1. Introduction
Gliomas are the most common primary brain tumors, and remain associated with a poor prognosis. Among them, diffuse gliomas are known to be highly infiltrative tumors in which invading tumor cells can be found as far as 4
from the gross tumor [
1]. However, the limited sensibility of magnetic resonance imaging (MRI)—the modality of choice for glioma imaging—to changes occurring at the cellular level makes the delineation of the whole tumor extent tedious and often leads to sub-optimal treatment plannings.
Knowledge of the whole tumor cell distribution, within and beyond the outlines of the tumor visible on MRI, would instead allow us to refine surgery or radiation therapy planning. Current standards for the planning of such therapies indeed rely on the addition of a fixed margin to account for tumor infiltration, defining the clinical target volume [
2,
3]. From an estimated tumor cell distribution, dose deposition could instead be redistributed, with a higher dose delivered to areas more likely to contain tumor cells and, on the other hand, a lower dose delivered to surrounding healthy tissues, while keeping the total dose unchanged [
2]. Furthermore, evaluation of the tumor growth dynamics from repeated medical imaging data would also be of great interest to better characterize the tumor, anticipate its growth, and identify its probable migration pathways as well as areas prone to recurrence.
Reaction-diffusion tumor growth models have been studied for decades to circumvent the limitations of current medical imaging techniques and improve treatment planning in gliomas [
2,
4,
5,
6,
7,
8,
9,
10]. These models rely on partial differential equations (PDEs) to capture the spatio-temporal evolution of a tumor cell-density function over the brain domain, driven by tumor cell migration and proliferation. The most commonly used form involves a logistic reaction term and is referred to as the Fisher equation [
11]:
where
is the tumor cell-density at location
and time
t normalized by the maximum carrying capacity
of brain tissues (i.e.,
),
d is the tumor cell diffusion coefficient, and
is the tumor cell proliferation rate. A property of the well-studied Equation (
1) is that, under certain conditions and for constant coefficients, it admits a traveling wave solution on the infinite cylinder with propagation speed
, whose profile decays exponentially with decay constant
as the distance to the origin tends to infinity and for large but finite times
t [
10,
12].
Since their first introduction by Murray and colleagues in the early 1990s [
4], reaction-diffusion growth models have been continuously improved to successively integrate (i) a variable tumor cell diffusion rate in white versus gray matter [
13] and (ii) an anisotropic diffusion tensor field accounting for the preferred migration of tumor cells along white matter tracts, whose orientation can be assessed by diffusion tensor imaging (DTI) [
14]. These improvements led to the formulation that is used throughout this work, presented in
Section 2.1. Tumor-induced mass effect [
6,
7], necrosis, and hypoxia [
15,
16], as well as the effects of surgery [
5], chemotherapy [
4,
17,
18], and radiotherapy [
18,
19] have also been introduced into reaction-diffusion glioma growth models, but are not considered in this work. For a more detailed overview of reaction-diffusion glioma growth modeling, the reader is referred to [
2,
4,
5,
6,
7,
8,
9,
10].
Although reaction-diffusion models have shown promising results for patient follow-up and improved radiotherapy planning [
2], their clinical application is still prone to severe limitations. Indeed, the estimation of the parameter values and the tumor cell-density distribution at diagnosis time—required to predict the tumor evolution at later times—but also the validation of such models in vivo, implies to extract information on the tumor at the cellular level from medical imaging data. To address this issue, Swanson and colleagues have proposed in [
8] to model the imaging function of MRI sequences
—indicating whether a tumor-induced abnormality is visible at location
and time
t on the image—as a simple tumor cell-density threshold function:
where
is the tumor cell-density detectability threshold of the sequence. The abnormalities considered in [
8] were the hyper-intense enhancing tumor core visible on T1-weighted sequences with injection of gadolinium-based contrast agent (T1Gd) and the peritumor vasogenic edema visible on T2-weighted sequences with or without fluid-attenuated inversion-recovery (T2/T2 FLAIR). Based on these assumptions, the authors suggested that the outlines of these abnormalities would correspond to iso-contours of the tumor cell-density function—i.e., hyper-surfaces along which
c has a constant value:
where
is the boundary of the visible abnormality.
Building upon this work, Konukoglu and colleagues proposed in [
10] a fast marching approach to construct an approximate solution of Equation (
1) at imaging time which satisfies Equation (
3). This approach has the interesting property of not attempting to dynamically solve the model but seeks to extrapolate the tumor invasion beyond its MR-visible margins within the reaction-diffusion framework, with applications for radiotherapy planning. It has, nevertheless, two major limitations: First, it requires the ability to extract a tumor cell-density iso-contour from the image, from which the whole tumor cell distribution is built. However, we showed in a previous work based on histological data that the outlines of the edema visible on T2 FLAIR MR images do not coincide with a cell-density iso-contour [
20]. The proposed explanation is that, due to spatial discontinuities of the tumor cell-density function at interfaces between white and gray matter as well as along the brain domain boundary, Equation (
2) does not necessarily imply Equation (
3). The second limitation of this approach is that the method still needs to specify the diffusivity and proliferation rate of the tumor, which are unknown at imaging time and need to be adjusted to each tumor.
The estimation of the model parameter values from medical imaging data has also been addressed previously. In [
21], the definition of the asymptotic speed of the tumor front
is used to estimate the tumor cell diffusivity
and
in white and gray matter using a fast marching approach. However, the method does not allow us to separate the individual contributions of
d and
to
v, hence
is supposed constant for all tumors. Furthermore, this estimation is only valid for large times for which the traveling-wave approximation holds. The approach was then further extended in [
9] to take into account the transient speed evolution and the curvature of the tumor front, but still considers a constant
value for all tumors. Besides, these fast marching formulations make the assumption that the outlines of the peritumor vasogenic edema visible on T2 MR images correspond to an iso-contour of the traveling wave arrival time function. However, this hypothesis might not be verified due to discontinuities appearing at the brain boundary voxels, which could have been reached long before the imaging time. In [
22], a Bayesian approach is used to estimate both the diffusion and proliferation parameters of the model from two imaging contours obtained by Equation (
2) at two different times. The method was found to accurately estimate the infiltration length
of the tumor, but less accurately the tumor front propagation speed
, based on synthetic and real glioblastoma (GBM) MRI data. In [
18], parameter values of a two-species reaction-diffusion model incorporating tumor-induced mass effect and response to chemoradiation are estimated based on tumor cell-density distributions derived from longitudinal T1Gd, T2 FLAIR, and diffusion-weighted (DW) MR data, with promising results. However, the cell-density distributions used to initialize the model and fit the parameters were built piecewise from the enhancing/non-enhancing tumor regions delineated on T1Gd/T2 FLAIR images as well as average diffusion coefficient (ADC) maps derived from DW-MR data, and are therefore not guaranteed to be solution of Equation (
1) nor to reflect the actual tumor cell distribution.
Tumor source localization is another widely addressed problem in reaction-diffusion glioma growth modeling. In [
23], an inverse problem approach is used to estimate the tumor source location from a given tumor cell-density distribution, with promising results. However, to be applicable in clinical practice, the method still requires the ability to derive a whole tumor cell-density distribution from medical imaging data.
