
Single-Cell Analyses of a Novel Mouse Urothelial Carcinoma Model Reveal a Role of Tumor-Associated Macrophages in Response to Anti-PD-1 Therapy

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
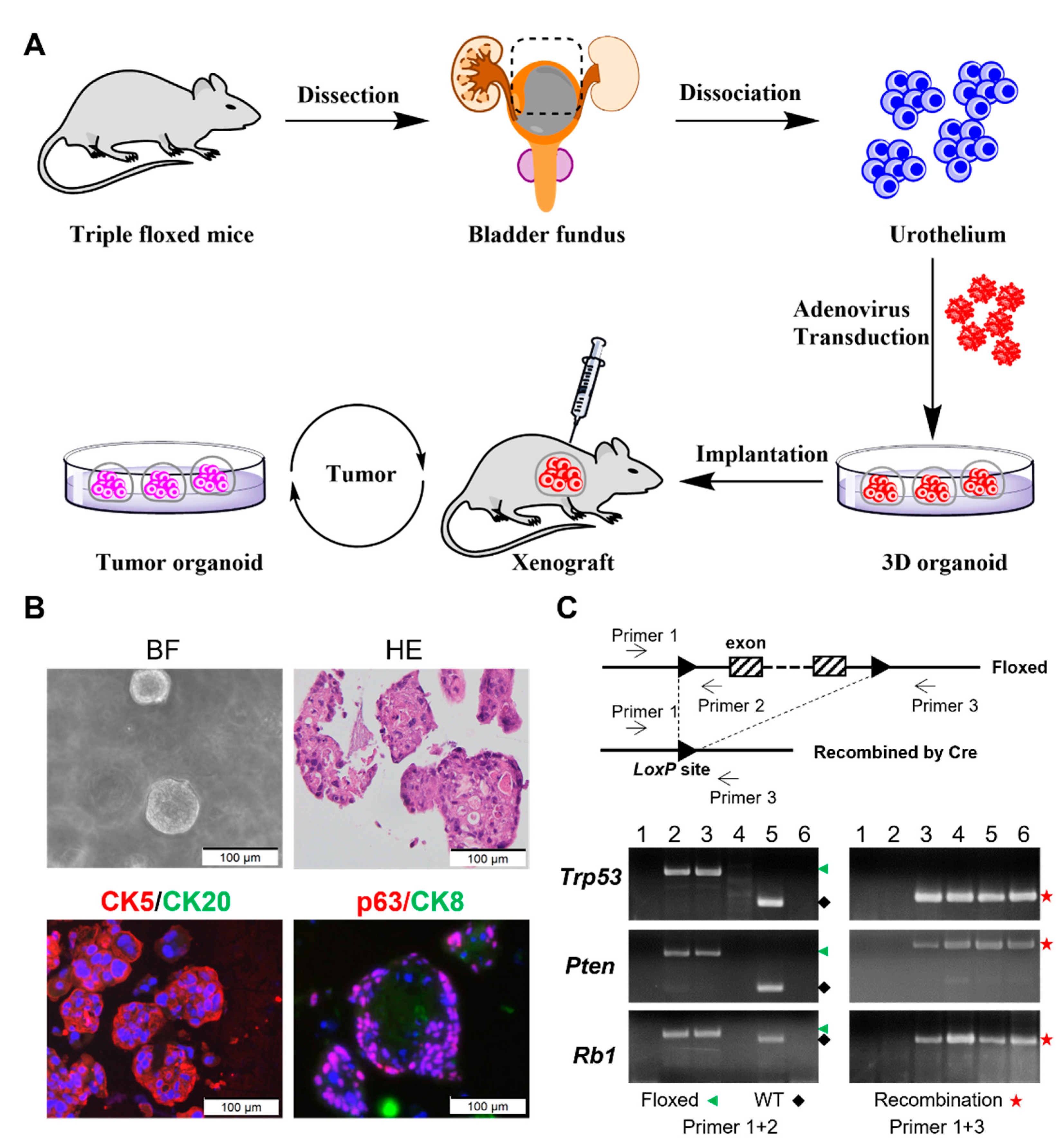
2.1. Isolation of Primary Urothelial Cells
2.2. Adenoviral Transduction and Organoid Culturing
2.3. PCR Validation
2.4. Histology, Immunohistochemistry, and Immunofluorescence
2.5. Transcriptional Profiling of Urothelial Tumors
2.6. Intravesical and Orthotopic Injections and Drug Response
2.7. Single-Cell Sequencing
2.8. Statistical Analysis
3. Results
3.1. Ex Vivo Inactivation of Trp53/Rb1/Pten Results in High-Grade Urothelial Carcinoma Organoids
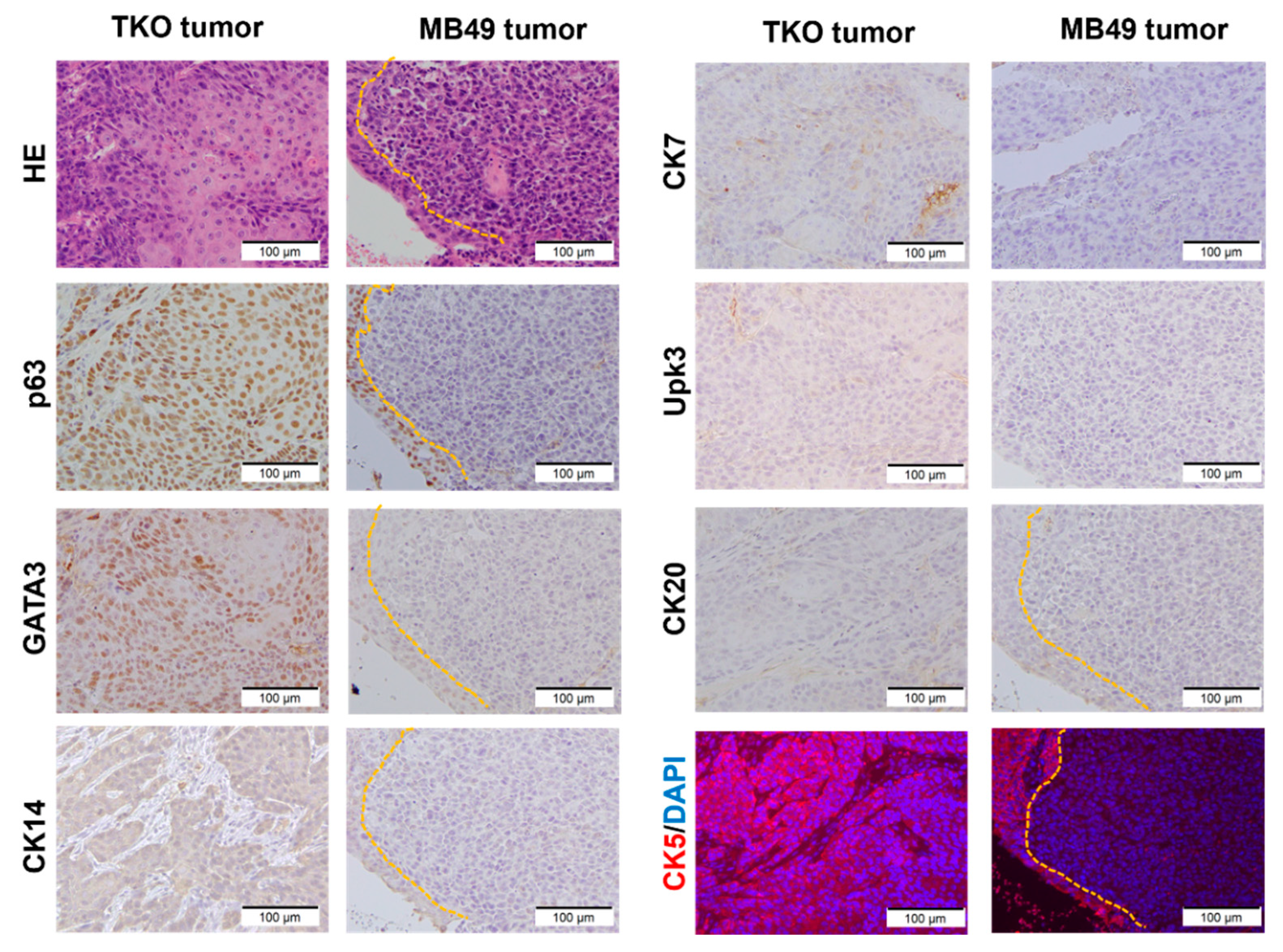
3.2. TKO Is a Basal-like Bladder Cancer Model
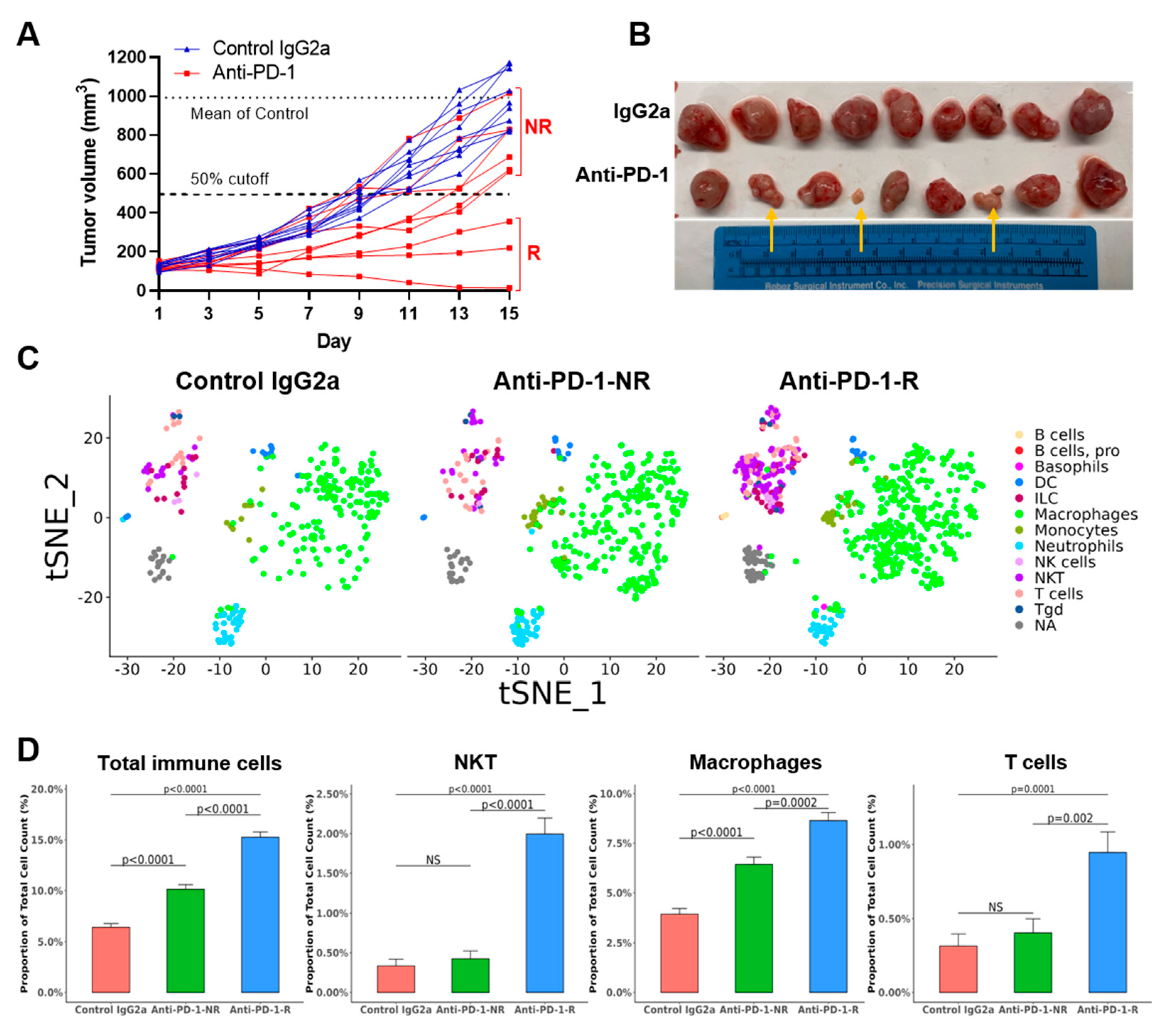
3.3. TKO Tumors Demonstrate Heterogeneous Responses to Anti-PD-1 Treatment
3.4. Anti-PD-1 Treatment Responders Exhibit Increased Immune Cell Infiltration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Heney, N.M. Natural history of superficial bladder cancer. Prognostic features and long-term disease course. Urol. Clin. N. Am. 1992, 19, 429–433. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cimadamore, A.; Blanca, A.; Massari, F.; Vau, N.; Scarpelli, M.; Cheng, L.; Montironi, R. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers 2021, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Rhea, L.P.; Aragon-Ching, J.B. Advances and Controversies with Checkpoint Inhibitors in Bladder Cancer. Clin. Med. Insights Oncol. 2021, 15, 11795549211044963. [Google Scholar] [CrossRef]
- Andreev-Drakhlin, A.Y.; Egoryan, G.; Shah, A.Y.; Msaouel, P.; Alhalabi, O.; Gao, J. The evolving treatment landscape of advanced urothelial carcinoma. Curr. Opin. Oncol. 2021, 33, 221–230. [Google Scholar] [CrossRef]
