
Preclinical Head and Neck Squamous Cell Carcinoma Models for Combined Targeted Therapy Approaches

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Sample Preparation
2.2. Ethical Statement
2.2.1. In Vivo Study
2.2.2. Patient-Derived Xenograft (PDX) Generation
2.2.3. Experimental Protocol
2.3. Collagenase Digestion for Ex Vivo Primary Cell Cultures
2.4. Spheroid Formation
2.5. Mycoplasma and Fibroblast Contamination Test
2.6. Histology and Immunohistochemistry
2.7. Tumor Microenvironment
2.8. Molecular Pathology
2.9. In Vitro Growth Kinetics
2.10. Phenotyping
2.11. Radiation Response
2.12. Migration and Invasiveness
2.13. In Vitro Drug Response
2.14. Cell Death and Cell Cycle
2.15. Assessment of Viability, Acidic Compartments, Cytoskeleton and ROS
2.16. RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR
2.17. Allogenic Co-Culture
2.18. Statistics
3. Results
3.1. Clinicopathological Patients’ Data, and Cell Line Establishment
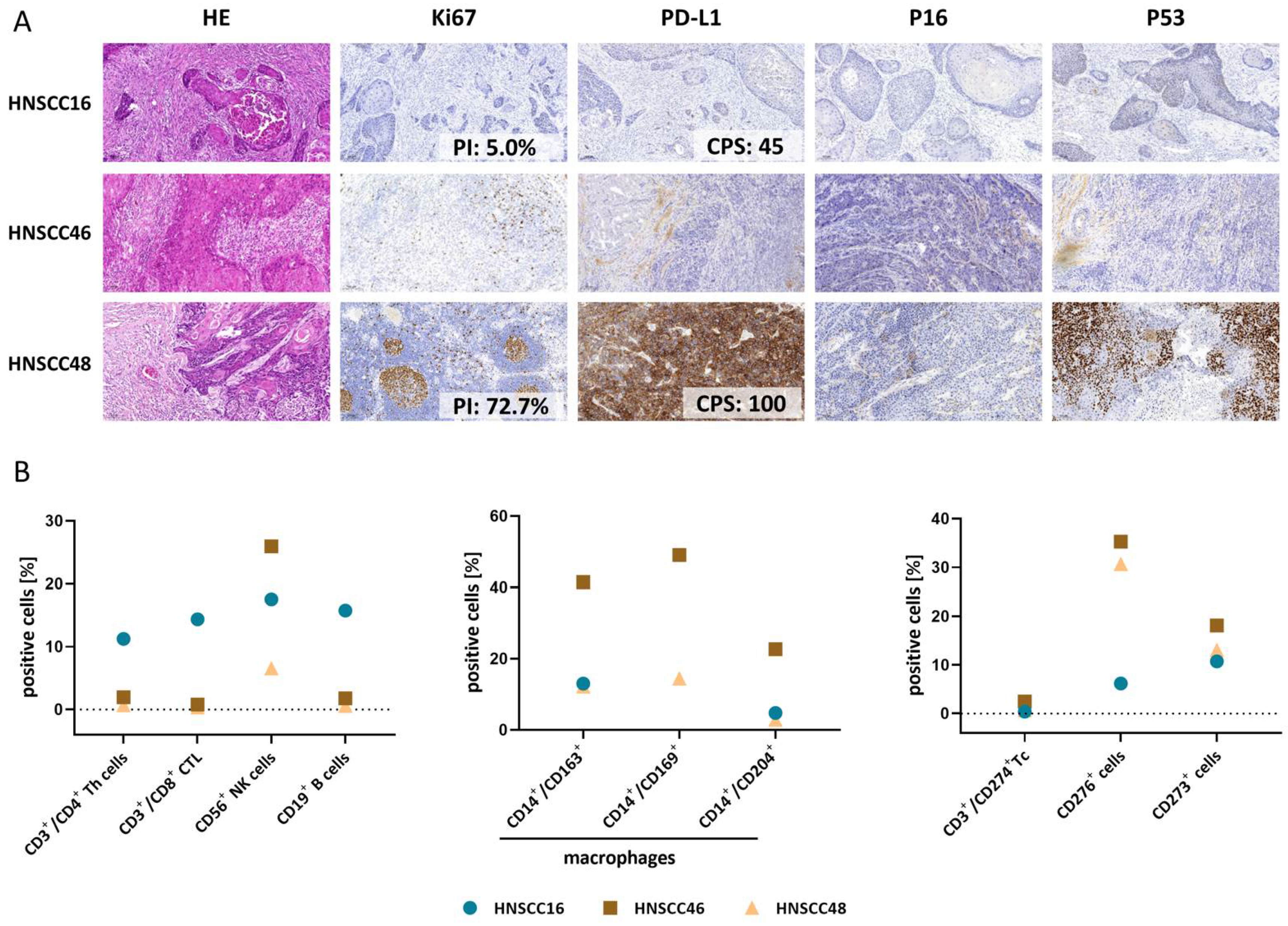
3.2. Histopathology and Tumor Microenvironment of Primary Tumors
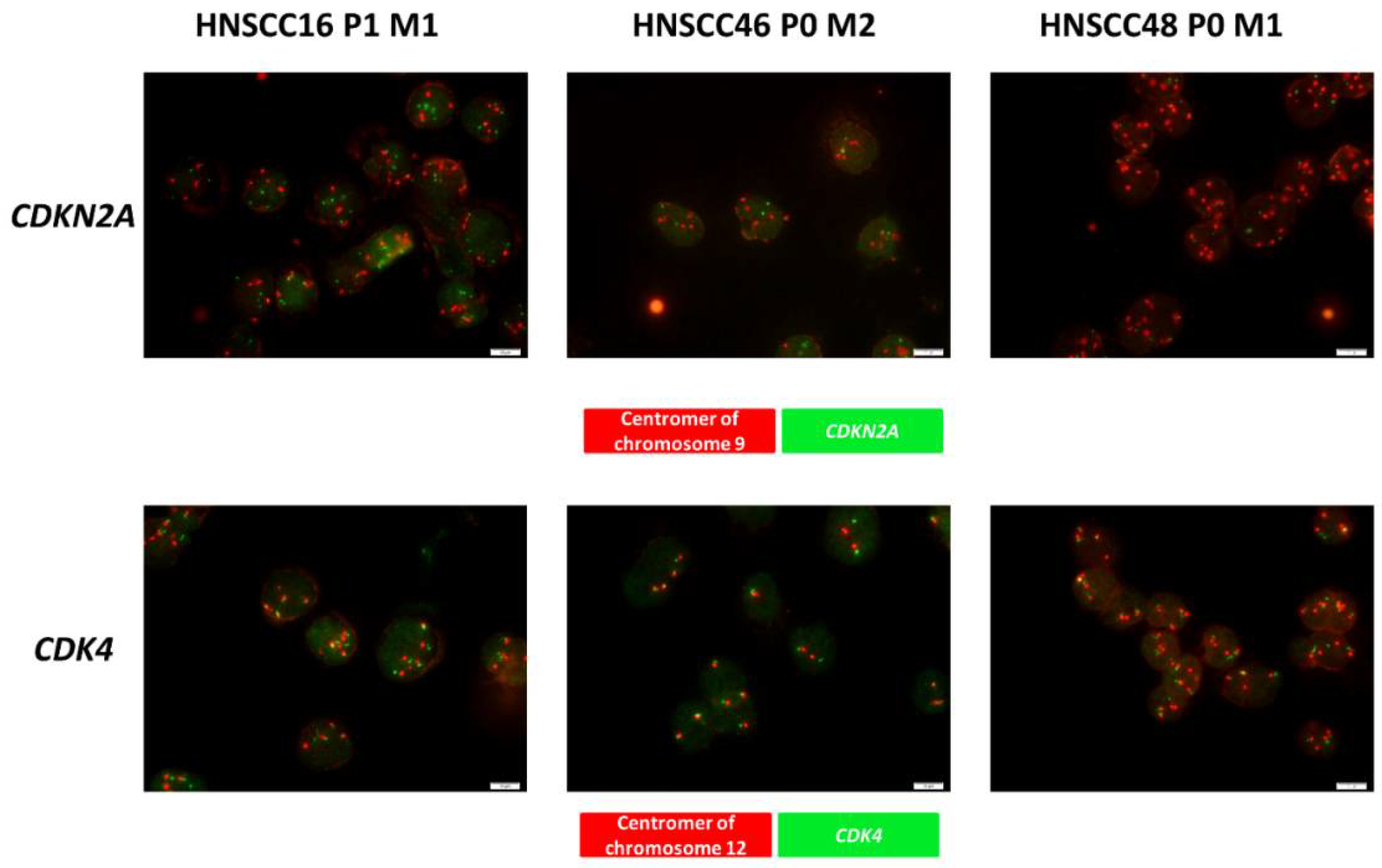
3.3. Quality Control, Comparative Analysis of the Molecular Profile & Detection of Gene Specific Alteration of CDK4 and CDKN2A
3.4. Cell Morphology and Spheroid Formation
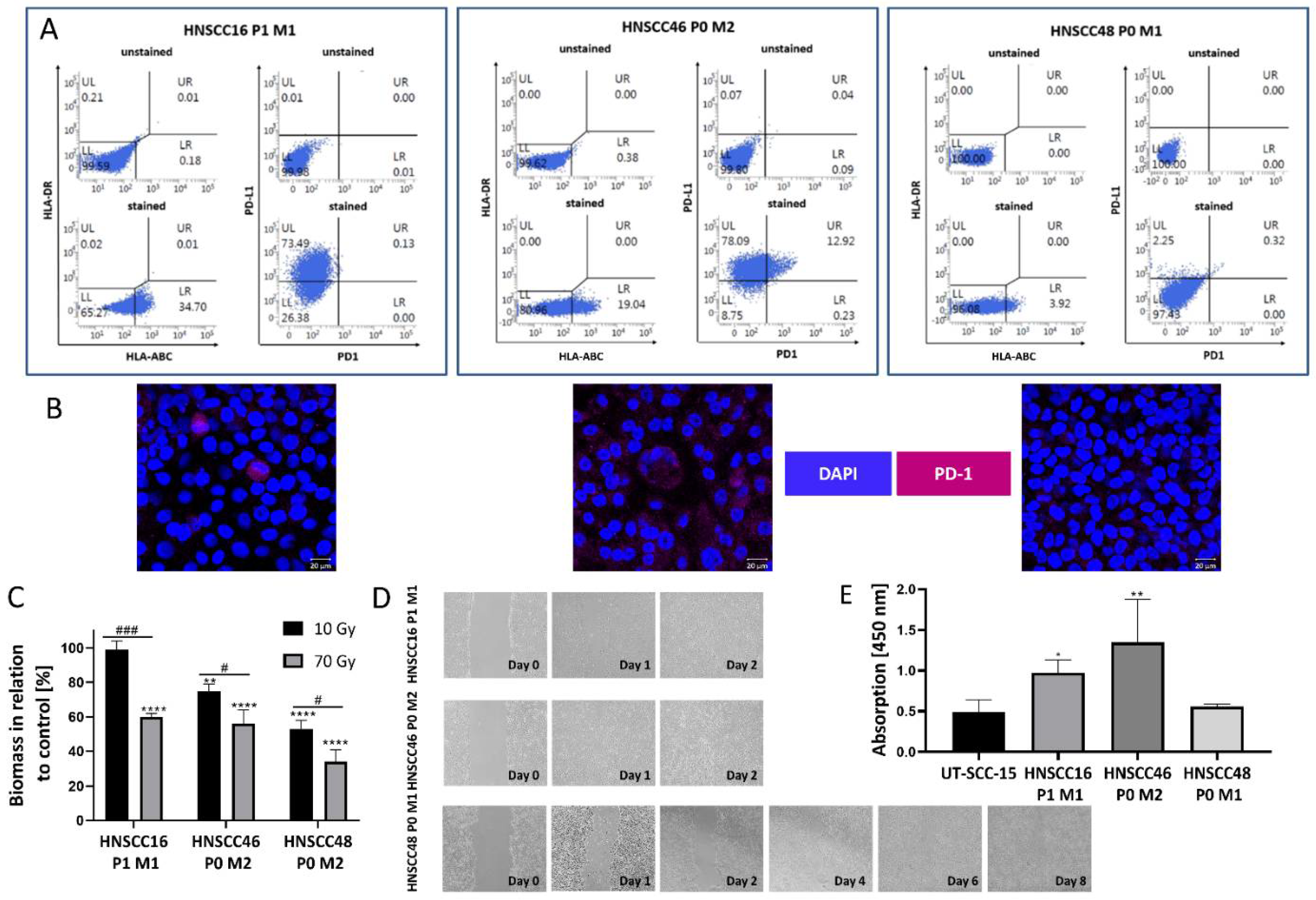
3.5. Cell Line Characterization
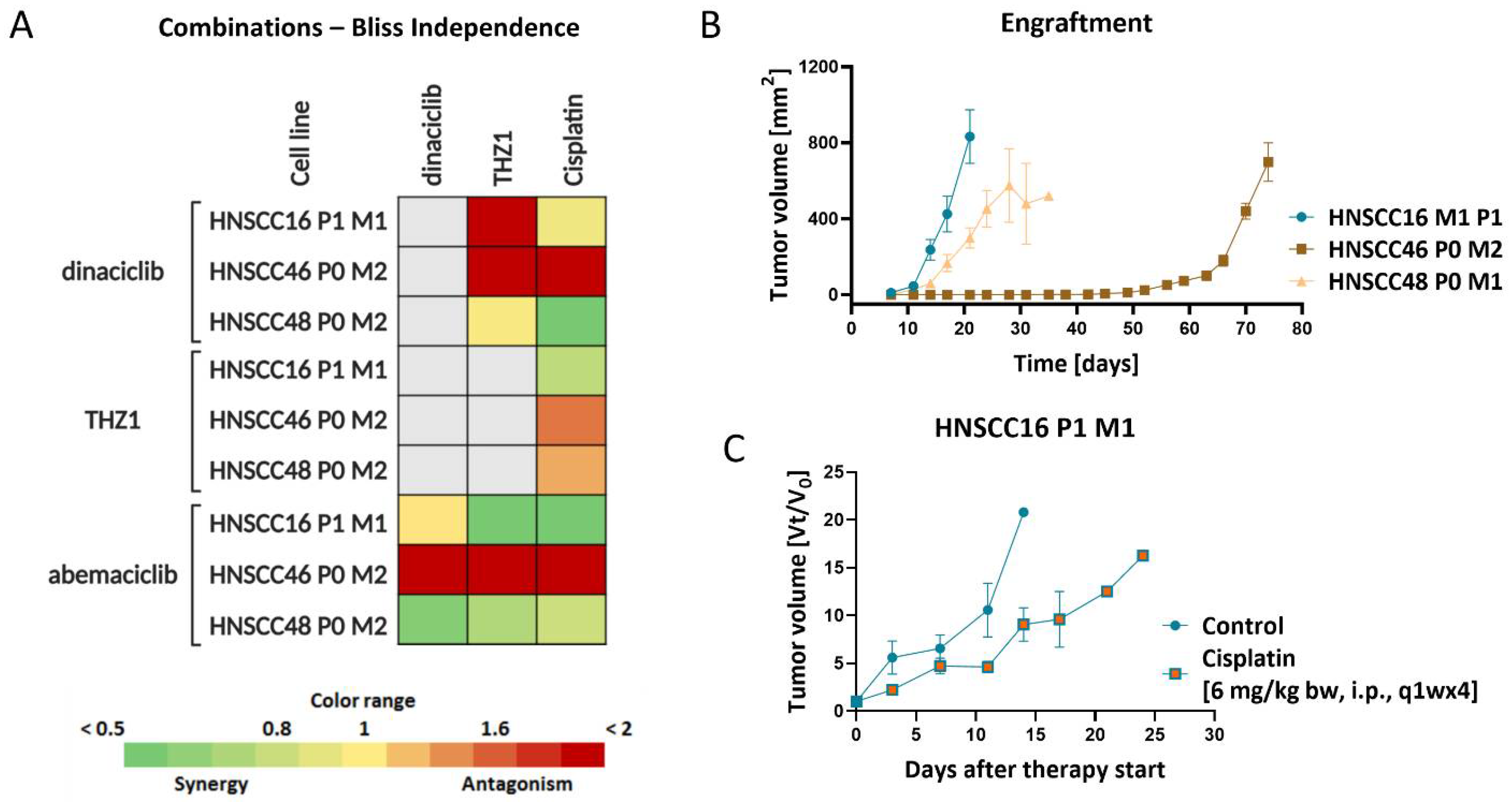
3.6. In Vitro and In Vivo Drug Response
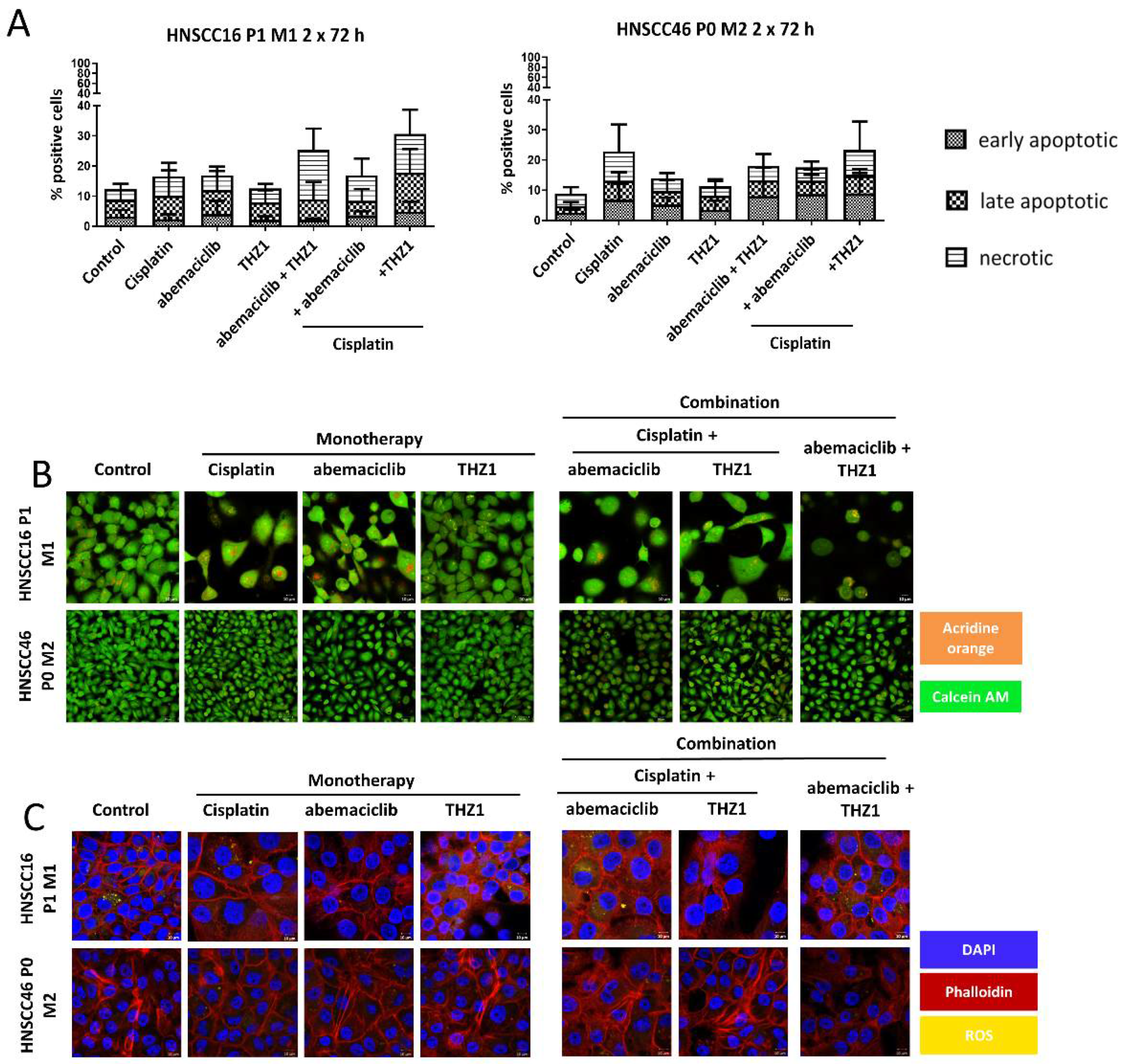
3.7. Molecular Alterations Predict CDKI Resistance, Which Is Partially Reversible by Combined Targeted Therapy In Vitro
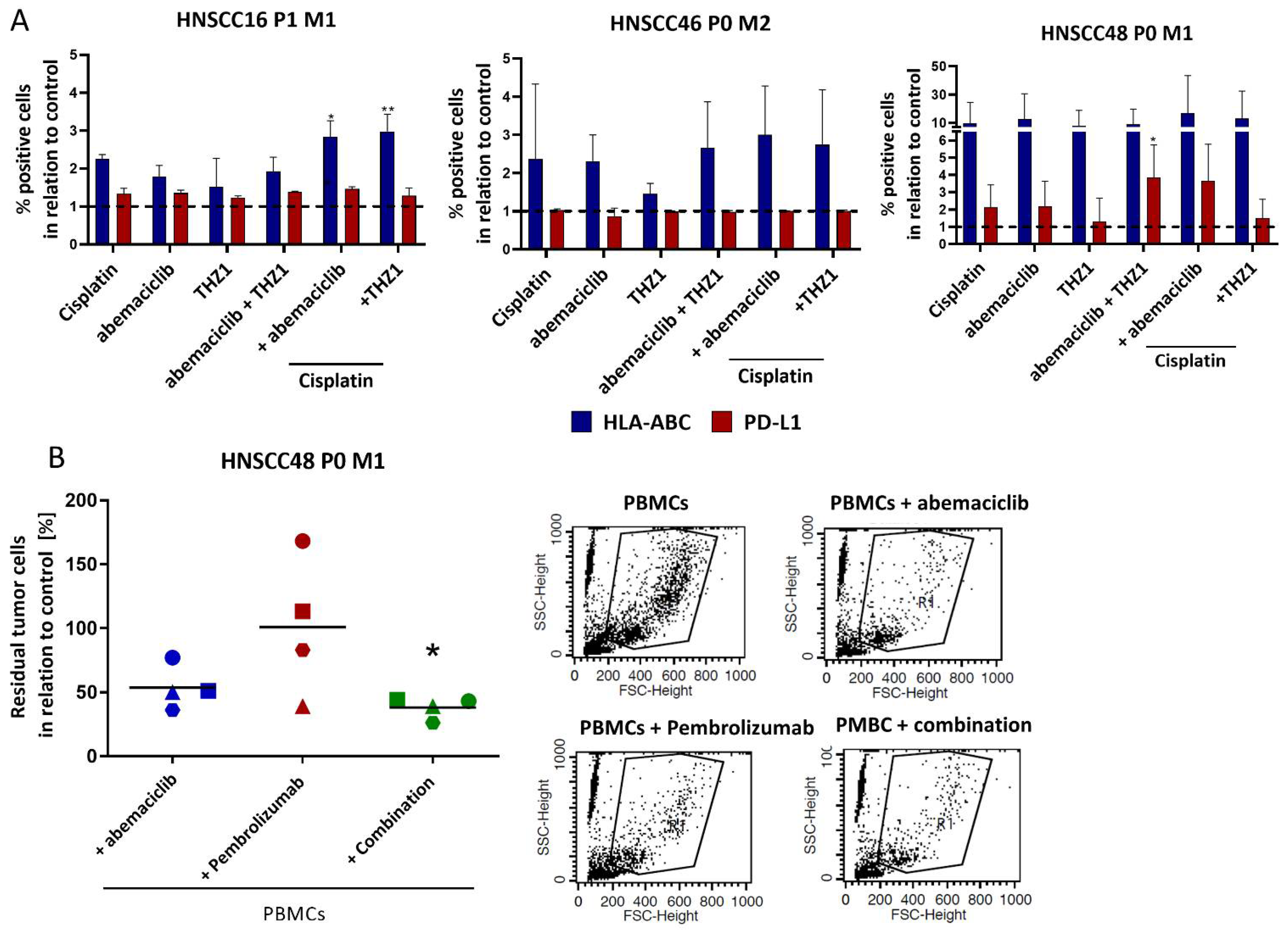
