
Mechanistic Basis for In Vivo Therapeutic Efficacy of CK2 Inhibitor CX-4945 in Acute Myeloid Leukemia

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Cells and Cell Culture
2.2. Drugs and Reagents
2.3. Human Leukemia Mouse Xenograft Models
2.4. Apoptosis Assay
2.5. Proliferation and Cytotoxicity Assays
2.6. Western Blot
2.7. In Vitro Phospho-IKAROS Labeling
2.8. Colony Formation Assay
2.9. Gene Expression Analysis by qRT-PCR
2.10. Luciferase Assay
2.11. Quantitative Chromatin Immunoprecipitation
2.12. Statistical Analysis
3. Results
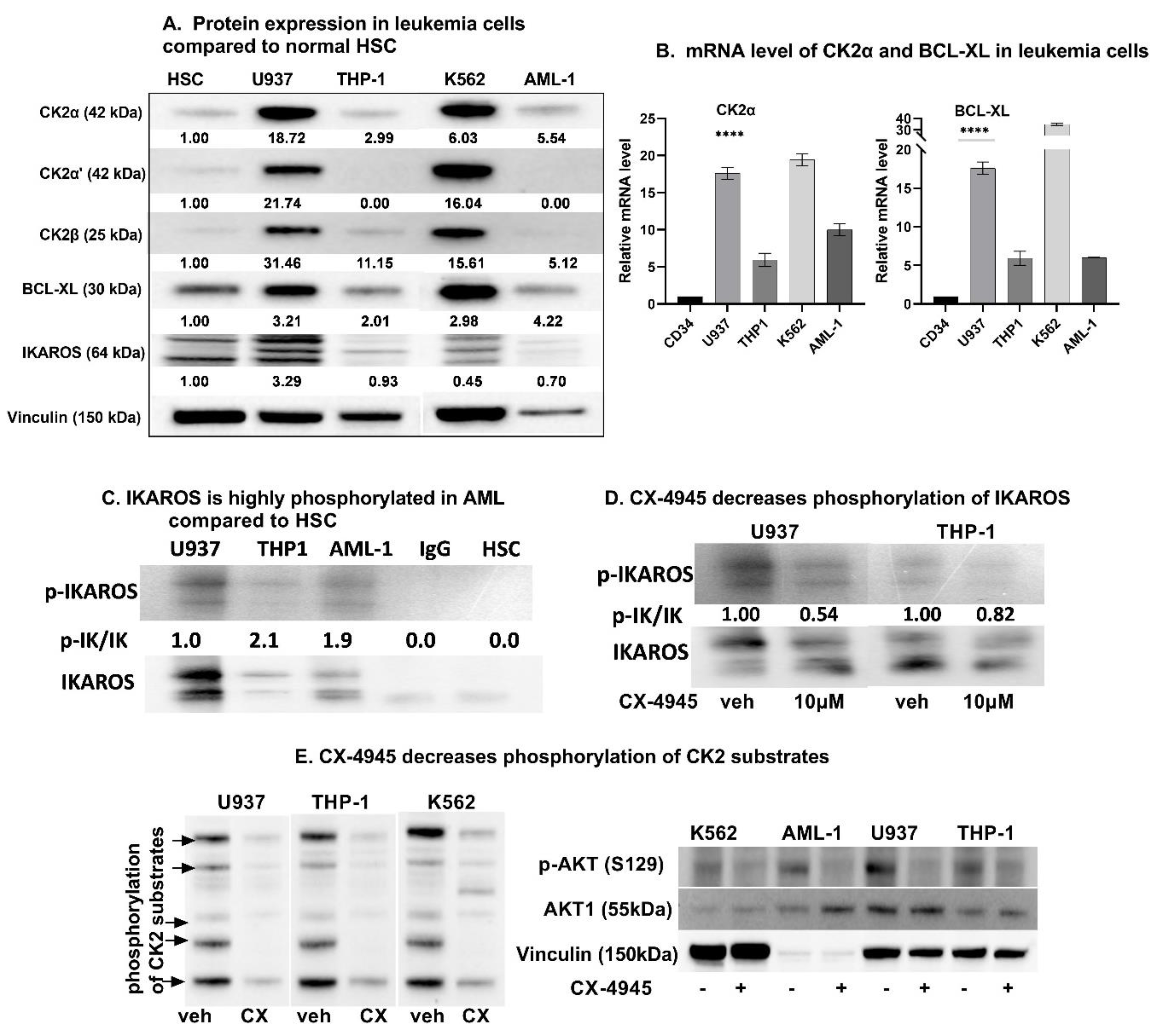
3.1. High Baseline Expression of CK2, Bcl-xl, and p-IKAROS in Myeloid Leukemia Cells Compared to Normal Hematopoietic Stem Cells
3.2. CK2 Inhibition Decreases IKAROS Phosphorylation and CK2 Cellualr Activity
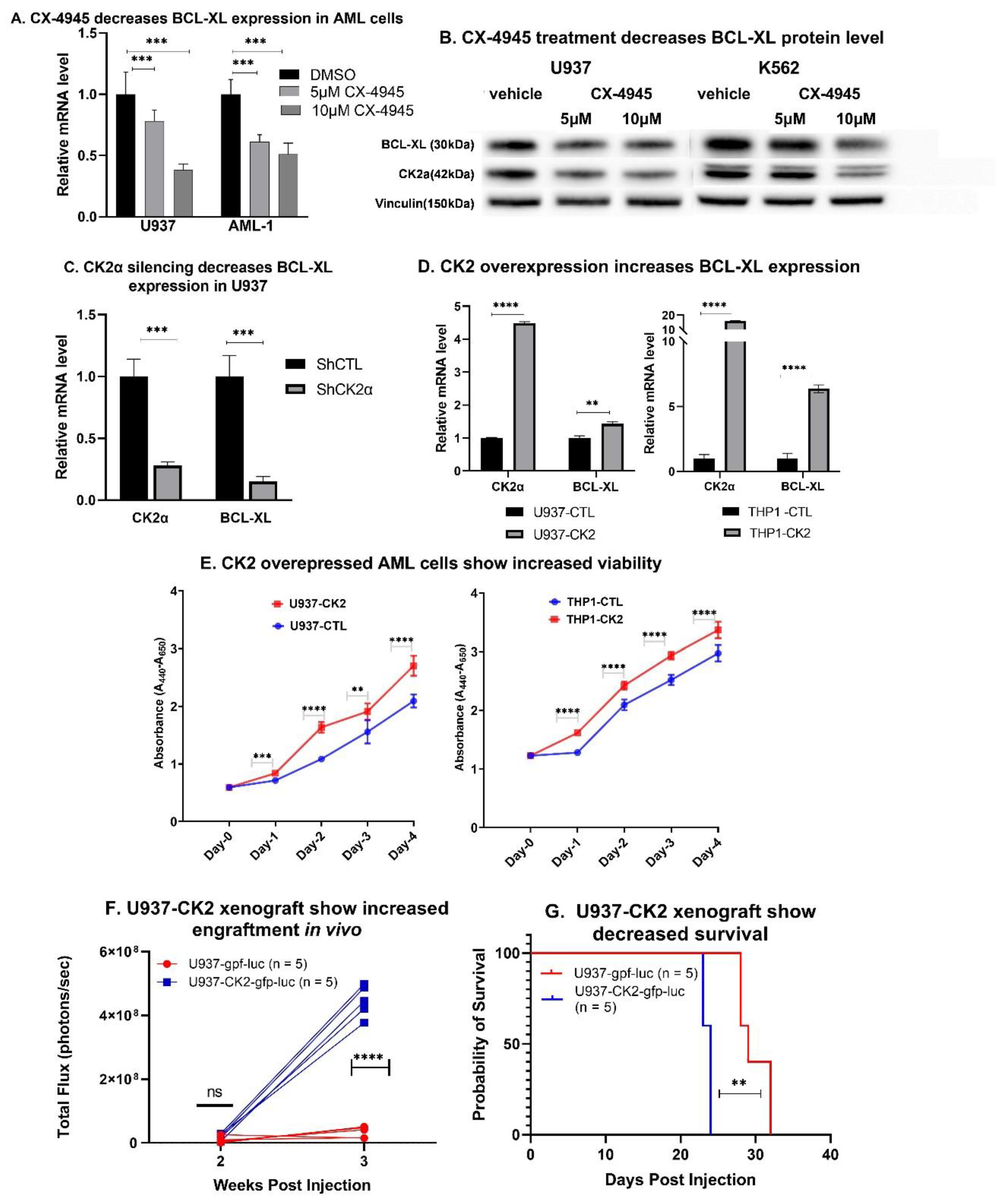
3.3. Inhibition of CK2 Represses BCL-XL Expression
3.4. CX-4945 Treatment Shows Therapeutic Efficacy in AML Patient-Derived Xenografts
3.5. CX-4945 Treatment Decreases BCL-XL Expression In Vivo
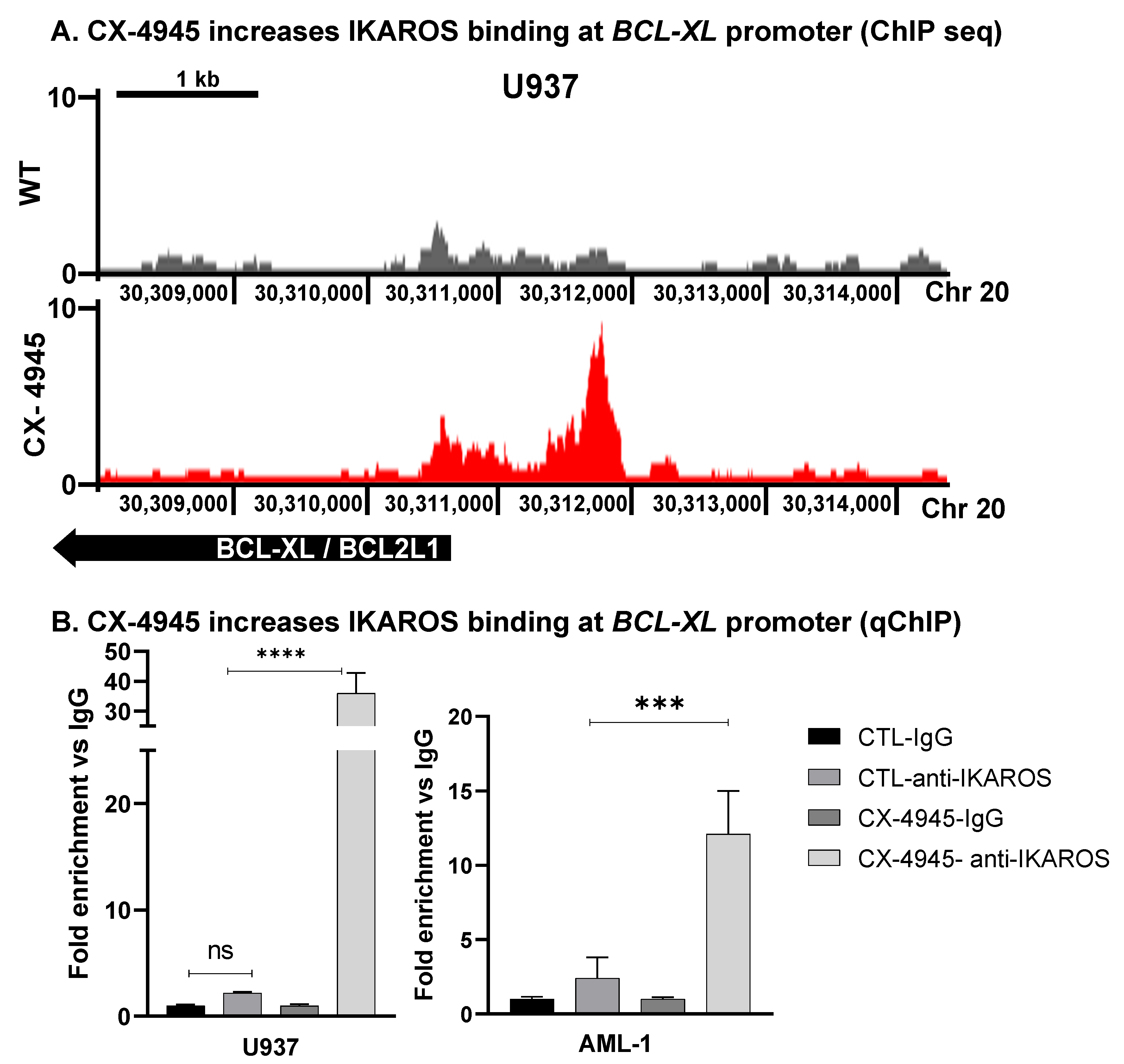
3.6. CK2 Inhibitor Increases IKAROS DNA-Binding to BCL-XL
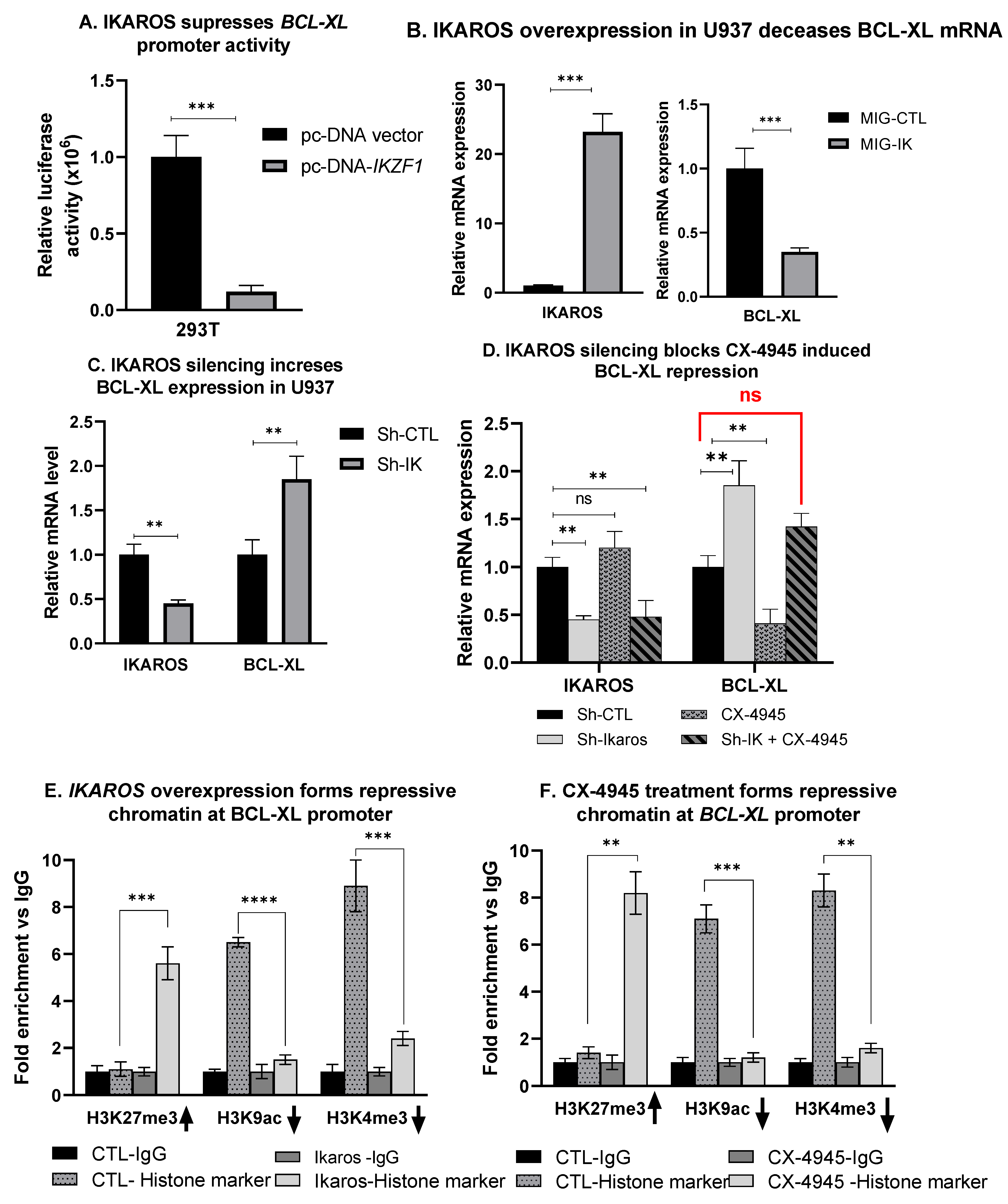
3.7. IKAROS Represses BCL-XL Expression
3.8. CX-4945-Induced BCL-XL Repression Is Mediated via IKAROS
3.9. IKAROS Represses BCL-XL via the Formation of Repressive Chromatin
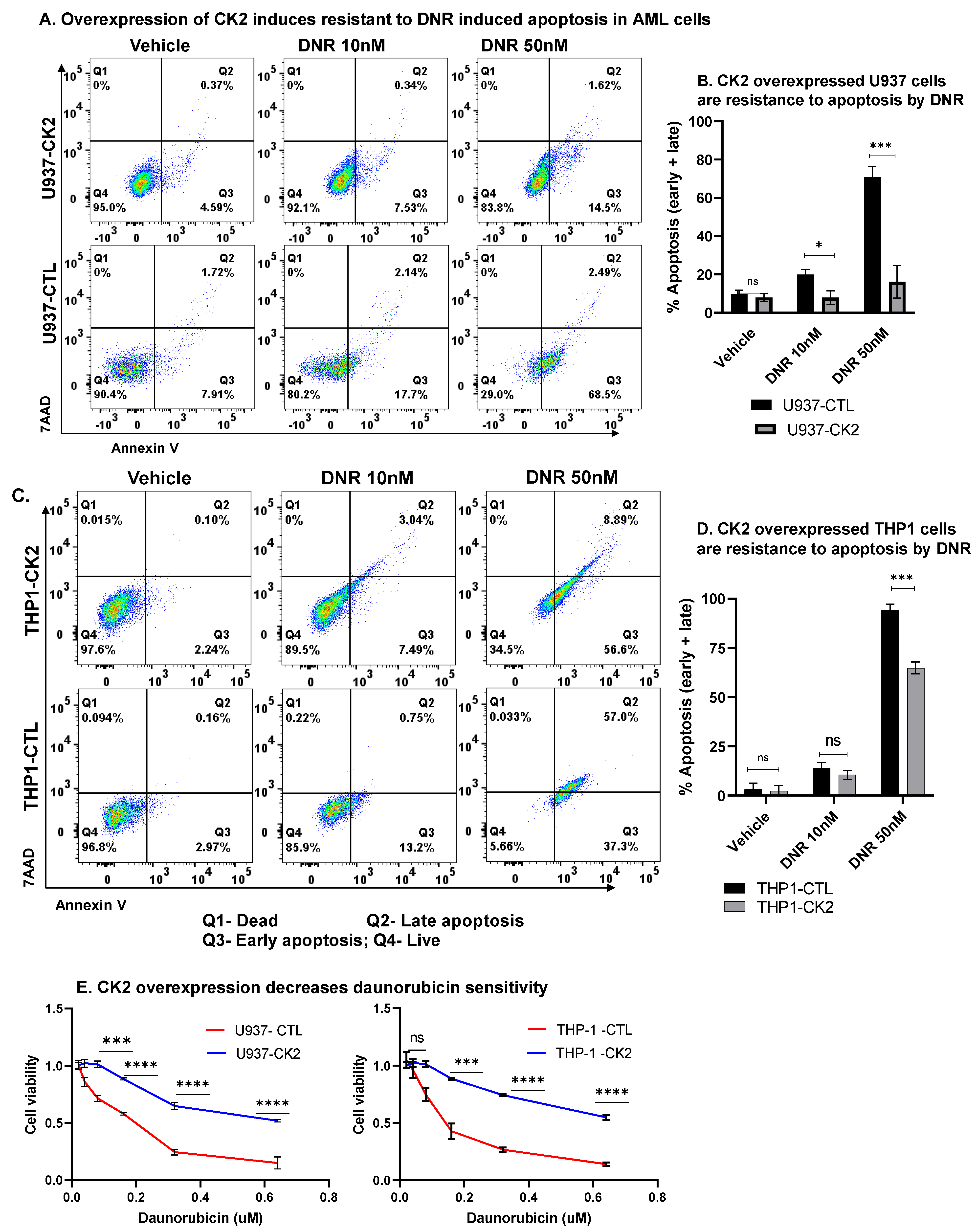
3.10. CK2 Regulate AML Cell Sensitivity towards Daunorubicin
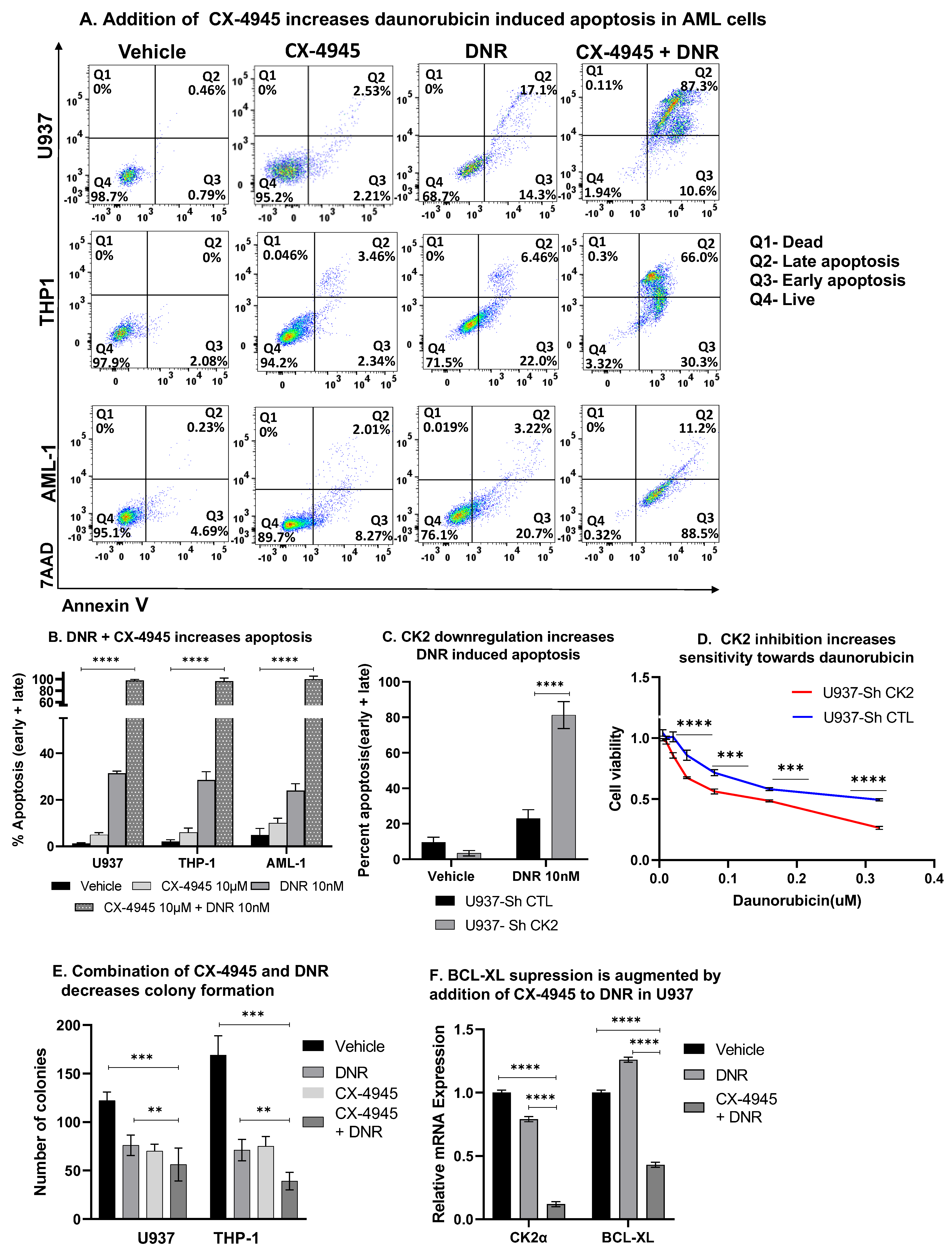
3.11. CK2 Inhibition Augments Daunorubicin Drug Response
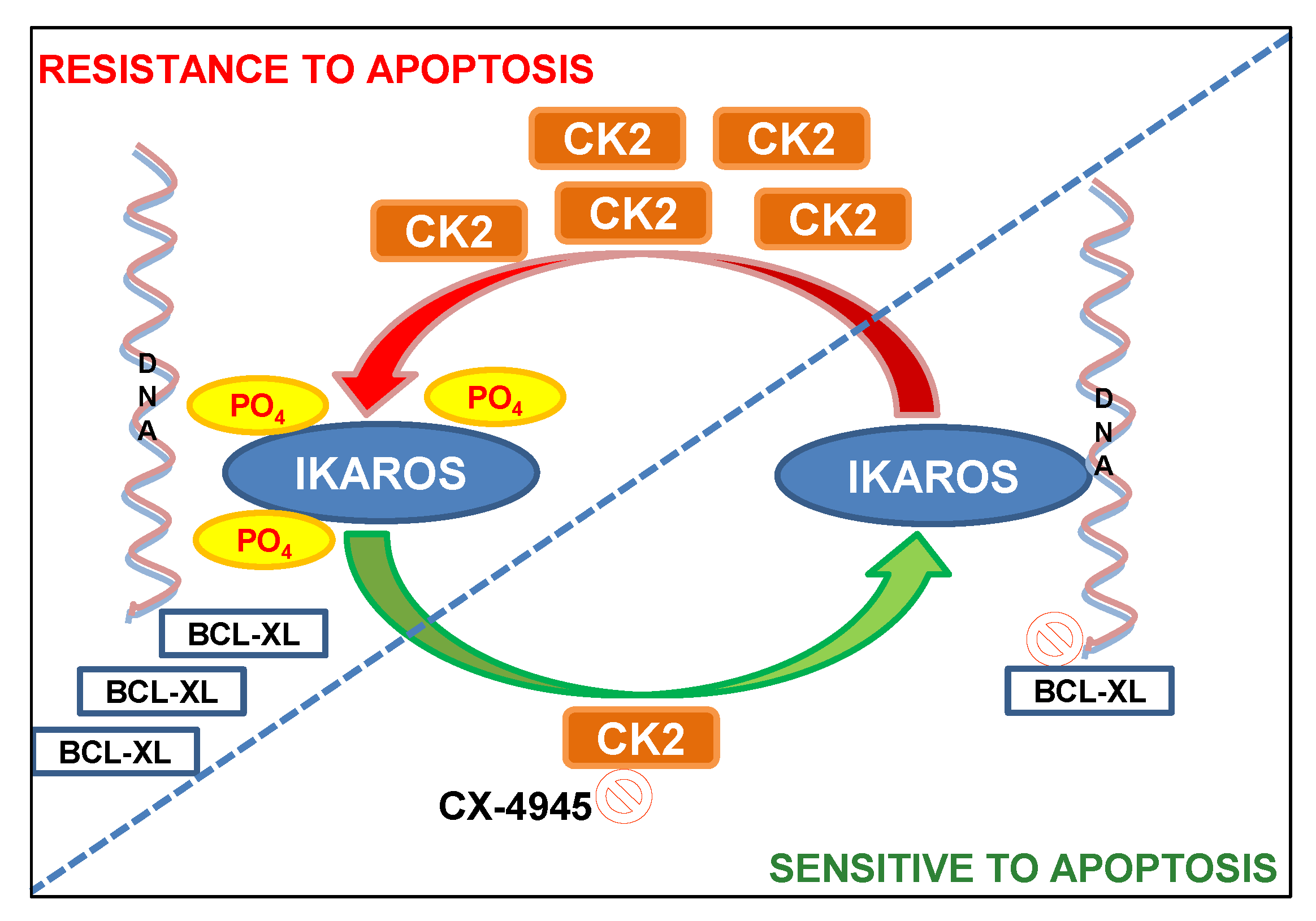
