry, T.S.; Done, M.; Samowitz, W.S.; DiSario, J.A.; Luba, D.G.; et al. Active BRAF-V600E Is the Key Player in Generation of a Sessile Serrated Polyp-Specific DNA Methylation Profile. PLoS ONE 2018, 13, e0192499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaron, C.; Lopez, R.; Pai, R.K.; Burke, C.A. Expression of Annexin A10 in Serrated Polyps Predicts the Development of Metachronous Serrated Polyps. Clin. Transl. Gastroenterol. 2016, 7, e205. [Google Scholar] [CrossRef]
- Taupin, D.; Lam, W.; Rangiah, D.; McCallum, L.; Whittle, B.; Zhang, Y.; Andrews, D.; Field, M.; Goodnow, C.C.; Cook, M.C. A Deleterious RNF43 Germline Mutation in a Severely Affected Serrated Polyposis Kindred. Hum. Genome Var. 2015, 2, 2014–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.H.N.; Lai, J.C.W.; Ho, S.L.; Leung, W.K.; Law, W.L.; Lee, J.F.Y.; Chan, A.K.W.; Tsui, W.Y.; Chan, A.S.Y.; Lee, B.C.H.; et al. RNF43 Germline and Somatic Mutation in Serrated Neoplasia Pathway and Its Association with BRAF Mutation. Gut 2017, 66, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like Mutational Processes in Human Somatic Cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.J.; Spring, K.J.; Wynter, C.V.A.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF Mutation Is Associated with DNA Methylation in Serrated Polyps and Cancers of the Colorectum. Gut 2004, 53, 1137–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horpaopan, S.; Kirfel, J.; Peters, S.; Kloth, M.; Hüneburg, R.; Altmüller, J.; Drichel, D.; Odenthal, M.; Kristiansen, G.; Strassburg, C.; et al. Exome Sequencing Characterizes the Somatic Mutation Spectrum of Early Serrated Lesions in a Patient with Serrated Polyposis Syndrome (SPS). Hered. Cancer Clin. Pract. 2017, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Esteban-Jurado, C.; Vila-Casadesús, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Muñoz, J.; Ocaña, T.; Balaguer, F.; López-Cerón, M.; et al. Whole-Exome Sequencing Identifies Rare Pathogenic Variants in New Predisposition Genes for Familial Colorectal Cancer. Genet. Med. 2015, 17, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM Mapper: Fast, Accurate and Versatile Alignment by Filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Moncunill, V.; Gonzalez, S.; Beà, S.; Andrieux, L.O.; Salaverria, I.; Royo, C.; Martinez, L.; Puiggròs, M.; Segura-Wang, M.; Stütz, A.M.; et al. Comprehensive Characterization of Complex Structural Variations in Cancer by Directly Comparing Genome Sequence Reads. Nat. Biotechnol. 2014, 32, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Franch-Expósito, S.; Esteban-Jurado, C.; Garre, P.; Quintanilla, I.; Duran-Sanchon, S.; Díaz-Gay, M.; Bonjoch, L.; Cuatrecasas, M.; Samper, E.; Muñoz, J.; et al. Rare Germline Copy Number Variants in Colorectal Cancer Predisposition Characterized by Exome Sequencing Analysis. J. Genet. Genom. 2018, 45, 41–45. [Google Scholar] [CrossRef]
- Krumm, N.; Sudmant, P.H.; Ko, A.; O’Roak, B.J.; Malig, M.; Coe, B.P.; Quinlan, A.R.; Nickerson, D.A.; Eichler, E.E. Copy Number Variation Detection and Genotyping from Exome Sequence Data. Genome Res. 2012, 22, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A Robust Model for Read Count Data in Exome Sequencing Experiments and Implications for Copy Number Variant Calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC Browser: Displaying Reference Data Information from over 60 000 Exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. DbNSFP: A Lightweight Database of Human Nonsynonymous SNPs and Their Functional Predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. DbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Hartline, E.; et al. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Supek, F.; Lehner, B. Systematic Discovery of Germline Cancer Predisposition Genes through the Identification of Somatic Second Hits. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.M.A.; Wu, Y.; Díaz-Gay, M.; Bergstrom, E.N.; He, Y.; Barnes, M.; Vella, M.; Wang, J.; Teague, J.W.; Clapham, P.; et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. bioRxiv 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Family | ID | Onset Age | Gender | Diagnosis | Criteria SPS | CRC | MMR +/− | Family Cancer History |