3.8. Enhanced Immunogenicity and Confirmation of Immune-Stimulatory Potential upon Co-Culture with Abemaciclib and Pembrolizumab
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lum, D.H.; Matsen, C.; Welm, A.L.; Welm, B.E. Overview of Human Primary Tumorgraft Models: Comparisons with Traditional Oncology Preclinical Models and the Clinical Relevance and Utility of Primary Tumorgrafts in Basic and Translational Oncology Research. Curr. Protoc. Pharmacol. 2012, 59, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures—Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef] [PubMed]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, V.; Chirovsky, D.; Cheung, W.Y.; Bertolini, F.; Ahn, M.-J.; Yang, M.-H.; Castro, G.; Berrocal, A.; Sjoquist, K.; Kuyas, H.; et al. Global treatment patterns and outcomes among patients with recurrent and/or metastatic head and neck squamous cell carcinoma: Results of the GLANCE H&N study. Oral Oncol. 2020, 102, 104526. [Google Scholar] [CrossRef]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Morris, L.G.T.; Chandramohan, R.; West, L.; Zehir, A.; Chakravarty, D.; Pfister, D.G.; Wong, R.J.; Lee, N.Y.; Sherman, E.J.; Baxi, S.S.; et al. The Molecular Landscape of Recurrent and Metastatic Head and Neck Cancers: Insights from a precision oncology sequencing platform. JAMA Oncol. 2017, 3, 244–255. [Google Scholar] [CrossRef]
- Niehr, F.; Eder, T.; Pilz, T.; Konschak, R.; Treue, D.; Klauschen, F.; Bockmayr, M.; Türkmen, S.; Jöhrens, K.; Budach, V.; et al. Multilayered Omics-Based Analysis of a Head and Neck Cancer Model of Cisplatin Resistance Reveals Intratumoral Heterogeneity and Treatment-Induced Clonal Selection. Clin. Cancer Res. 2017, 24, 158–168. [Google Scholar] [CrossRef] [Green Version]