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Tallman, M.S.; Wang, E.S.; Altman, J.K.; Appelbaum, F.R.; Bhatt, V.R.; Bixby, D.; Coutre, S.E.; De Lima, M.; Fathi, A.T.; Fiorella, M.; et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019, 17, 721–749. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.E. The role of targeted therapy in the management of patients with AML. Blood Adv. 2017, 1, 2281–2294. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.S. Boldly Targeting Kinases without mutations. Blood 2014, 123, 1119–1121. [Google Scholar] [CrossRef] [Green Version]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef]
- Litchfield, D.W.; Bosc, D.G.; Canton, D.A.; Saulnier, R.B.; Vilk, G.; Zhang, C. Functional specialization of CK2 isoforms and characterization of isoform-specific binding partners. Mol. Cell. Biochem. 2001, 227, 21–29. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Lee, M.; Dominguez, I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE 2017, 12, e0188854. [Google Scholar] [CrossRef] [Green Version]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef]
- Piazza, F.; Manni, S.; Ruzzene, M.; Pinna, L.A.; Gurrieri, C.; Semenzato, G. Protein kinase CK2 in hematologic malignancies: Reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia 2012, 26, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Chandrika, G.; Mansi, S.; Sunil, M.; Malika, K.; Lidija, P.-D.; Melanie, H.; Yali, D.; Chunhua, S.; Jonathon, L.P.; Tan BH, S.D. Casein kinase II (CK2) as a therapeutic target for hematological malignancies. Curr. Pharm. Des. 2016, 22, 1–13. [Google Scholar] [CrossRef]
- Buontempo, F.; McCubrey, J.A.; Orsini, E.; Ruzzene, M.; Cappellini, A.; Lonetti, A.; Evangelisti, C.; Chiarini, F.; Barata, J.T.; Martelli, A.M. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia 2018, 32, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mandato, E.; Manni, S.; Zaffino, F.; Semenzato, G.; Piazza, F. Targeting CK2-driven non-oncogene addiction in B-cell tumors. Oncogene 2016, 35, 6045–6052. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Quotti Tubi, L.; Canovas Nunes, S.; Brancalion, A.; Doriguzzi Breatta, E.; Manni, S.; Mandato, E.; Zaffino, F.; Macaccaro, P.; Carrino, M.; Gianesin, K.; et al. Protein kinase CK2 regulates AKT, NF-κB and STAT3 activation, stem cell viability and proliferation in acute myeloid leukemia. Leukemia 2017, 31, 292–300. [Google Scholar] [CrossRef]
- Quotti Tubi, L.; Gurrieri, C.; Brancalion, A.; Bonaldi, L.; Bertorelle, R.; Manni, S.; Pavan, L.; Lessi, F.; Zambello, R.; Trentin, L.; et al. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by RNA interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J. Hematol. Oncol. 2013, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Eom, J.I.; Cheong, J.W.; Choi, A.J.; Lee, J.K.; Yang, W.I.; Min, Y.H. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin. Cancer Res. 2007, 13, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- McKendrick, L.; Milne, D.; Meek, D. Protein kinase CK2-dependent regulation of p53 function: Evidence that the phosphorylation status of the serine 386 (CK2) site of p53 is constitutive and stable. Mol. Cell. Biochem. 1999, 191, 187–199. [Google Scholar] [CrossRef]
- Dovat, S.; Song, C.; Payne, K.J.; Li, Z. Ikaros, CK2 kinase, and the road to leukemia. Mol. Cell. Biochem. 2011, 356, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Pulido, R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Y.; Wang, C.; Liu, X.; Du, Y.; Liu, S.; Liu, K.; Zhou, J.; Zhou, C. CK2-mediated CCDC106 phosphorylation is required for p53 degradation in cancer progression. J. Exp. Clin. Cancer Res. 2019, 38, 131. [Google Scholar] [CrossRef] [Green Version]