- Kobayashi, T.; Owczarek, T.B.; McKiernan, J.M.; Abate-Shen, C. Modelling bladder cancer in mice: Opportunities and challenges. Nat. Rev. Cancer 2015, 15, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Fantini, D.; Glaser, A.P.; Rimar, K.J.; Wang, Y.; Schipma, M.; Varghese, N.; Rademaker, A.; Behdad, A.; Yellapa, A.; Yu, Y.; et al. A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene 2018, 37, 1911–1925. [Google Scholar] [CrossRef]
- Van Batavia, J.; Yamany, T.; Molotkov, A.; Dan, H.; Mansukhani, M.; Batourina, E.; Schneider, K.; Oyon, D.; Dunlop, M.; Wu, X.R.; et al. Bladder cancers arise from distinct urothelial sub-populations. Nat. Cell Biol. 2014, 16, 982–991. [Google Scholar] [CrossRef]
- Summerhayes, I.C.; Franks, L.M. Effects of donor age on neoplastic transformation of adult mouse bladder epithelium in vitro. J. Natl. Cancer Inst. 1979, 62, 1017–1023. [Google Scholar]
- Horinaga, M.; Fukuyama, R.; Nishiyama, T.; Harsch, K.M.; Cicek, M.; Heston, W.; Sizemore, N.; Casey, G.; Larchian, W. Novel enhanced lung-colonizing variant of murine MBT-2 bladder cancer cells. Urology 2005, 66, 676–681. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillere, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smith, C.C.; Utsumi, T.; Bixby, L.M.; Kardos, J.; Wobker, S.E.; Stewart, K.G.; Chai, S.; Manocha, U.; Byrd, K.M.; et al. Molecular Subtype-Specific Immunocompetent Models of High-Grade Urothelial Carcinoma Reveal Differential Neoantigen Expression and Response to Immunotherapy. Cancer Res. 2018, 78, 3954–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullenders, J.; de Jongh, E.; Brousali, A.; Roosen, M.; Blom, J.P.A.; Begthel, H.; Korving, J.; Jonges, T.; Kranenburg, O.; Meijer, R.; et al. Mouse and human urothelial cancer organoids: A tool for bladder cancer research. Proc. Natl. Acad. Sci. USA 2019, 116, 4567–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Cao, Z.; Lim, P.X.; Zhao, H.; Luo, H.; Mao, N.; Lee, Y.S.; Rivera, A.A.; Choi, D.; Wu, C.; et al. Rapid interrogation of cancer cell of origin through CRISPR editing. Proc. Natl. Acad. Sci. USA 2021, 118, e2110344118. [Google Scholar] [CrossRef]