- Tinhofer, I.; Braunholz, D.; Klinghammer, K. Preclinical models of head and neck squamous cell carcinoma for a basic understanding of cancer biology and its translation into efficient therapies. Cancers Head Neck 2020, 5, 9. [Google Scholar] [CrossRef]
- Ferris, R.; Gonçalves, A.; Baxi, S.; Martens, U.; Gauthier, H.; Langenberg, M.; Spanos, W.; Leidner, R.; Kang, H.; Russell, J.; et al. An open-label, multicohort, phase 1/2 study in patients with virus-associated cancers (CheckMate 358): Safety and efficacy of neoadjuvant nivolumab in squamous cell carcinoma of the head and neck (SCCHN). Ann. Oncol. 2017, 28, v628–v629. [Google Scholar] [CrossRef] [Green Version]
- Wise-Draper, T.M.; Old, M.O.; Worden, F.P.; O’Brien, P.E.; Cohen, E.E.; Dunlap, N.; Mierzwa, M.L.; Casper, K.; Palackdharry, S.; Hinrichs, B.; et al. Phase II multi-site investigation of neoadjuvant pembrolizumab and adjuvant concurrent radiation and pembrolizumab with or without cisplatin in resected head and neck squamous cell carcinoma. J. Clin. Oncol. 2018, 36, 6017. [Google Scholar] [CrossRef]
- Reinius, M.A.V.; Smyth, E. Anti-cancer therapy with cyclin-dependent kinase inhibitors: Impact and challenges. Expert Rev. Mol. Med. 2021, 23, e6. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef] [PubMed]
- Poratti, M.; Marzaro, G. Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Strüder, D.; Momper, T.; Irmscher, N.; Krause, M.; Liese, J.; Schraven, S.; Zimpfer, A.; Zonnur, S.; Burmeister, A.-S.; Schneider, B.; et al. Establishment and characterization of patient-derived head and neck cancer models from surgical specimens and endoscopic biopsies. J. Exp. Clin. Cancer Res. 2021, 40, 246. [Google Scholar] [CrossRef] [PubMed]