- Arriazu, E.; Vicente, C.; Pippa, R.; Peris, I.; Martínez-Balsalobre, E.; García-Ramírez, P.; Marcotegui, N.; Igea, A.; Alignani, D.; Rifón, J.; et al. A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia. Blood Cancer J. 2020, 10, 3. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Gowda, C.; Pan, X.; Ding, Y.; Tong, Y.; Tan, B.H.; Wang, H.; Muthusami, S.; Ge, Z.; Sachdev, M.; et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood 2015, 126, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Pan, X.; Ge, Z.; Gowda, C.; Ding, Y.; Li, H.; Li, Z.; Yochum, G.; Muschen, M.; Li, Q.; et al. Epigenetic regulation of gene expression by Ikaros, HDAC1 and Casein Kinase II in leukemia. Leukemia 2016, 30, 1436–1440. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Zhang, B.; Payne, J.L.; Song, C.; Ge, Z.; Gowda, C.; Iyer, S.; Dhanyamraju, P.K.; Dorsam, G.; Reeves, M.E.; et al. Ikaros tumor suppressor function includes induction of active enhancers and super-enhancers along with pioneering activity. Leukemia 2019, 33, 2720–2731. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Li, Z.; Erbe, A.K.; Savic, A.; Dovat, S. Regulation of Ikaros function by casein kinase 2 and protein phosphatase 1. World J. Biol. Chem. 2011, 2, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Gowda, C.; Song, C.; Ding, Y.; Iyer, S.; Dhanyamraju, P.K.; McGrath, M.; Bamme, Y.; Soliman, M.; Kane, S.; Payne, J.L.; et al. Cellular signaling and epigenetic regulation of gene expression in leukemia. Adv. Biol. Regul. 2020, 75, 100665. [Google Scholar] [CrossRef]