- Wang, L.; Smith, B.A.; Balanis, N.G.; Tsai, B.L.; Nguyen, K.; Cheng, M.W.; Obusan, M.B.; Esedebe, F.N.; Patel, S.J.; Zhang, H.; et al. A genetically defined disease model reveals that urothelial cells can initiate divergent bladder cancer phenotypes. Proc. Natl. Acad. Sci. USA 2020, 117, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Marino, S.; Vooijs, M.; van Der Gulden, H.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000, 14, 994–1004. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, L.; Wieczorek, K.; Wang, Y.; Zhang, X.; Goodrich, D.W.; Li, Q. Ex Vivo Organoid Model of Adenovirus-Cre Mediated Gene Deletions in Mouse Urothelial Cells. J. Vis. Exp. 2022, e63855. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, E.; Yamazaki, M.; Kawai, S.; Ohtani, Y.; Watanabe, T.; Kato, A.; Suzuki, M. A simple method for histopathological evaluation of organoids. J. Toxicol. Pathol. 2018, 31, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Cao, Q.; Wang, L.; Wang, J.; Xu, B.; Attwood, K.; Wei, L.; Wu, Y.; Smith, G.J.; Katsuta, E.; et al. A Preclinical Study to Repurpose Spironolactone for Enhancing Chemotherapy Response in Bladder Cancer. Mol. Cancer Ther. 2022, 21, 786–798. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.-L.; Apelo, C.A.; Torres, B.; Thai, K.H.; Hsieh, M.H. Mouse Bladder Wall Injection. JoVE 2011, e2523. [Google Scholar] [CrossRef] [PubMed]
- Raven, P.A.; D’Costa, N.M.; Moskalev, I.; Tan, Z.; Frees, S.; Chavez-Munoz, C.; So, A.I. Development of a murine intravesical orthotopic human bladder cancer (mio-hBC) model. Am. J. Clin. Exp. Urol. 2018, 6, 245–259. [Google Scholar] [PubMed]
- Wu, Q.; Mahendran, R.; Esuvaranathan, K. Nonviral cytokine gene therapy on an orthotopic bladder cancer model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 4522–4528. [Google Scholar]
- Merz, M.; Merz, A.M.A.; Wang, J.; Wei, L.; Hu, Q.; Hutson, N.; Rondeau, C.; Celotto, K.; Belal, A.; Alberico, R.; et al. Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nat. Commun. 2022, 13, 807. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y. Bladder Cancer and Genetic Mutations. Cell Biochem. Biophys. 2015, 73, 65–69. [Google Scholar] [CrossRef]