Finally, several works have attempted to jointly estimate the tumor source location and model parameters from patient imaging data. In [
24], a PDE-constrained optimization approach is used to assess the source location and parameter values of a reaction-diffusion glioma growth model including an additional advection term. The tumor growth model is coupled to a linear elastic model for the tumor-induced mass effect. Two optimization criteria are used in the study: (i) the squared difference between the true and estimated cell-density fields at given imaging times and (ii) the squared distance between the true and estimated position of manually defined landmarks on staggered scans, that are displaced as the surrounding brain tissues are deformed under mass effect. However, the first criterion requires the knowledge of the true tumor cell-density field, which again cannot be derived directly from imaging data. Promising results were obtained for the landmark-based criterion on a real glioma case but strong assumptions are made on the initial cell-density field—supposedly Gaussian—and no ground truth was available to assess the model parameter estimation. In [
25], the fast marching approach of Konukoglu and colleagues [
9,
21] is used to assess the diffusivity ratio
along with the tumor source location, but a fixed proliferation rate
was again considered. More recently, a Bayesian framework has been proposed to simultaneously estimate the tumor source, emergence time, diffusivity, and proliferation rate from a combination of T1Gd, T2 FLAIR, and [
18F]fluoroethyl-L-tyrosine ([
18F]FET) positron emission tomography (PET) images in [
26]. However, the study reported that these last three parameters cannot be individually assessed from a single imaging time point. Finally, in [
27] a numerical approach is proposed to solve the ill-posed problem of estimating both the tumor initial location as well as its diffusivity and proliferation rate, and is further applied on 206 GBM cases from the BraTS dataset [
28] in [
29], for which actual parameter values are however not known. Although the method has shown promising results and only requires a single imaging time point, it still has three major limitations: (i) it only allows for the estimation of unscaled dimensionless diffusion and proliferation parameters as the time between tumor emergence and imaging is unknown, which restricts the absolute comparison of the estimated parameter values between tumors and prevents the use of the model as a personalized prediction tool for the tumor evolution over time, (ii) it makes strong assumptions regarding the initial tumor cell-density distribution—a sparse set of Gaussian distributions with identical standard deviation and a maximum density value of 1 over the set—and (iii) it relies on many user-defined parameters, to some of which the method is reported sensitive [
27,
29].
In addition to their various limitations highlighted hereabove, none of the aforementioned works have jointly addressed the estimation of the tumor cell-density distribution at diagnosis time and individual diffusion and proliferation parameters of the model, which could however made it possible to anticipate the growth of the tumor at further times using the model and thus adapt treatment strategies. Besides, most of these works considered a spatially constant diffusion coefficient in white matter and/or an identical proliferation rate for all tumors, which is not realistic. The introduction of an arbitrary diffusion tensor field and a tumor-specific proliferation rate would make the addressed problems even more challenging.
Over the last five years, the advent of deep-learning techniques—and in particular deep convolutional neural networks (DCNNs)—has opened tremendous opportunities in the field of medical imaging, achieving state-of-the-art performance in many image classification and segmentation challenges [
30]. One interesting property of deep neural networks is their ability to approximate any function under certain conditions [
31]. This property makes the technique attractive for the problems addressed in this work. DCNNs may indeed be used to approximate solutions of PDEs such as Equation (
1) over complex domains, and for spatially variable coefficients, as well as to estimate their parameter values from partial observations provided in the form of threshold-like imaging contours.
In this work, we investigate the ability of DCNNs to address common pitfalls encountered in the clinical application of reaction-diffusion glioma growth models. In particular, we focus on the following two tasks:
Reconstructing a whole brain tumor cell-density distribution compatible with the reaction-diffusion model from a pair of contours obtained through a threshold-like imaging process as in Equation (
2) for two different detectability threshold values at a given imaging time. These contours may for example correspond to the outlines of the enhancing core and peritumor vasogenic edema on T1Gd and T2/T2 FLAIR MR images, respectively.
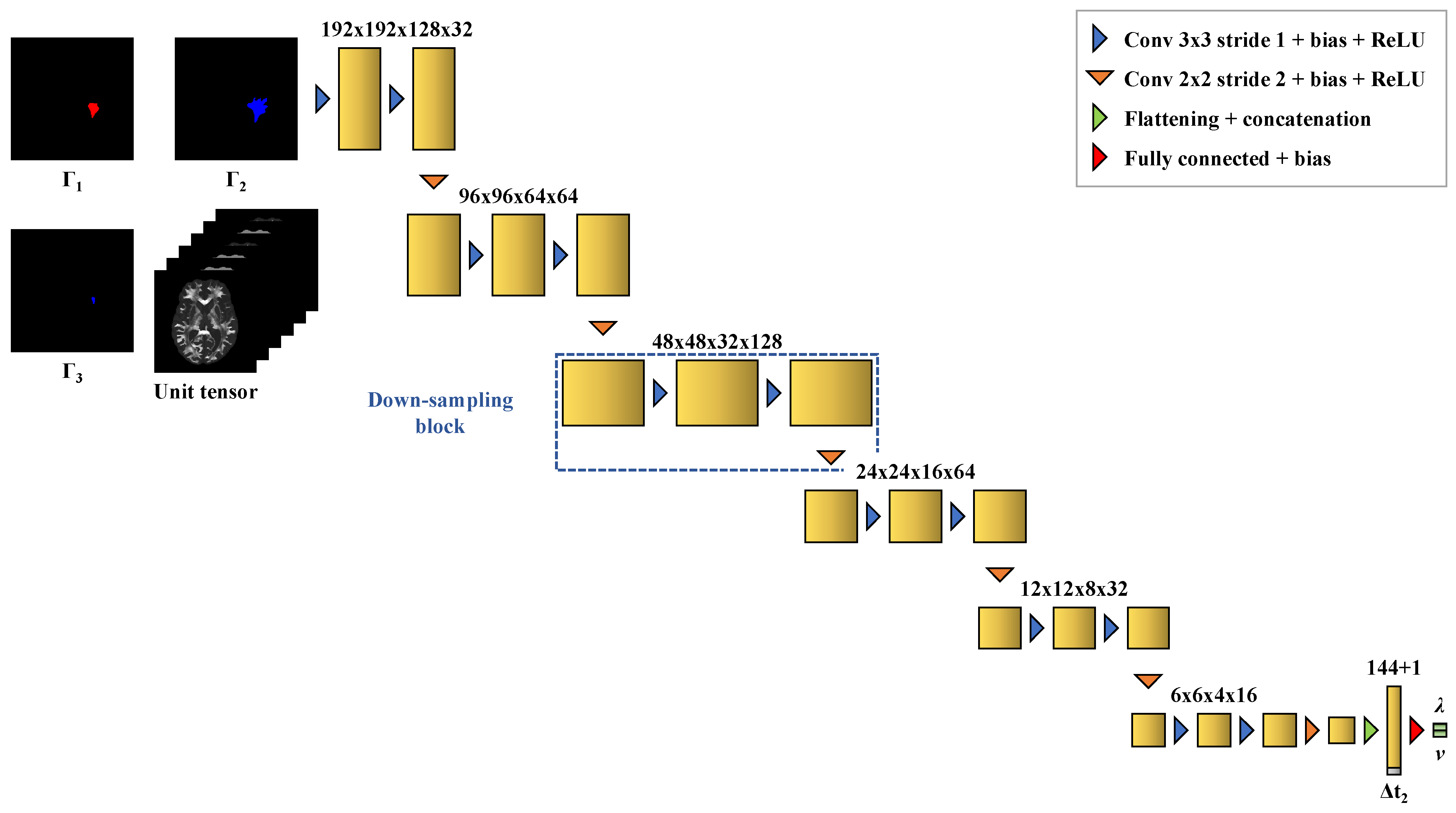
Estimating the values of the diffusion and proliferation parameters of the model from three imaging contours: (i) two contours obtained for a first detectability threshold value (e.g., the vasogenic edema outlines) at two different imaging times and (ii) a third contour obtained for a second detectability threshold value (e.g., the enhancing core outlines) at the second imaging time.