- Song, C.; Ge, Z.; Ding, Y.; Tan, B.H.; Desai, D.; Gowda, K.; Amin, S.G.; Gowda, R.; Robertson, G.; Yue, F.; et al. IKAROS and CK2 regulate expression of BCL-XL and chemosensitivity inhigh-risk B-cell acute lymphoblastic leukemia. Blood 2020. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Li, X.; Lv, Y.; Zhu, Y.; Wang, J.; Jin, J.; Yu, W. The specific distribution pattern of IKZF1 mutation in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Francis, O.L.; Payne, J.L.; Su, R.J.; Payne, K.J. Regulator of myeloid differentiation and function: The secret life of Ikaros. World J. Biol. Chem. 2011, 2, 119–125. [Google Scholar] [CrossRef]
- de Rooij, J.D.; Beuling, E.; van den Heuvel-Eibrink, M.M.; Obulkasim, A.; Baruchel, A.; Trka, J.; Reinhardt, D.; Sonneveld, E.; Gibson, B.E.; Pieters, R.; et al. Recurrent deletions of IKZF1 in pediatric acute myeloid leukemia. Haematologica 2015, 100, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Payne, K.J.; Huang, G.; Sahakian, E.; Zhu, J.Y.; Barteneva, N.S.; Barsky, L.W.; Payne, M.A.; Crooks, G.M. Ikaros isoform x is selectively expressed in myeloid differentiation. J. Immunol. 2003, 170, 3091–3098. [Google Scholar] [CrossRef]
- Jäger, R.; Gisslinger, H.; Passamonti, F.; Rumi, E.; Berg, T.; Gisslinger, B.; Pietra, D.; Harutyunyan, A.; Klampfl, T.; Olcaydu, D.; et al. Deletions of the transcription factor Ikaros in myeloproliferative neoplasms. Leukemia 2010, 24, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.W.; Min, Y.H.; Eom, J.I.; Kim, S.J.; Jeung, H.K.; Kim, J.S. Inhibition of CK2{alpha} and PI3K/Akt synergistically induces apoptosis of CD34+CD38- leukaemia cells while sparing haematopoietic stem cells. Anticancer Res. 2010, 30, 4625–4634. [Google Scholar]
