
NEDD9 Restrains dsDNA Damage Response during Non-Small Cell Lung Cancer (NSCLC) Progression

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transient Transfection
2.2. Cell Viability Assays and Determination of Cisplatin Effective Dose
2.3. Cell Migration (Wound Healing) Assays
2.4. Clonogenic Survival Assays and UV Irradiation Experiments
2.5. Flow Cytometry Assays for Cell Cycle Analysis
2.6. Immunofluorescent Detection of γ-H2AX
2.7. Western Blot Analysis
2.8. Immunohistochemistry and Human Samples Cohort
2.9. The Cancer Genome Atlas (TCGA) Analysis
2.10. Statistical Analysis
3. Results
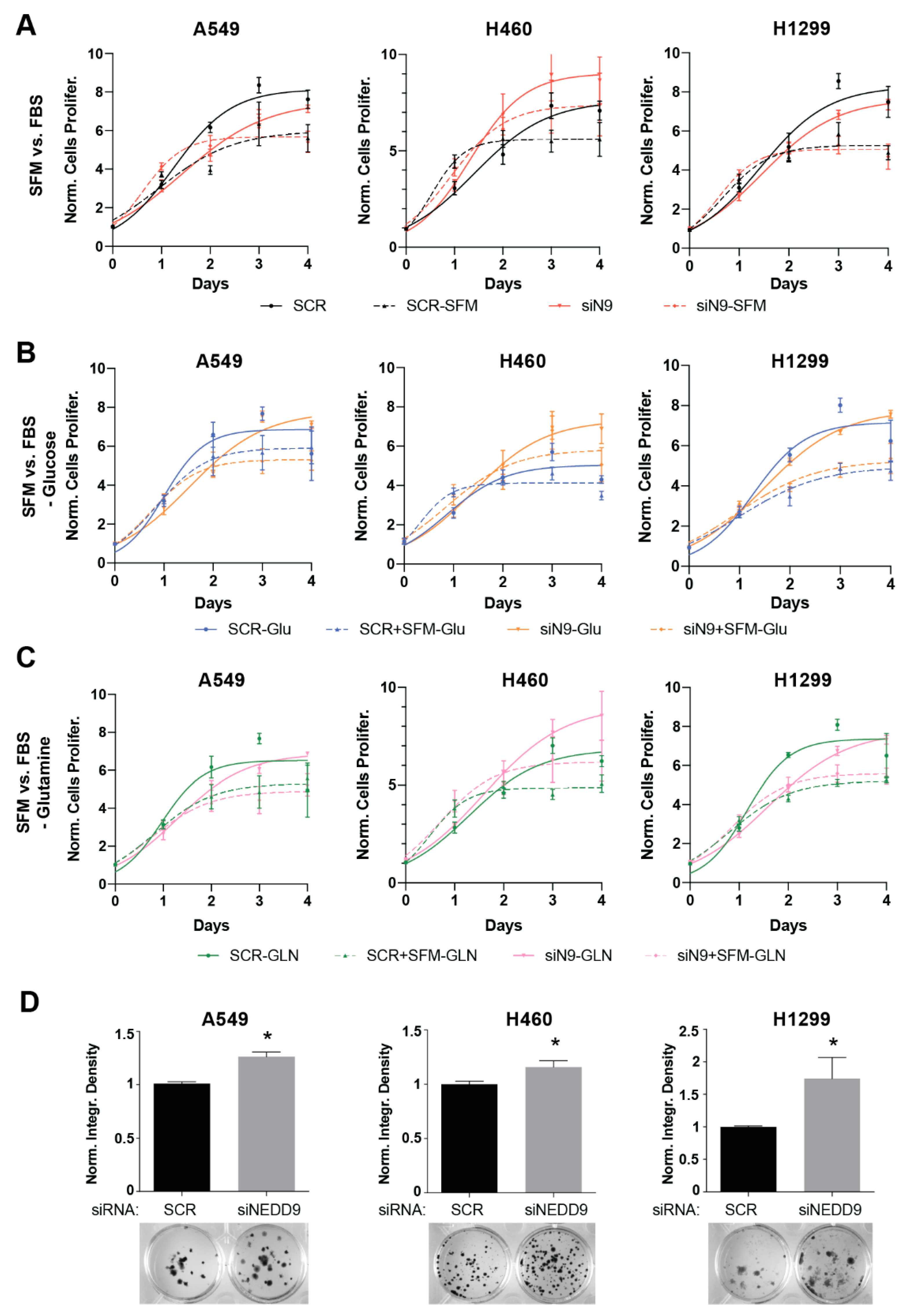
3.1. NEDD9 Depletion Is Associated with Increased Tumorigenic Capacity in Human and Murine NSCLC Cell Line Models