- Oppel, F.; Shao, S.; Schürmann, M.; Goon, P.; Albers, A.E.; Sudhoff, H. An Effective Primary Head and Neck Squamous Cell Carcinoma In Vitro Model. Cells 2019, 8, 555. [Google Scholar] [CrossRef] [Green Version]
- Maletzki, C.; Gock, M.; Randow, M.; Klar, E.; Huehns, M.; Prall, F.; Linnebacher, M. Establishment and characterization of cell lines from chromosomal instable colorectal cancer. World J. Gastroenterol. 2015, 21, 164–176. [Google Scholar] [CrossRef]
- Schoenwaelder, N.; Salewski, I.; Engel, N.; Krause, M.; Schneider, B.; Müller, M.; Riess, C.; Lemcke, H.; Skorska, A.; Grosse-Thie, C.; et al. The Individual Effects of Cyclin-Dependent Kinase Inhibitors on Head and Neck Cancer Cells—A Systematic Analysis. Cancers 2021, 13, 2396. [Google Scholar] [CrossRef]
- Maletzki, C.; Rosche, Y.; Riess, C.; Scholz, A.; William, D.; Classen, C.F.; Kreikemeyer, B.; Linnebacher, M.; Fiedler, T. Deciphering molecular mechanisms of arginine deiminase-based therapy—Comparative response analysis in paired human primary and recurrent glioblastomas. Chem. Interact. 2017, 278, 179–188. [Google Scholar] [CrossRef]
- Riess, C.; Schneider, B.; Kehnscherper, H.; Gesche, J.; Irmscher, N.; Shokraie, F.; Classen, C.F.; Wirthgen, E.; Domanska, G.; Zimpfer, A.; et al. Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 2020, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Welkoborsky, H.