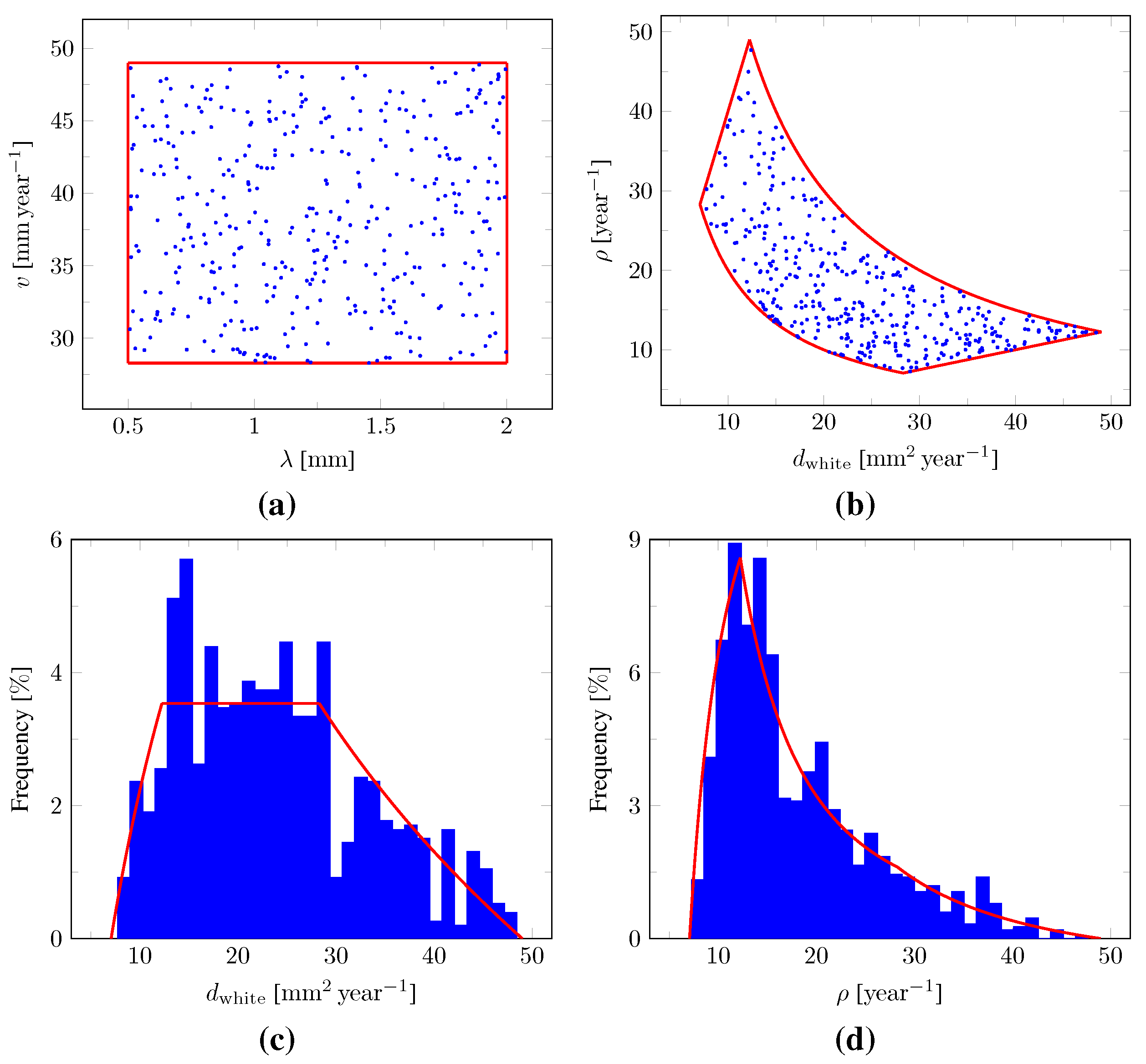
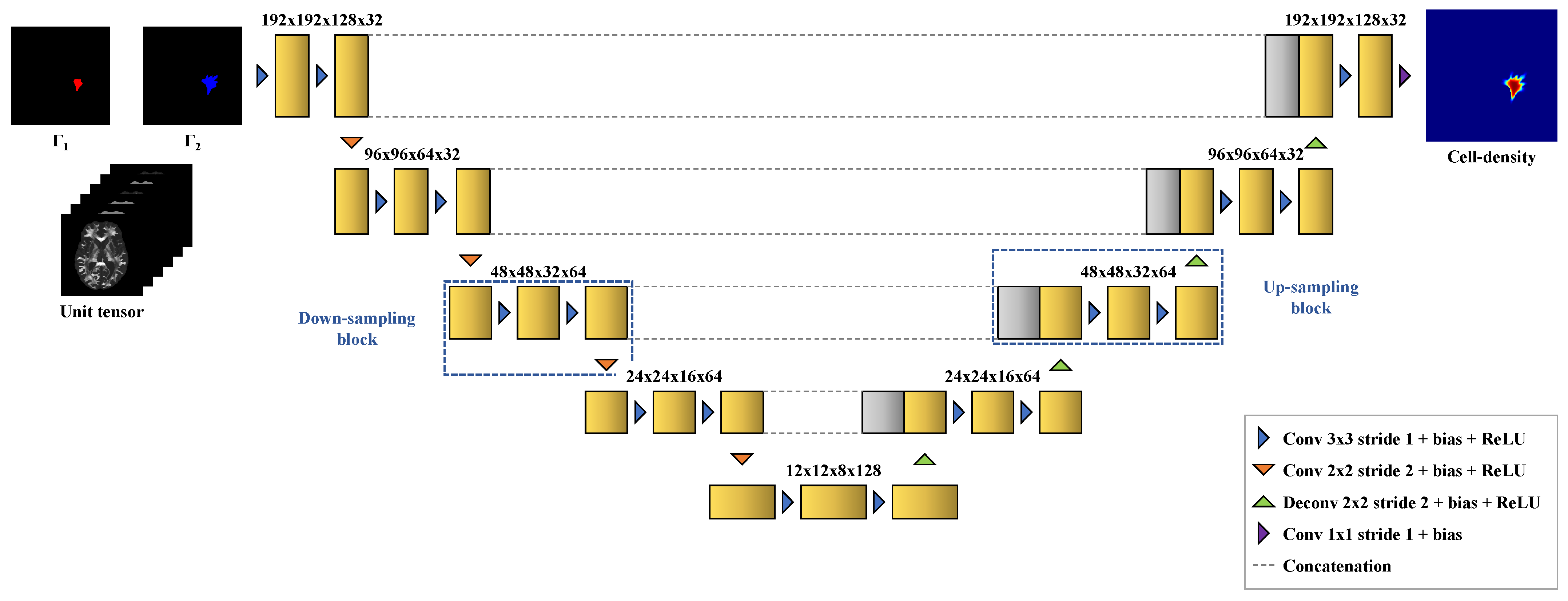
We demonstrate the ability of DCNNs to perform these tasks accurately based on 1200 synthetic tumors grown over brain geometries derived from the MR data of six healthy subjects. We also show the applicability of our approach on MR data of a real glioblastoma patient.