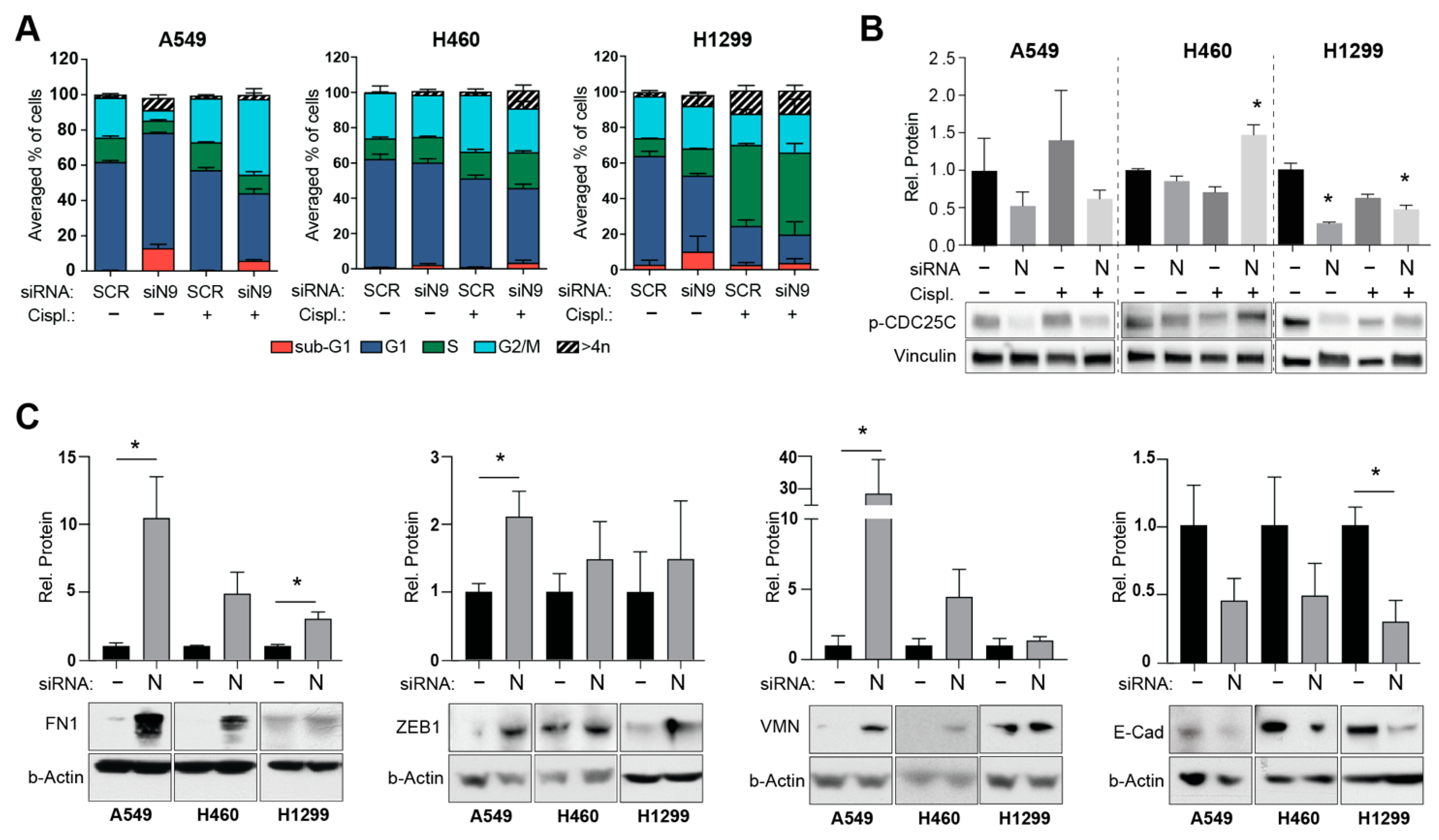
3.2. Upregulation of ATM Is Notable in NEDD9 Depleted and Short-Time Cisplatin Treated NSCLC Cells
3.3. NEDD9-Mediated ATM Activation Is Associated with Elevated Epithelial to Mesenchymal Transition (EMT) Rather Than Cell Cycle Alterations in NSCLC Cells
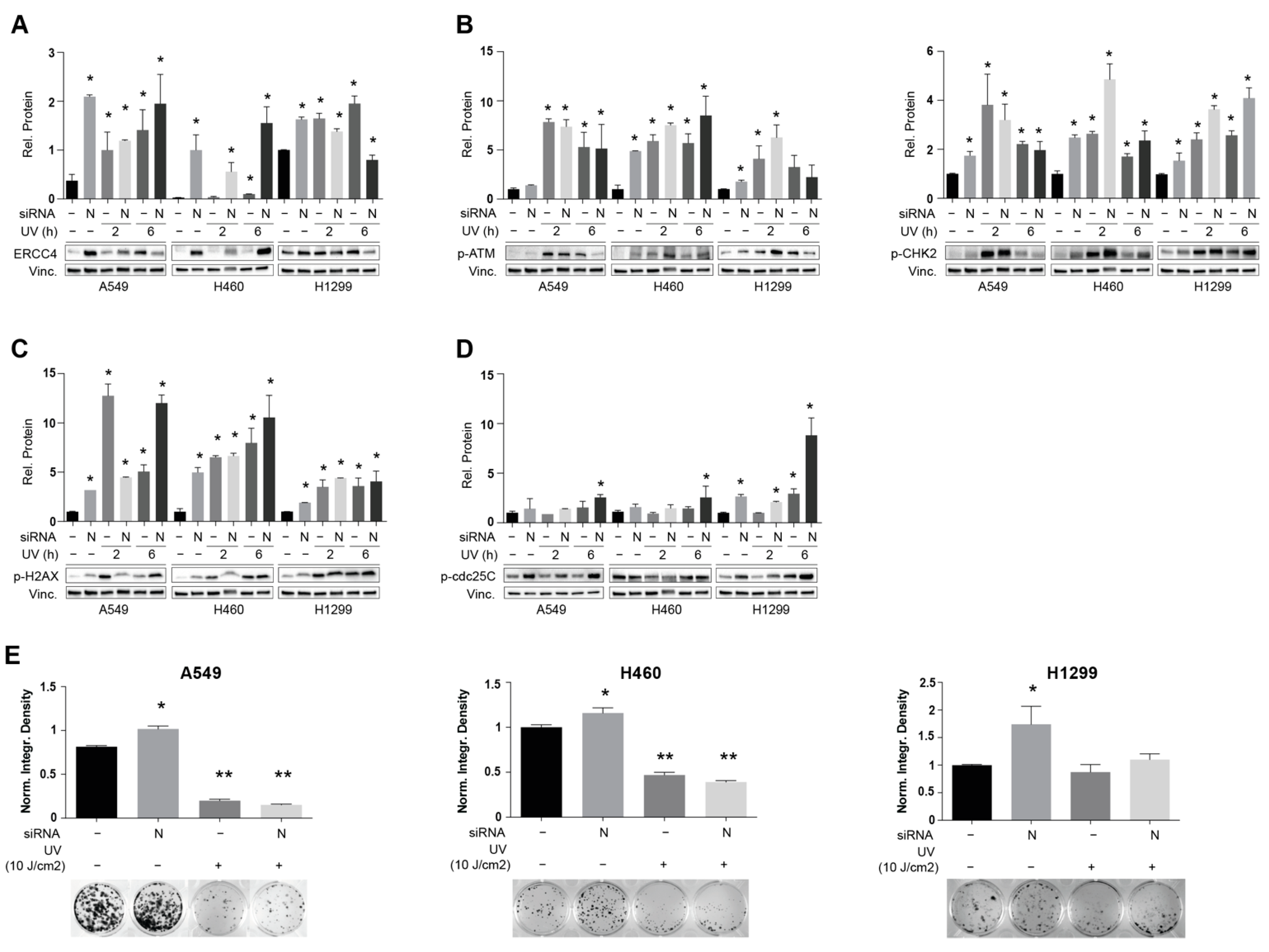