-J.; Jacob, R.; Riazimand, S.H.; Bernauer, H.S.; Mann, W.J. Molecular Biologic Characteristics of Seven New Cell Lines of Squamous Cell Carcinomas of the Head and Neck and Comparison to Fresh Tumor Tissue. Oncology 2003, 65, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farah, C.S. Molecular landscape of head and neck cancer and implications for therapy. Ann. Transl. Med. 2021, 9, 915. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Charuruks, N.; Lippman, S.M.; Lee, J.J.; Ro, J.Y.; Hong, W.K.; Hittelman, W.N. p53 protein accumulation and genomic instability in head and neck multistep tumorigenesis. Cancer Epidemiol. Biomark. Prev. 2001, 10, 603–609. [Google Scholar]
- Bs, M.Y.L.; Allen, C.T. Mechanisms of resistance to T cell-based immunotherapy in head and neck cancer. Head Neck 2020, 42, 2722–2733. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef]
- Van Gestel, D.; Weyngaert, D.V.D.; Schrijvers, D.; Weyler, J.; Vermorken, J.B. Intensity-modulated radiotherapy in patients with head and neck cancer: A European single-centre experience. Br. J. Radiol. 2011, 84, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Hino, H.; Iriyama, N.; Kokuba, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Aizawa, S.; Miyazawa, K. Abemaciclib induces atypical cell death in cancer cells characterized by formation of cytoplasmic vacuoles derived from lysosomes. Cancer Sci. 2020, 111, 2132–2145. [Google Scholar] [CrossRef]
- Riess, C.; Koczan, D.; Schneider, B.; Linke, C.; del Moral, K.; Classen, C.F.; Maletzki, C. Cyclin-dependent kinase inhibitors exert distinct effects on patient-derived 2D and 3D glioblastoma cell culture models. Cell Death Discov. 2021, 7, 54. [Google Scholar] [CrossRef]