- Han, K.S.; Jeong, I.G.; Joung, J.Y.; Yang, S.O.; Chung, J.; Seo, H.K.; Kwon, K.S.; Park, W.S.; Lee, K.H. Clinical value of PTEN in patients with superficial bladder cancer. Urol. Int. 2008, 80, 264–269. [Google Scholar] [CrossRef]
- Sfakianos, J.P.; Lin Gellert, L.; Maschino, A.; Gotto, G.T.; Kim, P.H.; Al-Ahmadie, H.; Bochner, B.H. The role of PTEN tumor suppressor pathway staining in carcinoma in situ of the bladder. Urol. Oncol. 2014, 32, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satyal, U.; Sikder, R.K.; McConkey, D.; Plimack, E.R.; Abbosh, P.H. Clinical implications of molecular subtyping in bladder cancer. Curr. Opin. Urol. 2019, 29, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Batista da Costa, J.; Gibb, E.A.; Bivalacqua, T.J.; Liu, Y.; Oo, H.Z.; Miyamoto, D.T.; Alshalalfa, M.; Davicioni, E.; Wright, J.; Dall’Era, M.A.; et al. Molecular Characterization of Neuroendocrine-like Bladder Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3908–3920. [Google Scholar] [CrossRef] [Green Version]
- Clinton, T.N. Defining Resistance to Immune Checkpoint Inhibitors in Urothelial Carcinoma: Insights from PURE-01. Eur. Urol. 2021, 80, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhou, X.M. A model of orthotopic murine bladder (MBT-2) tumor implants. Urol. Res. 1997, 25, 179–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, J.H.; Jurczok, A.; Wulf, T.; Brandau, S.; Deinert, I.; Jocham, D.; Böhle, A. Optimizing syngeneic orthotopic murine bladder cancer (MB49). Cancer Res. 1999, 59, 2834–2837. [Google Scholar] [PubMed]
- Chang, M.T.; Penson, A.; Desai, N.B.; Socci, N.D.; Shen, R.; Seshan, V.E.; Kundra, R.; Abeshouse, A.; Viale, A.; Cha, E.K.; et al. Small-Cell Carcinomas of the Bladder and Lung Are Characterized by a Convergent but Distinct Pathogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1965–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wei, D.; Wang, L.; Wang, L.; Jia, Z.; Le, X.; Gao, Y.; Huang, S.; Xie, K. Modeling liver metastasis using a tumor cell line derived from an enhanced green fluorescent protein transgenic mouse. Clin. Exp. Metastasis 2010, 27, 11–18. [Google Scholar] [CrossRef]