- Gurel, Z.; Ronni, T.; Ho, S.; Kuchar, J.; Payne, K.J.; Turk, C.W.; Dovat, S. Recruitment of ikaros to pericentromeric heterochromatin is regulated by phosphorylation. J. Biol. Chem. 2008, 283, 8291–8300. [Google Scholar] [CrossRef] [Green Version]
- Popescu, M.; Gurel, Z.; Ronni, T.; Song, C.; Hung, K.Y.; Payne, K.J.; Dovat, S. Ikaros stability and pericentromeric localization are regulated by protein phosphatase 1. J. Biol. Chem. 2009, 284, 13869–13880. [Google Scholar] [CrossRef] [Green Version]
- Lertsuwan, J.; Lertsuwan, K.; Sawasdichai, A.; Tasnawijitwong, N.; Lee, K.Y.; Kitchen, P.; Afford, S.; Gaston, K.; Jayaraman, P.S.; Satayavivad, J. CX-4945 Induces Methuosis in Cholangiocarcinoma Cell Lines by a CK2-Independent Mechanism. Cancers 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Oshima, T.; Niwa, Y.; Kuwata, K.; Srivastava, A.; Hyoda, T.; Tsuchiya, Y.; Kumagai, M.; Tsuyuguchi, M.; Tamaru, T.; Sugiyama, A.; et al. Cell-based screen identifies a new potent and highly selective CK2 inhibitor for modulation of circadian rhythms and cancer cell growth. Sci. Adv. 2019, 5, eaau9060. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Park, K.H.; Kim, H.M.; Kim, T.S.; Zhang, X.; Park, S.M.; Beom, S.H.; Kim, H.S.; Cheong, J.H.; Chung, H.C.; et al. Inhibiting casein kinase 2 overcomes paclitaxel resistance in gastric cancer. Gastric. Cancer 2019, 22, 1153–1163. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, J.; Miyoshi, C.; Li, Y.; Sato, M.; Ogawa, Y.; Lou, T.; Ma, C.; Gao, X.; Lee, C.; et al. Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature 2018, 558, 435–439. [Google Scholar] [CrossRef]