- Billard-Sandu, C.; Tao, Y.G.; Sablin, M.P.; Dumitrescu, G.; Billard, D.; Deutsch, E. CDK4/6 Inhibitors in P16/HPV16-Negative Squamous Cell Carcinoma of the Head and Neck. Eur. Arch. Oto-Rhino Laryngol. 2020, 277, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Karamboulas, C.; Bruce, J.P.; Hope, A.J.; Meens, J.; Huang, S.H.; Erdmann, N.; Hyatt, E.; Pereira, K.; Goldstein, D.P.; Weinreb, I.; et al. Patient-Derived Xenografts for Prognostication and Personalized Treatment for Head and Neck Squamous Cell Carcinoma. Cell Rep. 2018, 25, 1318–1331.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, S.P.; Generali, D. Abemaciclib: A CDK4/6 inhibitor for the treatment of HR+/HER2– advanced breast cancer. Drug Des. Dev. Ther. 2018, 12, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, K.E.; Wu, F.; Keysar, S.B.; Morton, J.J.; Miller, B.; Chimed, T.-S.; Le, P.N.; Nieto, C.; Chowdhury, F.N.; Tyagi, A.; et al. Cancer cell CD44 mediates macrophage/monocyte-driven regulation of head and neck cancer stem cells. Cancer Res. 2020, 80, 4185–4198. [Google Scholar] [CrossRef] [PubMed]
- Alexa-Stratulat, T.; Pešić, M.; Gašparović, A.Č.; Trougakos, I.P.; Riganti, C. What sustains the multidrug resistance phenotype beyond ABC efflux transporters? Looking beyond the tip of the iceberg. Drug Resist. Updates 2019, 46, 100643. [Google Scholar] [CrossRef] [PubMed]