- Park, S.; Rong, L.; Owczarek, T.B.; Bernardo, M.D.; Shoulson, R.L.; Chua, C.W.; Kim, J.Y.; Lankarani, A.; Chakrapani, P.; Syed, T.; et al. Novel Mouse Models of Bladder Cancer Identify a Prognostic Signature Associated with Risk of Disease Progression. Cancer Res. 2021, 81, 5161–5175. [Google Scholar] [CrossRef]
- Sfakianos, J.P.; Daza, J.; Hu, Y.; Anastos, H.; Bryant, G.; Bareja, R.; Badani, K.K.; Galsky, M.D.; Elemento, O.; Faltas, B.M.; et al. Epithelial plasticity can generate multi-lineage phenotypes in human and murine bladder cancers. Nat. Commun. 2020, 11, 2540. [Google Scholar] [CrossRef]
- Santos, C.P.; Lapi, E.; Martínez de Villarreal, J.; Álvaro-Espinosa, L.; Fernández-Barral, A.; Barbáchano, A.; Domínguez, O.; Laughney, A.M.; Megías, D.; Muñoz, A.; et al. Urothelial organoids originating from Cd49f(high) mouse stem cells display Notch-dependent differentiation capacity. Nat. Commun. 2019, 10, 4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, M.; Enting, D. Immune Responses in Bladder Cancer-Role of Immune Cell Populations, Prognostic Factors and Therapeutic Implications. Front. Oncol. 2019, 9, 1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Zhu, Y.; Wang, Y.; Liu, Z.; Zhang, J.; Xie, H.; Fu, Q.; Dai, B.; Ye, D.; Xu, J. Identification and Validation of Stromal Immunotype Predict Survival and Benefit from Adjuvant Chemotherapy in Patients with Muscle-Invasive Bladder Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3069–3078. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [Green Version]
- Tzetzo, S.L.; Abrams, S.I. Redirecting macrophage function to sustain their “defender” antitumor activity. Cancer Cell 2021, 39, 734–737. [Google Scholar] [CrossRef]