|---|---|---|---|---|---|---|---|---|
| SPS.1 | AA3519 * | 58 | M | SPS | 1 + 3 | N | N/A | Prostate (father, 82) |
| AA3520 | 62 | F | SPS | 2 | N | |||
| SPS.2 | AA3521 * | 62 | M | SPS | 1 + 3 | N | N/A | Pancreas (father, 59), Colon (mother, 78) |
| AA3522 | 59 | M | SPS | 2 | N | |||
| SPS.3 | AA3523 | 32 | M | CRC | - | Y | + | CRC (mom, 56), CRC (brother, 56), Pancreas (aunt, 60), CRC (uncle, 53), CRC (uncle, 71), CRC (cousin, 32), Brain (cousin, 32) |
| AA3524 * | 64 | F | SPS | 3 | N | |||
| SPS.4 | AA3525 | 58 | M | SPS | 3 | N | + | CRC (father, 86), Laryngeal (father, 85), Breast (aunt, 61), Breast (aunt, 56), CRC (grandfather, 61) |
| AA3526 * | 53 | F | SPS | 2 | N | |||
| SPS.5 | AA3527 | 22 | F | SPS | 3 | N | N/A | Laryngeal (grandfather, unknown age) |
| AA3528 * | 20 | M | SPS | 2 | N | |||
| SPS.6 | AA3529 | 61 | F | SPS | 3 | N | + | CRC (sister, 43), CRC (aunt, 70), Gastrointestinal (father, 54) |
| AA3530 * | 53 | F | SPS | 2 | N | |||
| SPS.7 | AA3531 * | 57 | F | SPS | 1 + 3 | Y | MLH1-/ | Lymphoma (sister, 46), Gastric (grandfather, 48) |
| AA3532 | 56 | M | SPS | 2 | N | PMS2- | ||
| SPS.8 | AA3533 * | 62 | M | SPS | 3 | N | N/A | - |
| AA3534 | 65 | M | SPS | 2 | N | |||
| SPS.9 | AA3535 | 65 | F | SPS/CRC | 2 + 3 | Y | N/A | CRC (father, 62) |
| AA3536 | 70 | F | SPS/mixed polyposis | 2 | N | |||
| AA3537 * | 64 | M | Mixed polyposis | 3 | N | |||
| SPS.10 | AA3538 | 58 | F | CRC | - | Y | + | CRC (mother, 58), Lung (mother, 70), CRC (uncle, 77), CRC (grandfather, 83), Thyroid (sister, 57), Skin (sister, 42), Liver (cousin, 28), Liver (cousin, unknown age), Skin (grandmother, unknown age) |
| AA3539 | 60 | M | SPS | 2 | N | |||
| AA3540 | 53 | F | SPS | 2 | N | |||
| AA3541 | 40 | F | SPS | 1 + 3 | N | |||
| SPS.11 | AA3559 | 39 | F | SPS | 2 | N | + | Skin (grandmother, 91), CRC (mother, 54), CRC (father, 64), Endometrial (aunt, 72), Endometrial (grandmother, 70), Ovarian teratoma (niece, 7) |
| AA3560 | 46 | F | SPS | 1 | N | |||
| AA3561 | 38 | F | SPS | 2 | N | |||
| AA3562 | 44 | M | SPS | 2 | N | |||
| AA3563 * | 54 | F | CRC | 1 | Y | |||
| AA3564 | 48 | M | SPS/adenomas | 2 | N | |||
| SPS.12 | AA3565 | 55 | M | SPS | 2 + 3 | N | + | CRC (sister, 46) |
| AA3566 * | 59 | M | SPS | 1 + 2 + 3 | N | |||
| SPS.13 | AA3567 * | 59 | M | SPS | 2 + 3 | N | N/A | Thyroid (mother, 66), Endometrial (mother, 76), Unknown cancer (father, 75) |
| AA3568 | 55 | F | SPS | 1 + 2 + 3 | N | |||
| SPS.14 | AA3569 * | 68 | M | SPS/CRC | 2 + 3 | Y | N/A | CRC (uncle, 85), Pancreas (father, 75) |
| AA3570 | 74 | M | SPS | 2 + 3 | N | |||
| SPS.15 | AA3571 | 67 | M | SPS | 2 | N | N/A | Sarcoma (father 51), Thyroid (cousin, 21), Glioma (cousin, 23) |
| AA3572 * | 38 | M | SPS | 1 + 3 | N | |||
| SPS.16 | AA3573 | 47 | F | SPS | 2 + 3 | N | MLH1-/PMS2- | Gastric (father, 72), Brain (mother, 71), Pancreas (uncle, 61), Lung (uncle, 67), Bile duct (uncle, unknown age), Gastric (grandfather, 61) |
| AA3574 | 51 | M | SPS | 2 | N |
| Gene | Family | Variant | Pred. Tools | gnomAD | Protein Domain Location | Biological Process | Analysis Approach | Selection Criteria |
|---|---|---|---|---|---|---|---|---|
| ANXA10 | SPS.12 | c.692A>G p.(Asp231Gly) | 6 | 0.000036 | Annexin 3 (171–243 AA) | growth regulation; apoptosis; cell division | G | Histological marker for serrated pathway |
| ASXL1 | SPS.13 | c.2110G>A p.(Gly704Arg) | 6 | 0.000668 | No | transcription; cell morphogenesis; hemopoiesis; homeostasis of number of cells; protein deubiquitination; | G | Involvement in cancer development |