3. Results
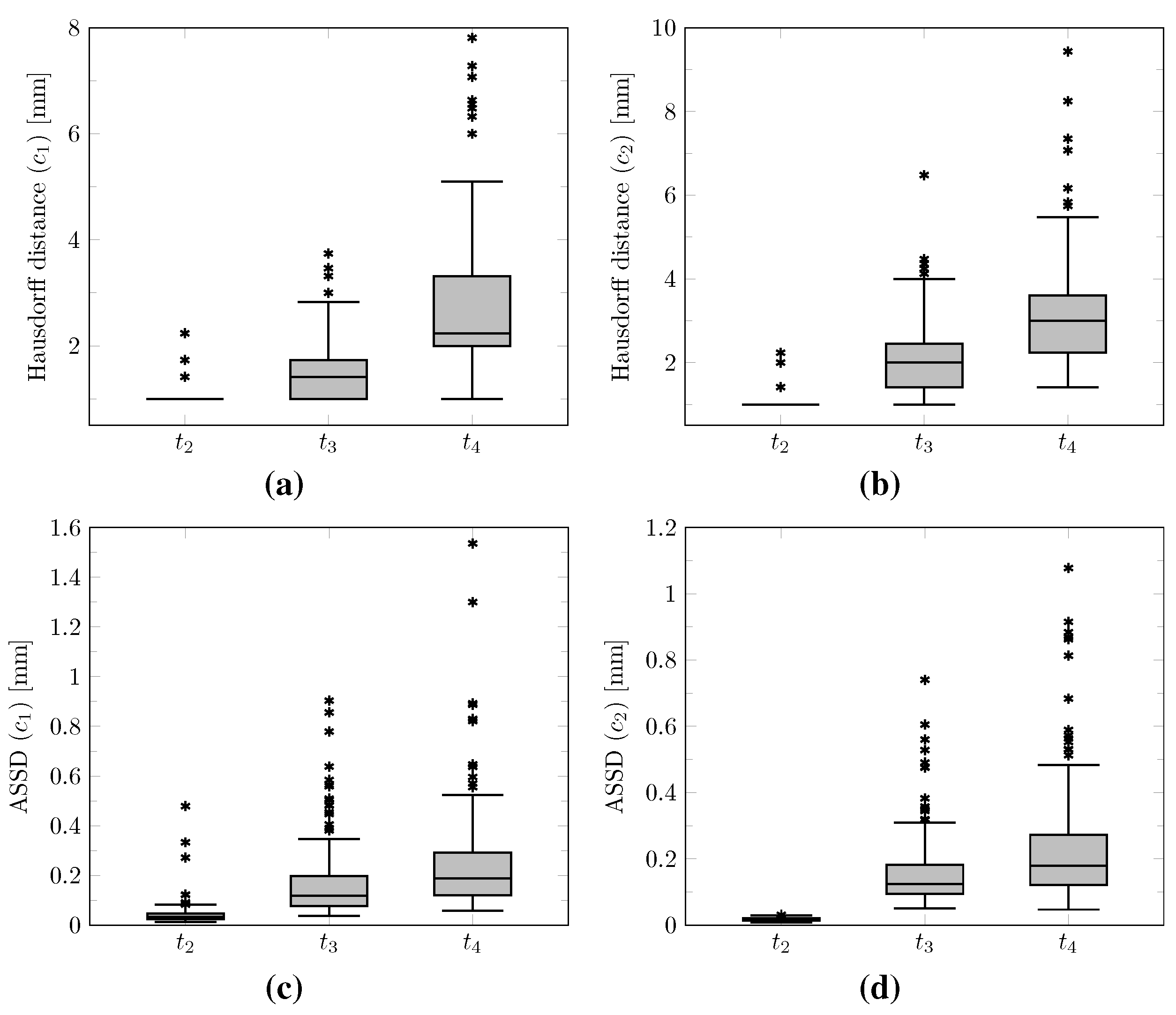
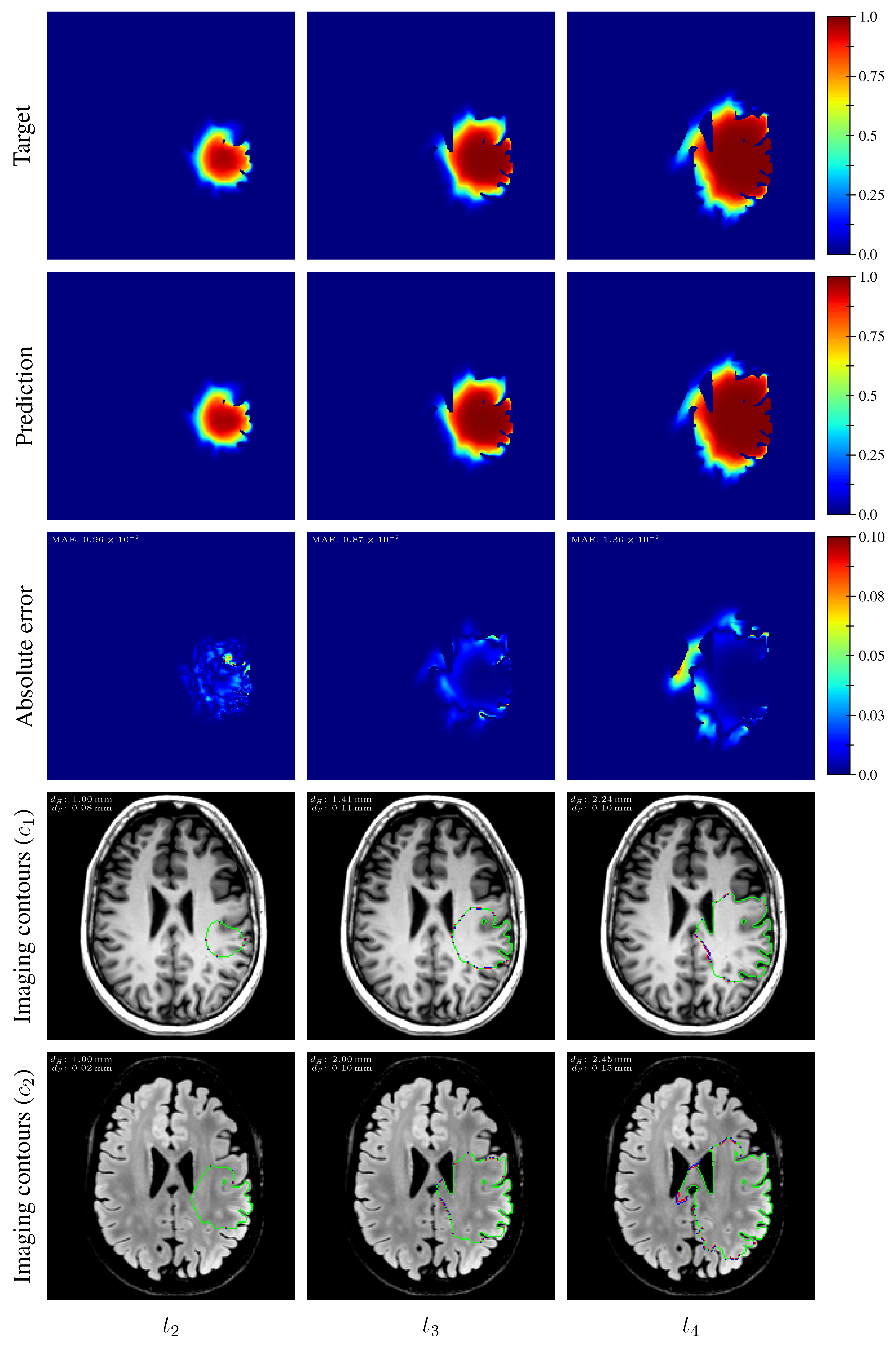
The distribution of the mean absolute error computed over the test set between the true and estimated tumor cell-density distributions at time
within the
contour is summarized by a boxplot in
Figure 7 (first plot). Boxplots of the Hausdorff distance and ASSD distributions computed over the test set between the true and estimated imaging contours at time
for threshold values
and
are provided in
Figure 8 (first plots). The corresponding median values are provided in