- Leblond, M.M.; Zdimerova, H.; Desponds, E.; Verdeil, G. Tumor-Associated Macrophages in Bladder Cancer: Biological Role, Impact on Therapeutic Response and Perspectives for Immunotherapy. Cancers 2021, 13, 4712. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Control IgG2a Cell Count | Anti-PD-1-NR Cell Count | Anti-PD-1-R Cell Count | Control IgG2a % in Total Cell | Anti-PD-1-NR % in Total Cell | Anti-PD-1-R % in Total Cell |
|---|---|---|---|---|---|---|
| Basophils | 0 | 0 | 2 | 0.00 | 0.00 | 0.04 |
| B cells | 0 | 0 | 3 | 0.00 | 0.00 | 0.06 |
| B cells, pro | 0 | 0 | 1 | 0.00 | 0.00 | 0.02 |
| DC | 14 | 14 | 20 | 0.29 | 0.31 | 0.41 |
| ILC | 12 | 15 | 33 | 0.25 | 0.34 | 0.68 |
| Macrophages | 188 | 287 | 420 | 3.95 | 6.44 | 8.64 |
| Monocytes | 10 | 24 | 24 | 0.21 | 0.54 | 0.49 |
| Neutrophils | 28 | 46 | 25 | 0.59 | 1.03 | 0.51 |
| NK cells | 3 | 2 | 11 | 0.06 | 0.04 | 0.23 |
| NKT | 16 | 19 | 97 | 0.34 | 0.43 | 2.00 |
| T cells | 15 | 18 | 46 | 0.31 | 0.40 | 0.95 |
| Tgd | 2 | 5 | 6 | 0.04 | 0.11 | 0.12 |
| NA | 18 | 22 | 54 | 0.38 | 0.49 | 1.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.; Wang, L.; Wieczorek, K.; Zhang, Y.; Wang, Z.; Wang, J.; Xu, B.; Singh, P.K.; Wang, Y.; Zhang, X.; et al. Single-Cell Analyses of a Novel Mouse Urothelial Carcinoma Model Reveal a Role of Tumor-Associated Macrophages in Response to Anti-PD-1 Therapy. Cancers 2022, 14, 2511. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102511
Xu D, Wang L, Wieczorek K, Zhang Y, Wang Z, Wang J, Xu B, Singh PK, Wang Y, Zhang X, et al. Single-Cell Analyses of a Novel Mouse Urothelial Carcinoma Model Reveal a Role of Tumor-Associated Macrophages in Response to Anti-PD-1 Therapy. Cancers. 2022; 14(10):2511. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102511
Chicago/Turabian StyleXu, Dongbo, Li Wang, Kyle Wieczorek, Yali Zhang, Zinian Wang, Jianmin Wang, Bo Xu, Prashant K. Singh, Yanqing Wang, Xiaojing Zhang, and et al. 2022. "Single-Cell Analyses of a Novel Mouse Urothelial Carcinoma Model Reveal a Role of Tumor-Associated Macrophages in Response to Anti-PD-1 Therapy" Cancers 14, no. 10: 2511. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102511