3.4. NEDD9 Depletion Is Associated with Increased Sensitivity to UV-Induced dsDNA Breaks
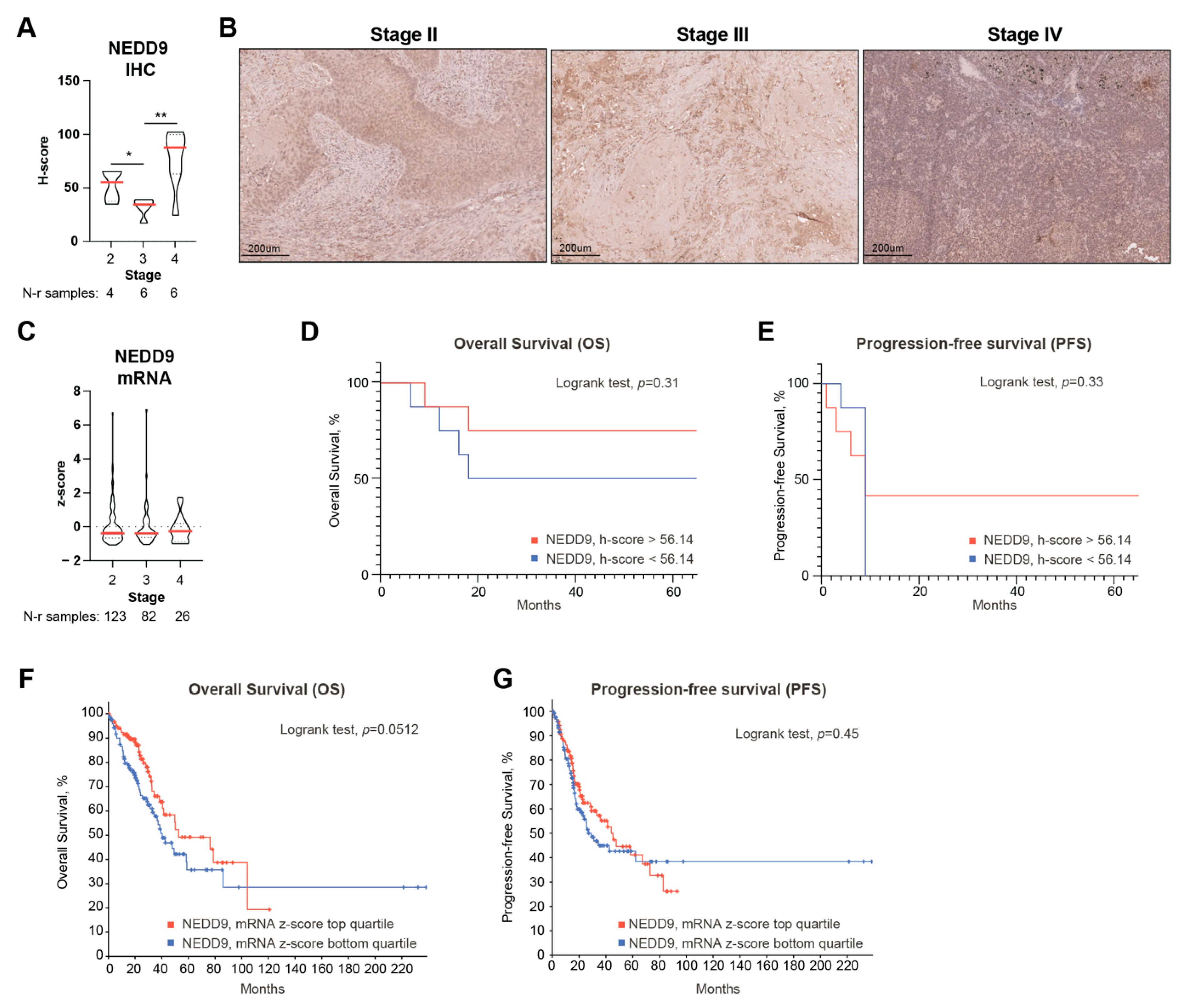
3.5. Decreased NEDD9 Levels Precede Human NSCLC Metastasis and Are Associated with Decreased Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Facts and Figures 2020; American Cancer Society Annual Report; American Cancer Society: Atlanta, GA, USA, 2020.