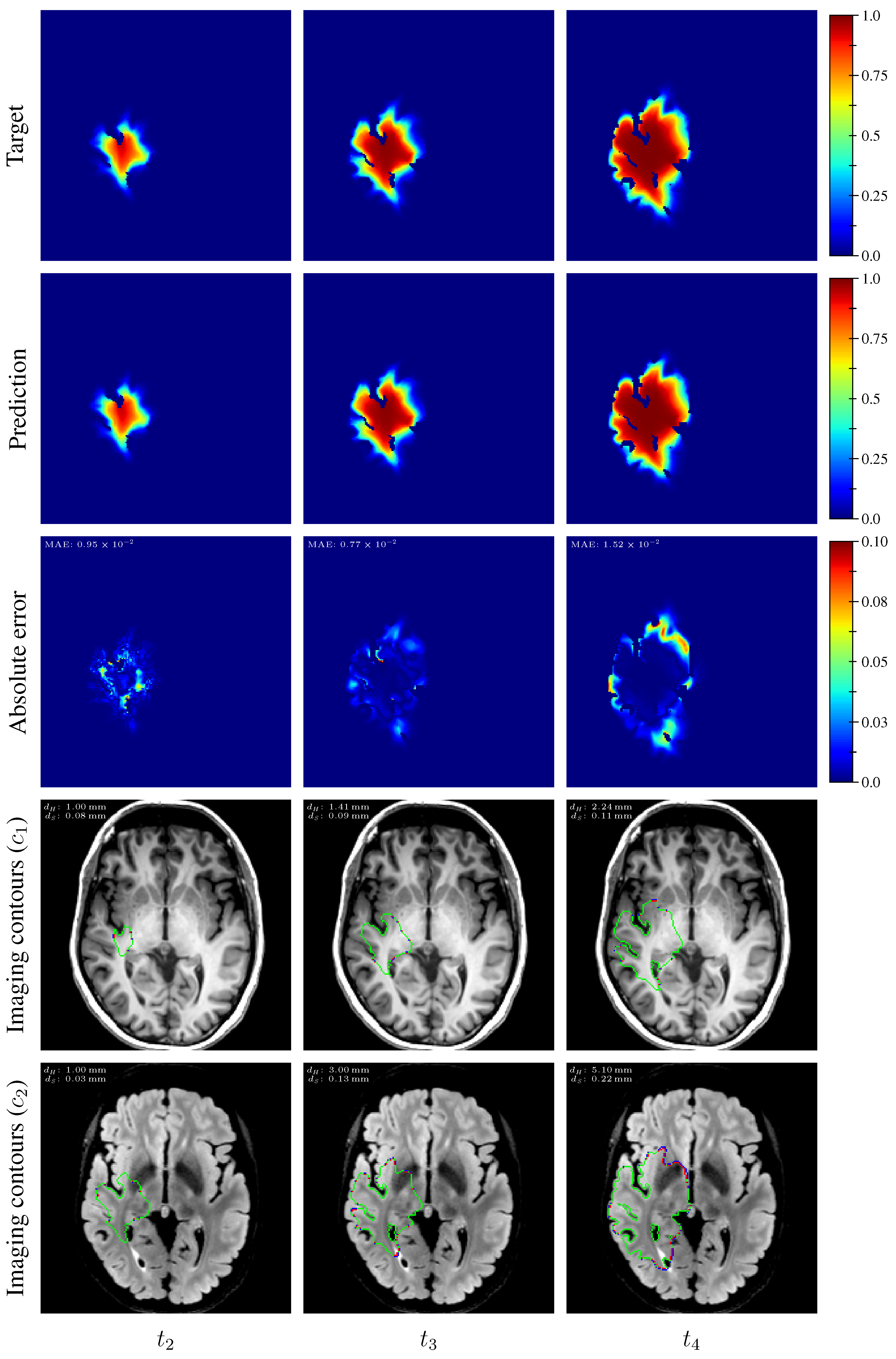
Table 3. An example of true and estimated tumor cell-density distributions at time
from the test set is depicted in
Figure 9 (first column), along with the corresponding absolute error map as well as the true and estimated imaging contours for threshold values
and
. Additional examples are provided in
Appendix C. All predicted tumor cell-density distributions at time
used in
Figure 7,
Figure 8 and
Figure 9 were provided by the first network (
Figure 5).
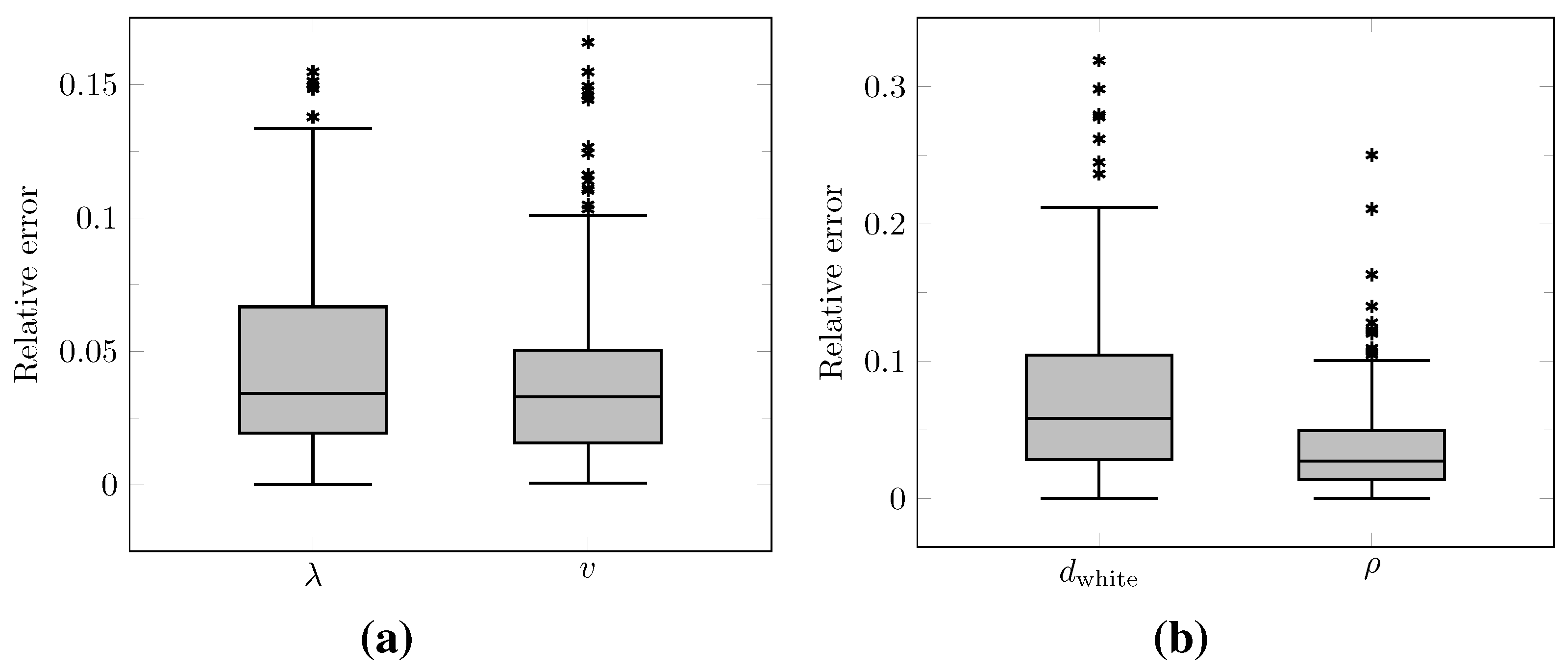
The distributions of the relative error on the values of