| CFTR | SPS.3 | c.3995C>A p.(Pro1332His) | 6 | N/A | Nucleotide -Binding 2 (1207–1436 AA) | transmembrane transport; positive regulation of voltage-gated chloride channel activity; protein deubiquitination; | G | Involvement in cancer development |
| SPS.14 | c.1601C>A p.(Ala534Glu) | 3 | 0.0000159 | Nucleotide -Binding 1 (390–645 AA) | ||||
| DOT1L | SPS.11 | c.3307G>A p.(Val1103Met) | 5 | 0.0000349 | No | chromatin silencing; histone H3-K79 methylation; regulation of JAK-STAT cascade; telomere organization; DNA repair | G | Senescence and epigenetics regulator candidate |
| HIC1 | SPS.14 | c.1295A>G p.(Gln432Arg) | 3 | N/A | Zinc finger 1 (439–459 AA) | regulation of transcription; positive regulation of DNA damage response, signal transduction by p53 class mediator | G-S paired analysis | Involvement in cancer development |
| INO80 | SPS.6 | c.4271G>C p.(Arg1424Pro) | 5 | 0.00022 | No | mitotic sister chromatid segregation; double-strand break repair via homologous recombination | G | Senescence candidate |
| KLF3 | SPS.11 | c.275C>T p.(Ser92Leu) | 4 | 0.000203 | No | regulation of transcription; cellular response to peptide | G | Involvement in cancer development |
| MCM3AP | SPS.4 | c.1592A>G p.(Glu531Gly) | 4 | 0.000502 | No | mRNA transport; nucleosome organization; somatic hypermutation of immunoglobulin genes | G | Epigenetics regulator candidate |
| SPS.7 | c.5327T>C p.(Val1776Ala) | 6 | 0.000191 | No | ||||
| MCM8 | SPS.6 | c.876-1delG | N/A | N/A | No | G1/S transition of mitotic cell cycle; mitotic cell cycle; double-strand break repair via homologous | G | Involvement in cancer development |
| PDLIM2 | SPS.7 | p.(Pro130Leu) | 3 | N/A | No | stability of Nuclear Factor kappa-B (NFκB) and other transcription factors; polarized cell migration | G-S paired analysis | Involvement in cancer development |
| POLD1 | SPS.7 | c.1941delG p.(Lys648fs) | N/A | N/A | Polymerase domain (581–910 AA) | mitotic cell cycle; DNA replication; DNA repair; DNA replication proofreading; DNA damage response | G | Involvement in cancer development |
| TP53BP1 | SPS.13 | c.3835G>A p.(Glu1279Lys) | 6 | 0.000008 | No | DNA repair; positive regulation of transcription; DNA damage checkpoint; protein sumoylation | G | Senescence candidate |
| WNK2 | SPS.4 | c.4820C>T p.(Ala1607Val) | 5 | 0.0000788 | No | protein phosphorylation; negative regulation of cell proliferation; regulation of ion homeostasis | G | Involvement in cancer development |
| SPS.6 | c.6157G>A p.(Val2053Ile) | 5 | 0.0000434 | No | ||||
| WRN | SPS.1 | c.2023G>C p.(Glu675Gln) | 4 | 0.00000795 | Helicase ATP-binding (558–724 AA) | telomere maintenance: DNA synthesis involved in DNA repair; replicative cell aging; DNA metabolic process | G | Senescence candidate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares de Lima, Y.; Arnau-Collell, C.; Díaz-Gay, M.; Bonjoch, L.; Franch-Expósito, S.; Muñoz, J.; Moreira, L.; Ocaña, T.; Cuatrecasas, M.; Herrera-Pariente, C.; et al. Germline and Somatic Whole-Exome Sequencing Identifies New Candidate Genes Involved in Familial Predisposition to Serrated Polyposis Syndrome. Cancers 2021, 13, 929. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13040929
Soares de Lima Y, Arnau-Collell C, Díaz-Gay M, Bonjoch L, Franch-Expósito S, Muñoz J, Moreira L, Ocaña T, Cuatrecasas M, Herrera-Pariente C, et al. Germline and Somatic Whole-Exome Sequencing Identifies New Candidate Genes Involved in Familial Predisposition to Serrated Polyposis Syndrome. Cancers. 2021; 13(4):929. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13040929
Chicago/Turabian StyleSoares de Lima, Yasmin, Coral Arnau-Collell, Marcos Díaz-Gay, Laia Bonjoch, Sebastià Franch-Expósito, Jenifer Muñoz, Leticia Moreira, Teresa Ocaña, Miriam Cuatrecasas, Cristina Herrera-Pariente, and et al. 2021. "Germline and Somatic Whole-Exome Sequencing Identifies New Candidate Genes Involved in Familial Predisposition to Serrated Polyposis Syndrome" Cancers 13, no. 4: 929. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13040929