- Abutorabi, E.S.; Irani, S.; Yaghmaie, M.; Ghaffari, S.H. Abemaciclib (CDK4/6 Inhibitor) Blockade Induces Cytotoxicity in Human Anaplastic Thyroid Carcinoma Cells. Rep. Biochem. Mol. Biol. 2020, 8, 438. [Google Scholar]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. OncoImmunology 2020, 9, 1777625. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Patnaik, A.; Yap, T.A.; Chung, H.C.; de Miguel, M.J.; Bang, Y.-J.; Lin, C.-C.; Su, W.-C.; Italiano, A.; Chow, K.H.; Szpurka, A.M.; et al. Safety and Clinical Activity of a New Anti-PD-L1 Antibody as Monotherapy or Combined with Targeted Therapy in Advanced Solid Tumors: The PACT Phase Ia/Ib Trial. Clin. Cancer Res. 2020, 27, 1267–1277. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Age Sex | TNM Grade | Localization | Origin | p16/HPV | Noxes | Treatment | Clinical Follow-Up |
|---|---|---|---|---|---|---|---|---|
| HNSCC16 P1 M1 | 82 m | rpT4a pN0 cM0 G2 | Larynx | Recurrence | -/- | non-smoker | surgery 2008, radiation 2015, surgery 2020 | alive |
| HNSCC46 P0 M2 | 69 m | pT3 pN3b cM0 G2 | Hypopharynx | primary tumor | +/- | nicotine, C2 | surgery (later adjuvant RCT) | † |
| HNSCC48 P0 M1 | 63 m | pT3 pN3b cM0 G2 | Lymph node (primary tumor: oral cavity (floor of moth)) | metastasis/ recurrence | -/- | non-smoker | RCT (64 Gy, Cisplatin), surgery (later Nivolumab) | † |
| Mutation | HNSCC16 | HNSCC46 | HNSCC48 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient | PDX | Cell Line | Patient | PDX | Cell Line | Patient | PDX | Cell Line | |
| TP53 | p.R306 * VAF 37% p.P72R VAF 32.7% | p.R306 * VAF 100% | p.R306 * VAF 99.5% | p.E294Sfs *51 VAF 64.5% p.P72R VAF 100% | p.E294Sfs *51 VAF 99.3% p.P72R VAF 99.3% | p.E294Sfs *51 VAF 99.9% p.P72R VAF 99.6% | p.R175H VAF 99.4% p.P72R VAF 99.5% | p.R175H VAF 60.5% p.P72R VAF 80.4% | p.R175H VAF 99.8% p.P72R VAF 99.9% |