and
v computed at time
over the test set as well as on the values of
and
derived with Equations (12) and (13) are summarized by boxplots in
Figure 10. The corresponding median relative errors were 3.41%, 3.30%, 5.86%, and 2.75% for
,
v,
, and
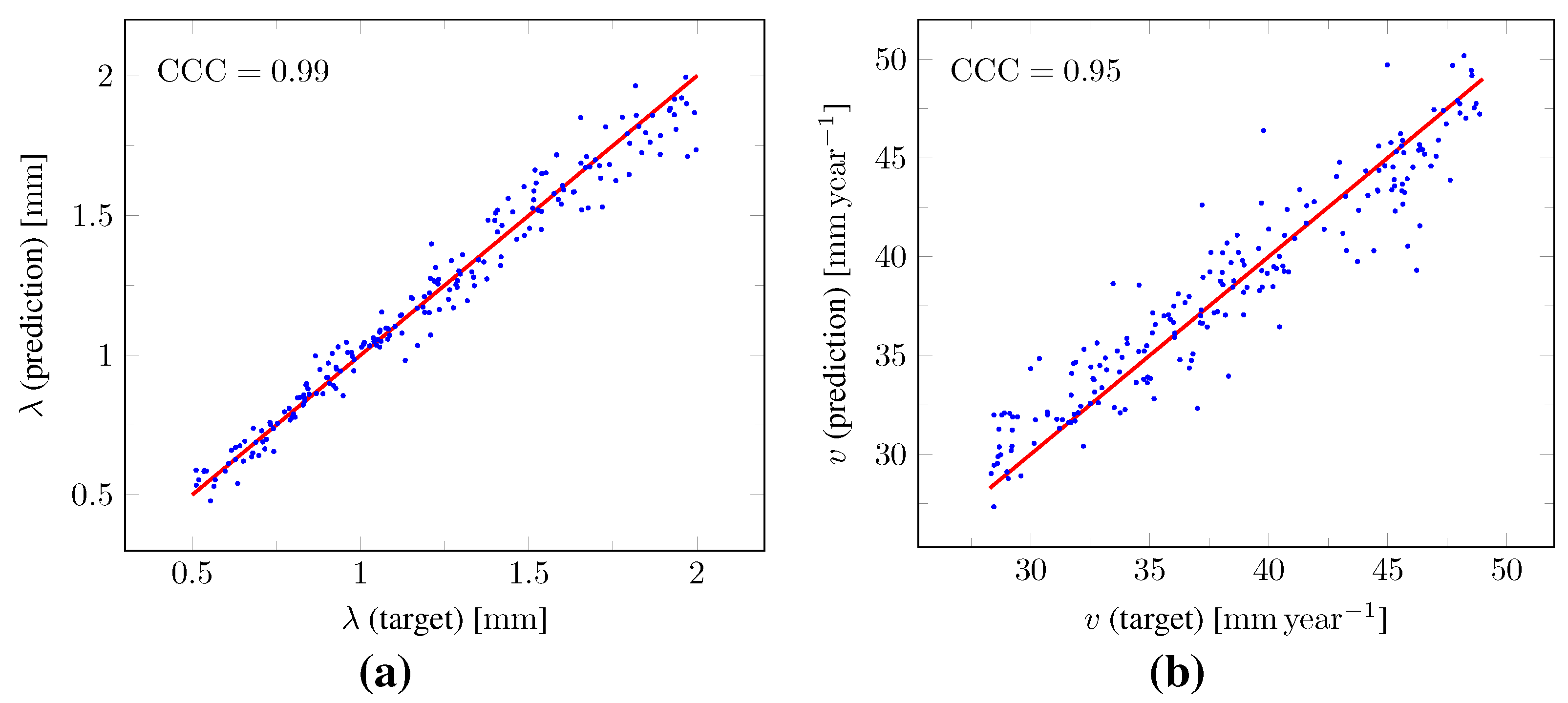
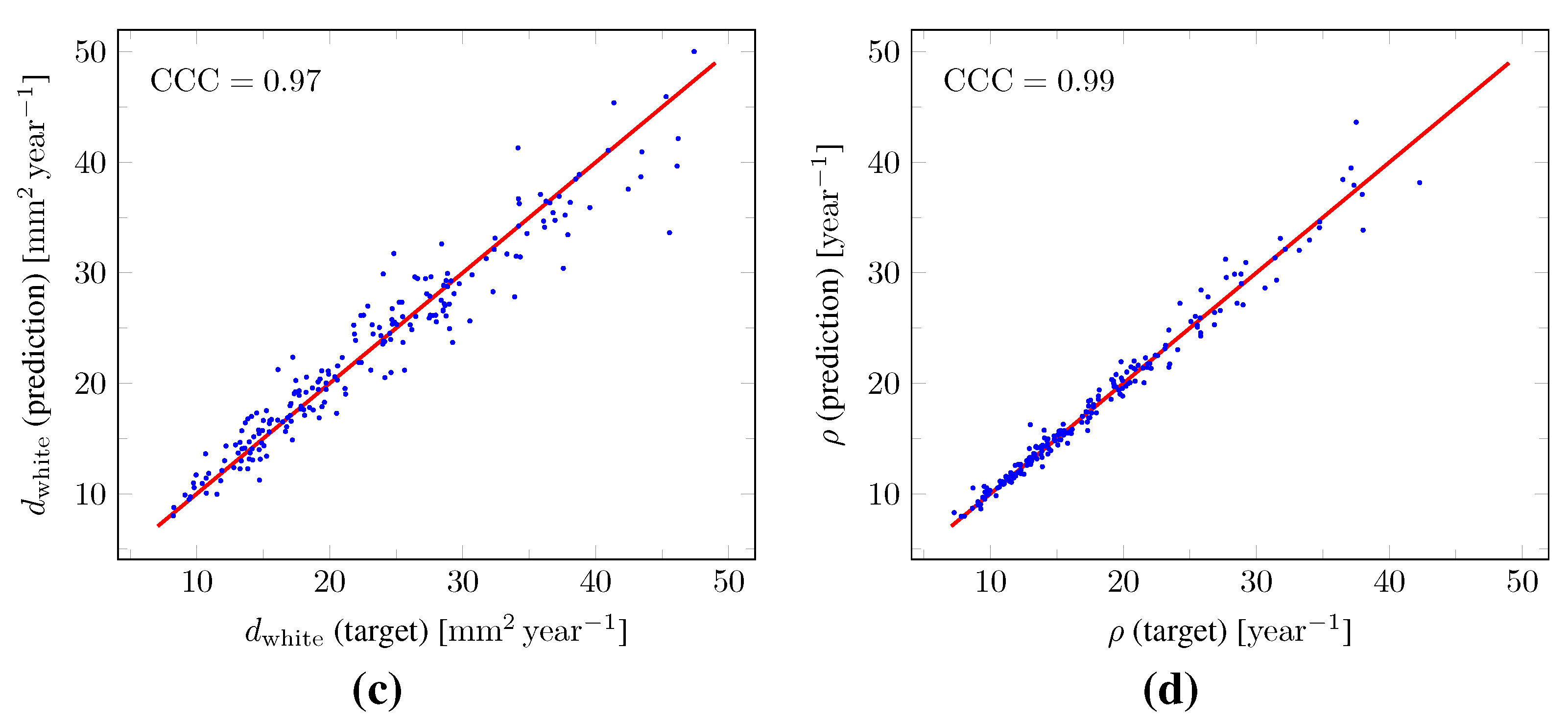
, respectively. The true versus predicted values of
and
v as well as of
and
from the test set are plotted in
Figure 11. The corresponding Lin’s concordance correlation coefficients (
) [
47] were 0.99, 0.95, 0.97, and 0.99 for
,
v,
, and
, respectively.