- von Morgen, P.; Burdova, K.; Flower, T.G.; O’Reilly, N.J.; Boulton, S.J.; Smerdon, S.J.; Macurek, L.; Hořejší, Z. MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene 2017, 36, 4943–4950. [Google Scholar] [CrossRef] [Green Version]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Di Maira, G.; Tosoni, K.; Pinna, L.A. Assessment of CK2 constitutive activity in cancer cells. Methods Enzymol. 2010, 484, 495–514. [Google Scholar] [CrossRef]
- Lou, D.Y.; Dominguez, I.; Toselli, P.; Landesman-Bollag, E.; O’Brien, C.; Seldin, D.C. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell. Biol. 2008, 28, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Paganelli, F.; Fazio, A.; Bazzichetto, C.; Conciatori, F.; McCubrey, J.A. The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers 2019, 11, 629. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Feng, C.; He, Y.; Ding, W.; Sheng, J.; Arshad, M.; Zhang, X.; Li, P. Phosphorylation of apoptosis repressor with caspase recruitment domain by protein kinase CK2 contributes to chemotherapy resistance by inhibiting doxorubicin induced apoptosis. Oncotarget 2015, 6, 27700–27713. [Google Scholar] [CrossRef] [Green Version]
- Borgo, C.; Ruzzene, M. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019, 38, 287. [Google Scholar] [CrossRef]
- Trembley, J.H.; Chen, Z.; Unger, G.; Slaton, J.; Kren, B.T.; Van Waes, C.; Ahmed, K. Emergence of protein kinase CK2 as a key target in cancer therapy. Biofactors 2010, 36, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, L.R.; Lúcio, P.; Silva, M.C.; Anderes, K.L.; Gameiro, P.; Silva, M.G.; Barata, J.T. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood 2010, 116, 2724–2731. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Melchionda, F.; Pession, A.