| CDKN2A | wt | wt | wt | wt | wt | wt | p.R58 * VAF 99.6% | p.R58 * VAF 55.8% | p.R58 * VAF 99.8% |
| SMAD4 | wt | wt | wt | wt | wt | wt | p.R135 * VAF 99.6% | p.R135 * VAF 48.2% | p.R135 * VAF 100% |
| Substance | HNSCC16 P1 M1 | HNSCC46 P0 M2 | HNSCC48 P0 M1 |
|---|---|---|---|
| Cisplatin [µg/mL] | 1.43 | 1.54 | 0.30 |
| 5-FU [µg/mL] | 0.31 | 0.17 | 0.04 |
| Cetuximab [µg/mL] | not reached | not reached | 137.26 |
| dinaciclib [nM] | 2.69 | 1.65 | 7.33 |
| THZ1 [nM] | 49.22 | 39.47 | 32.00 |
| abemaciclib [nM] | not reached | not reached | 696.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoenwaelder, N.; Krause, M.; Freitag, T.; Schneider, B.; Zonnur, S.; Zimpfer, A.; Becker, A.S.; Salewski, I.; Strüder, D.F.; Lemcke, H.; et al. Preclinical Head and Neck Squamous Cell Carcinoma Models for Combined Targeted Therapy Approaches. Cancers 2022, 14, 2484. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102484
Schoenwaelder N, Krause M, Freitag T, Schneider B, Zonnur S, Zimpfer A, Becker AS, Salewski I, Strüder DF, Lemcke H, et al. Preclinical Head and Neck Squamous Cell Carcinoma Models for Combined Targeted Therapy Approaches. Cancers. 2022; 14(10):2484. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102484
Chicago/Turabian StyleSchoenwaelder, Nina, Mareike Krause, Thomas Freitag, Björn Schneider, Sarah Zonnur, Annette Zimpfer, Anne Sophie Becker, Inken Salewski, Daniel Fabian Strüder, Heiko Lemcke, and et al. 2022. "Preclinical Head and Neck Squamous Cell Carcinoma Models for Combined Targeted Therapy Approaches" Cancers 14, no. 10: 2484. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102484