As for imaging time
, the distributions of the mean absolute error computed over the test set between the true and estimated tumor cell distributions at times
and
within the
contour are summarized by boxplots in
Figure 7 (second and third plot, respectively). Boxplots of the Hausdorff distance and ASSD distributions computed over the test set between the true and estimated imaging contours at times
and
for threshold values
and
are also provided in
Figure 8 (second and third plots). The corresponding median values are provided in
Table 3. The true and estimated tumor cell-density distributions at times
and
are depicted in
Figure 9 (second and third column, respectively) for the same test case as for time
, along with the corresponding absolute error maps as well as the true and estimated imaging contours for threshold values
and
. Additional examples are provided in
Appendix C. A loss of accuracy in the estimated tumor cell-density distributions over simulated time is observed in
Figure 7,
Figure 8 and
Figure 9 and
Table 3. The estimated tumor cell-density distributions at times
and
used in
Figure 7,
Figure 8 and
Figure 9 and
Table 3 were computed using the reaction-diffusion model as described in
Section 2.6 from (i) the cell-density distribution predicted at time
provided the first network (
Figure 5) and (ii) the predicted model parameter values provided by the second network (
Figure 6).
The results of the sensitivity analyses are summarized in
Table 4 and
Table 5. Performance indices are reported for all possible combinations of
perturbations on the
and
values used to generate the input
,
, and
imaging contours of both CNNs.
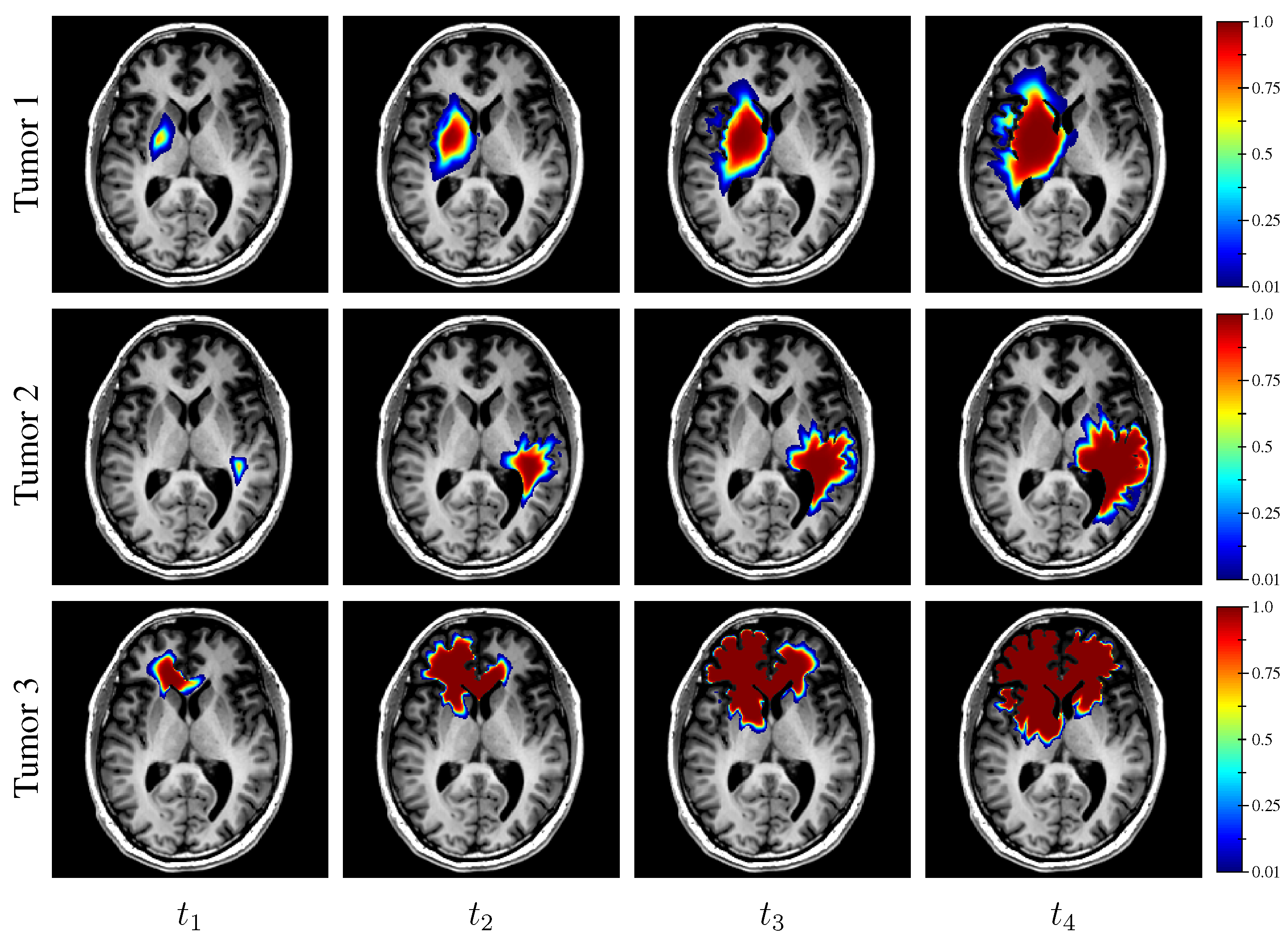
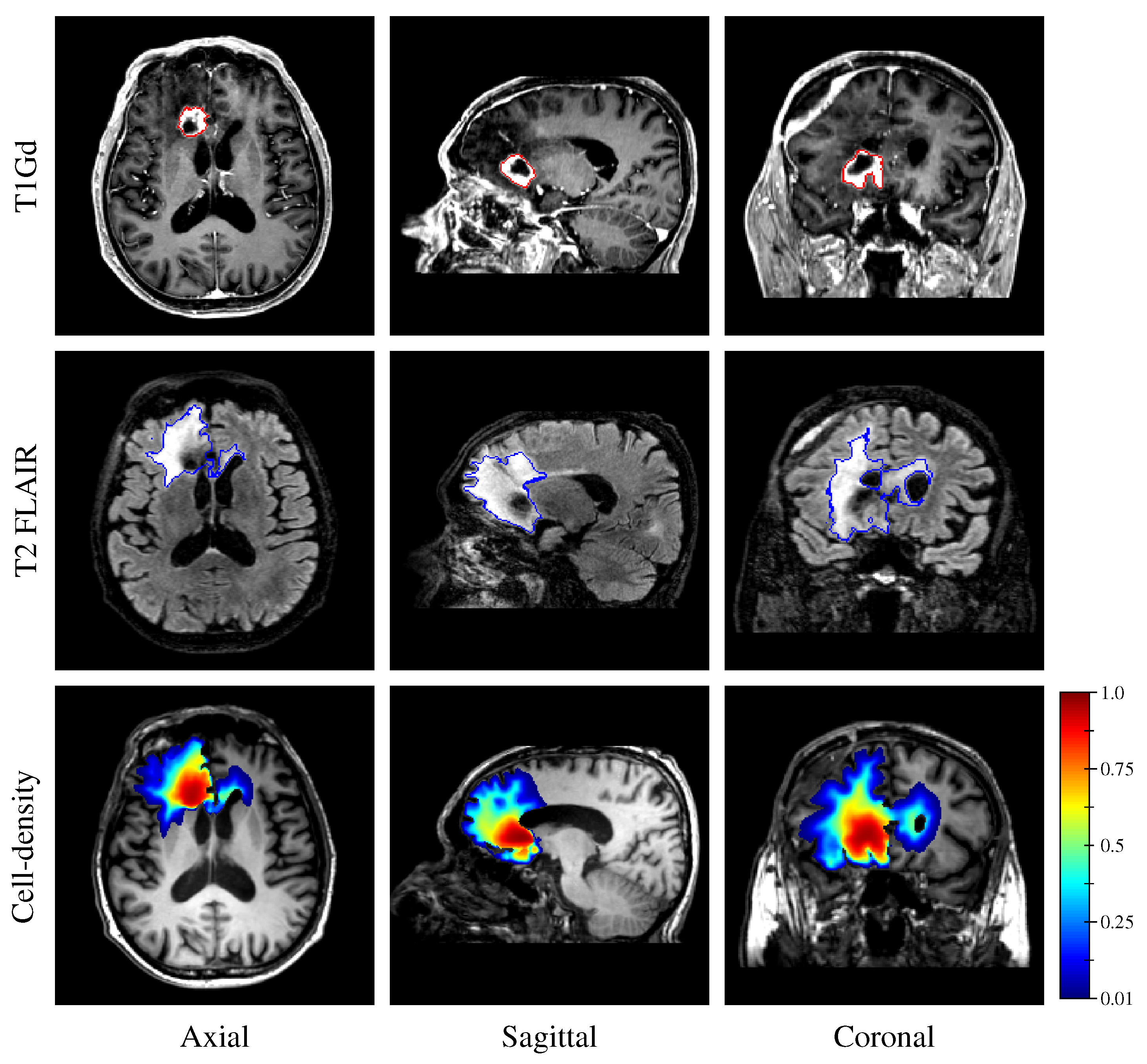
Finally, the estimated tumor cell-density distribution for the studied GBM patient provided by the first network (see
Figure 5) is depicted in
Figure 12 along with the T1Gd and T2 FLAIR images with superimposed segmented enhancing core and edema contours, respectively.