- US. National Institutes of Health; National Cancer Institute. SEER Cancer Statistics Review, 1975–2011; National Cancer Institute: Bethesda, MD, USA, 2014. [Google Scholar]
- Karachaliou, N.; Mayo, C.; Costa, C.; Magri, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Rosell, R. KRAS mutations in lung cancer. Clin. Lung Cancer 2013, 14, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. p53: A frequent target for genetic abnormalities in lung cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Jonna, S.; Deneka, A.; Patel, J.D.; Abazeed, M.E.; Golemis, E.; Borghaei, H.; Boumber, Y. Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies. Cancers 2021, 13, 3164. [Google Scholar] [CrossRef] [PubMed]
- Baldini, E.; Tibaldi, C.; Delli Paoli, C. Chemo-radiotherapy integration in unresectable locally advanced non-small-cell lung cancer: A review. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2020, 22, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Güthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Xu, Z.; Xu, W.; Jiang, K.; Zhang, F.; Ding, Q.; Xu, Z.; Chen, Y. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 149. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. Initiating Cellular Stress Responses. Cell 2004, 118, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Yang, D.-Q.; Kastan, M.B. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat. Cell Biol. 2000, 2, 893–898. [Google Scholar] [CrossRef]
- Tang, S.; Yang, L.; Tang, X.; Liu, M. The role of oxidized ATM in the regulation of oxidative stress-induced energy metabolism reprogramming of CAFs. Cancer Lett. 2014, 353, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tang, X.; Guo, X.; Niikura, Y.; Kitagawa, K.; Cui, K.; Wong, S.T.C.; Fu, L.; Xu, B. Aurora-B Mediated ATM Serine 1403 Phosphorylation Is Required for Mitotic ATM Activation and the Spindle Checkpoint. Mol. Cell 2011, 44, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Seventer, G.A.; Salman, H.J.; Law, S.F.; O’Neill, G.M.; Mullen, M.M.; Franz, A.A.; Kanner, S.B.; Golemis, E.A.; van Seventer, J.M. Focal adhesion kinase regulates beta1 integrin dependent migration through an HEF1 effector pathway. Eur. J. Imm. 2001, 31, 1417–1427. [Google Scholar] [CrossRef]
- Fashena, S.J.; Einarson, M.B.; O’Neill, G.M.; Patriotis, C.; Golemis, E.A. Dissection of HEF1-dependent functions in motility and transcriptional regulation. J. Cell Sci. 2002, 115, 99–111. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, G.M.; Golemis, E.A. Proteolysis of the docking protein HEF1 and implications for focal adhesion dynamics. Mol. Cell. Biol. 2001, 21, 5094–5108. [Google Scholar] [CrossRef] [Green Version]
- Astsaturov, I.; Ratushny, V.; Sukhanova, A.; Einarson, M.B.; Bagnyukova, T.; Zhou, Y.; Devarajan, K.; Silverman, J.S.; Tikhmyanova, N.; Skobeleva, N.; et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci. Signal. 2010, 3, ra67. [Google Scholar] [CrossRef] [Green Version]
- Izumchenko, E.; Singh, M.K.; Plotnikova, O.V.; Tikhmyanova, N.; Little, J.L.; Serebriiskii, I.G.; Seo, S.; Kurokawa, M.; Egleston, B.L.; Klein-Szanto, A.; et al. NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res. 2009, 69, 7198–7206. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.K.; Dadke, D.; Nicolas, E.; Serebriiskii, I.G.; Apostolou, S.; Canutescu, A.; Egleston, B.L.; Golemis, E.A. A novel Cas family member, HEPL, regulates FAK and cell spreading. Mol. Biol. Cell 2008, 19, 1627–1636. [Google Scholar] [CrossRef] [Green Version]