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, L.; Volk, A.G.; Birch, N.W.; Stoltz, K.L.; Bartom, E.T.; Marshall, S.A.; Rendleman, E.J.; Nestler, C.M.; Shilati, J.; et al. Regulation of MLL/COMPASS stability through its proteolytic cleavage by taspase1 as a possible approach for clinical therapy of leukemia. Genes Dev. 2019, 33, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.; Downing, J. Ikaros and acute leukemia. Leuk. Lymphoma 2008, 49, 847–849. [Google Scholar] [CrossRef]
- Minn, A.J.; Rudin, C.M.; Boise, L.H.; Thompson, C.B. Expression of bcl-xL can confer a multidrug resistance phenotype. Blood 1995, 86, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M.; Hamali, H.A.; Sun, X.M.; Bampton, E.T.; Dinsdale, D.; Snowden, R.T.; Dyer, M.J.; Goodall, A.H.; Cohen, G.M. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 2011, 117, 7145–7154. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klink, M.; Rahman, M.A.; Song, C.; Dhanyamraju, P.K.; Ehudin, M.; Ding, Y.; Steffens, S.; Bhadauria, P.; Iyer, S.; Aliaga, C.; et al. Mechanistic Basis for In Vivo Therapeutic Efficacy of CK2 Inhibitor CX-4945 in Acute Myeloid Leukemia. Cancers 2021, 13, 1127. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13051127
Klink M, Rahman MA, Song C, Dhanyamraju PK, Ehudin M, Ding Y, Steffens S, Bhadauria P, Iyer S, Aliaga C, et al. Mechanistic Basis for In Vivo Therapeutic Efficacy of CK2 Inhibitor CX-4945 in Acute Myeloid Leukemia. Cancers. 2021; 13(5):1127. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13051127
Chicago/Turabian StyleKlink, Morgann, Mohammad Atiqur Rahman, Chunhua Song, Pavan Kumar Dhanyamraju, Melanie Ehudin, Yali Ding, Sadie Steffens, Preeti Bhadauria, Soumya Iyer, Cesar Aliaga, and et al. 2021. "Mechanistic Basis for In Vivo Therapeutic Efficacy of CK2 Inhibitor CX-4945 in Acute Myeloid Leukemia" Cancers 13, no. 5: 1127. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13051127