- Nikonova, A.S.; Plotnikova, O.V.; Serzhanova, V.; Efimov, A.; Bogush, I.; Cai, K.Q.; Hensley, H.H.; Egleston, B.L.; Klein-Szanto, A.; Seeger-Nukpezah, T.; et al. Nedd9 restrains renal cystogenesis in Pkd1−/− mice. Proc. Natl. Acad. Sci. USA 2014, 111, 12859–12864. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Asai, T.; Saito, T.; Suzuki, T.; Morishita, Y.; Nakamoto, T.; Ichikawa, M.; Yamamoto, G.; Kawazu, M.; Yamagata, T. Crk-associated substrate lymphocyte type is required for lymphocyte trafficking and marginal zone B cell maintenance. J. Immunol. 2005, 175, 3492–3501. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Li, A.L.; Wang, L.; Fan, C.F.; Zhang, X.P.; Xu, H.T.; Yang, L.H.; Liu, Y.; Wang, E.H. Overexpression of NEDD9 is associated with altered expression of E-Cadherin, beta-Catenin and N-Cadherin and predictive of poor prognosis in non-small cell lung cancer. Pathol. Oncol. Res. 2013, 19, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Deneka, A.Y.; Kopp, M.C.; Nikonova, A.S.; Gaponova, A.V.; Kiseleva, A.A.; Hensley, H.H.; Flieder, D.B.; Serebriiskii, I.G.; Golemis, E.A. Nedd9 Restrains Autophagy to Limit Growth of Early Stage Non-Small Cell Lung Cancer. Cancer Res. 2021, 81, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.N.; Golemis, E.A. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat. Cell Biol. 2005, 7, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadke, D.; Jarnik, M.; Pugacheva, E.N.; Singh, M.K.; Golemis, E.A. Deregulation of HEF1 Impairs M-Phase Progression by Disrupting the RhoA Activation Cycle. Mol. Biol. Cell 2006, 17, 1204–1217. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Deneka, A.Y.; Einarson, M.B.; Bennett, J.; Nikonova, A.S.; Elmekawy, M.; Zhou, Y.; Lee, J.W.; Burtness, B.A.; Golemis, E.A. Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer. Cancers 2020, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Gaponova, A.V.; Deneka, A.Y.; Beck, T.N.; Liu, H.; Andrianov, G.; Nikonova, A.S.; Nicolas, E.; Einarson, M.B.; Golemis, E.A.; Serebriiskii, I.G. Identification of evolutionarily conserved DNA damage response genes that alter sensitivity to cisplatin. Oncotarget 2017, 8, 19156–19171. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Caiola, E.; Salles, D.; Frapolli, R.; Lupi, M.; Rotella, G.; Ronchi, A.; Garassino, M.C.; Mattschas, N.; Colavecchio, S.; Broggini, M.; et al. Base excision repair-mediated resistance to cisplatin in KRAS(G12C) mutant NSCLC cells. Oncotarget 2015, 6, 30072–30087. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.J.; Constanzo, J.D.; Venkateswaran, N.; Melegari, M.; Ilcheva, M.; Morales, J.C.; Skoulidis, F.; Heymach, J.V.; Boothman, D.A.; Scaglioni, P.P. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5851–5863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2016, 129, 1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, S.; Yan, H.; Cho, I.; Fan, H.Y.; Luo, B.; Gai, X.; Bodian, D.L.; Vockley, J.G.; Zhou, Y.; Handorf, E.A.; et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015, 149, 1872–1883.e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turinetto, V.; Giachino, C. Multiple facets of histone variant H2AX: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Topchu, Y.A.; Mazitova, A.M.; Tikhomirova, M.V.; Abramova, Z.I.; Deneka, A.Y. Nedd9 Regulates Metastasis of Non-Small Cell Lung Cancer through Activation of Epithelial-Mesenchymal Transition and Tumor Cells Migration. Uchenye Zap. Kazan. Univ. Seriya Estestv. Nauk. 2020, 162, 123–133. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Sanz-Moreno, V.; Marshall, C.J. The metastasis gene NEDD9 product acts through integrin β3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J. Cell Sci. 2012, 125, 1814–1826. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.C.; Kelley, L.C.; Loskutov, Y.V.; Marinak, K.M.; Kozyreva, V.K.; Smolkin, M.B.; Pugacheva, E.N. Dual Targeting of Mesenchymal and Amoeboid Motility Hinders Metastatic Behavior. Mol. Cancer Res. MCR 2017, 15, 670–682. [Google Scholar] [CrossRef] [Green Version]
- van Vuuren, A.J.; Appeldoorn, E.; Odijk, H.; Yasui, A.; Jaspers, N.G.; Bootsma, D.; Hoeijmakers, J.H. Evidence for a repair enzyme complex involving ERCC1 and complementing activities of ERCC4, ERCC11 and xeroderma pigmentosum group F. EMBO J. 1993, 12, 3693–3701. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Wang, Z.; Fang, Z.; Li, F.; Gao, Y.; Liu, H.; Xiao, T.; Li, F.; Zhou, Y.; et al. The CRTC1-NEDD9 signaling axis mediates lung cancer progression caused by LKB1 loss. Cancer Res. 2012, 72, 6502–6511. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, G.M.; Fashena, S.J.; Golemis, E.A. Integrin signalling: A new Cas(t) of characters enters the stage. Trends Cell Biol. 2000, 10, 111–119. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, K.; Xu, B.; Shi, J.; Qiang, J.; Shi, S.; Yi, Y.; Li, H.; Jin, T.; Guo, R.; et al. Epigenetic Input Dictates the Threshold of Targeting of the Integrin-Dependent Pathway in Non-small Cell Lung Cancer. Front. Cell Dev. Biol. 2020, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Shagisultanova, E.; Gaponova, A.V.; Gabbasov, R.; Nicolas, E.; Golemis, E.A. Preclinical and clinical studies of the NEDD9 scaffold protein in cancer and other diseases. Gene 2015, 567, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koudelková, L.; Brábek, J.; Rosel, D. Src kinase: Key effector in mechanosignalling. Int. J. Biochem. Cell Biol. 2021, 131, 105908. [Google Scholar] [CrossRef] [PubMed]
- Gąsiorkiewicz, B.M.; Koczurkiewicz-Adamczyk, P.; Piska, K.; Pękala, E. Autophagy modulating agents as chemosensitizers for cisplatin therapy in cancer. Investig. New Drugs 2021, 39, 538–563. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Xu, F.; Kong, Q.; Yang, T.; Tan, D.; Zhang, X.; Li, N.; Zhao, S.; Zhao, J.; Li, M. Inhibition of p62/SQSTM1 sensitizes small-cell lung cancer cells to cisplatin-induced cytotoxicity by targeting NEDD9 expression. Mol. Carcinog. 2020, 59, 967–979. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tikhomirova, M.; Topchu, I.; Mazitova, A.; Barmin, V.; Ratner, E.; Sabirov, A.; Abramova, Z.; Deneka, A.Y. NEDD9 Restrains dsDNA Damage Response during Non-Small Cell Lung Cancer (NSCLC) Progression. Cancers 2022, 14, 2517. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102517
Tikhomirova M, Topchu I, Mazitova A, Barmin V, Ratner E, Sabirov A, Abramova Z, Deneka AY. NEDD9 Restrains dsDNA Damage Response during Non-Small Cell Lung Cancer (NSCLC) Progression. Cancers. 2022; 14(10):2517. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102517
Chicago/Turabian StyleTikhomirova, Mariya, Iuliia Topchu, Aleksandra Mazitova, Vitaly Barmin, Ekaterina Ratner, Alexey Sabirov, Zinaida Abramova, and Alexander Y. Deneka. 2022. "NEDD9 Restrains dsDNA Damage Response during Non-Small Cell Lung Cancer (NSCLC) Progression" Cancers 14, no. 10: 2517. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102517