4. Discussion
Reaction-diffusion models have been studied for decades to capture the growth of gliomas, but the ill-posedness of their initialization at imaging time and estimation of their parameter values has restrained their use as a personalized predictive clinical tool. In this work, we showed the ability of DCNNs to circumvent these limitations, opening a wide range of opportunities in the field. Our approach only requires (i) deriving a unit diffusion tensor field from clinical DTI data as described herein, accounting for the preferential migration of tumor cells along white matter tracts and (ii) extracting three imaging contours obtained through a cell-density threshold-like process described by Equation (
2) for two different threshold values and time points.
Regarding the second requirement, the outlines of the peritumor vasogenic edema and enhancing core have been proposed previously [
8], visible on T2 FLAIR and T1Gd MR images acquired in routine for glioma follow-up, respectively. Nevertheless, it is worth noticing that peritumor vasogenic edema does not strictly speaking correspond to a region of tumor cell invasion but results from an accumulation of extracellular fluid originating from tumor-induced alterations of the blood–brain barrier [
48,
49] and changes in hydrodynamic pressure [
50]. Consequently, the T2 FLAIR imaging process might not be accurately described by Equation (
2), as also supported by our previous histological analysis in [
20]. Furthermore, anti-angiogenic drugs are known to dramatically reduce vasogenic edema without however stopping tumor progression [
48]. Therefore, other MR sequences or modalities could be better suited for the estimation of the tumor cell-density distribution and parameters of reaction-diffusion glioma growth models. For instance, ADC maps derived from DW-MRI data could more accurately reflect tumor cell invasion, as proposed in [
18,
51]. PET imaging with radio-labeled amino acids could also provide additional information to this extent, as suggested in [
26,
52].
Once the aforementioned prerequisites are met, our approach makes it possible to (i) extrapolate a whole brain-tumor cell-density distribution within and beyond the visible outlines of the tumor that is compatible with the reaction-diffusion model in Equations (4)–(6) and (ii) individually assess the value of the diffusion and proliferation parameters of the model. Extrapolating tumor invasion is of utmost interest for radiotherapy planning since it would allow us to define personalized margins which more accurately target the tumor while avoiding irradiation of the healthy tissues, as previously discussed in [
2,
10]. The independent assessment of the diffusivity and proliferation parameters of the model is for its part of great interest to better characterize the tumor [
22]. The combination of both gives access to a fully personalized tool, initialized from clinical imaging data and allowing us to anticipate the spatial–temporal growth of gliomas. Such a tool could, for example, be of considerable interest for dose fractionation optimization in radiotherapy using a reinforcement learning approach, as used in [
53]. Furthermore, as it only depends on post-processed data (binary segmentations and a DTI-derived water diffusion tensor) rather than raw MR data, the proposed approach may be robustly extended to other scanners and centers. In addition, the method is by design robust to variations in the time interval between the two required MR acquisitions since the interval is provided as an input of the second network for the estimation of the model parameters, which makes it well-adapted to the clinical reality.
The proposed method was found to provide accurate estimations of the three-dimensional tumor cell distribution from only two imaging contours at a single time point, with a median voxelwise MAE below 10
−2 within the
contour—as evaluated on 200 synthetic tumors grown over the real brain domain of a test subject not used for network training. Our method also provided accurate estimates of the individual diffusion and proliferation parameters of the model from three imaging contours extracted from two time points for the same test tumors, with median relative errors of 5.86% and 2.75%, respectively (see
Figure 10), and strong concordance (
with the true parameter values (see
Figure 11). Furthermore, we showed that the spatio-temporal evolution of the tumor cell-density distribution at later time points (90 d and 180 d later) can be accurately captured from the estimated distribution at imaging time and parameter values using the reaction-diffusion model. The ASSD between the true and estimated imaging contours obtained for threshold values of
and
were indeed found to be lower than or equal to the pixel spacing (
) in most cases (see
Figure 8). Nevertheless, a loss of accuracy in the estimated tumor cell-density over simulated time was observed (see
Figure 7,
Figure 8 and
Figure 9 and
Table 3), imputed to the amplification of errors originating from uncertainties in the estimated model parameter values and tumor cell-density distribution at imaging time. In particular, artefactual local maxima in the tumor cell-density distributions predicted by the CNN were found to give rise to new tumor foci over time. Post-processing steps were introduced to circumvent these effects (see
Section 2.6), but residual artefacts were still observed, resulting in a large Hausdorff distance though small ASSD values for a few isolated cases (see outliers in
Figure 8a,b). Our approach was also found to be robust to uncertainties in the tumor cell-density threshold values defining the input imaging contours of both CNNs. Indeed, all combinations of
perturbations on both threshold values used to generate the contours resulted in an increase in median relative error within reasonable ranges of 14.82–22.39% and 10.25–17.79% for the diffusivity and proliferation rate, respectively. Finally, we also demonstrated the applicability of our proposed method to actual MR data of a GBM patient, for which we were able to reconstruct a tumor cell-density distribution compatible with the imaging data. Nevertheless, the lack of biopsy samples combined with the multiple treatments undergone by the patient prevented the validation of the estimated distribution, which was left for a future prospective study.
Compared to the current state-of-the-art approach in [
27], our method appears to perform at least as well or better in most cases on the model parameter estimation problem, by comparison of the reported relative errors on the parameter values in [
27]. Nevertheless, both methods were not evaluated on the same synthetic tumor dataset and no open-source code was available in [
27] for further comparison. Besides, once trained, the CNNs used in this work no longer depend on any arbitrary parameters, as opposed to the method in [
27] for which adequate sparsity level and observation operator weighting need to be selected, in addition to the many parameters involved in the numerical scheme (Gaussian standard deviation, coarsening levels, initial solution guesses, tolerance thresholds, termination criteria, …) [
27,
29]. Our method also performs significantly faster: parameter estimation on a single case is performed within around
versus 1000
for [
27], which however remains largely sufficient for the application in both cases. Furthermore, the proposed approach allows for absolute parameter estimation, as opposed to [
27] which only provides non-dimensionalized estimates that could not be scaled as the time between tumor emergence and imaging is actually not known. Consequently, comparison of the estimated parameter values between tumors or their application to predict tumor evolution over time using the model are prevented. Nevertheless, contrary to [
27], our method requires two imaging time points to compensate for the ill-posedness of the problem and to allow for dimensionalized parameter estimation—but in return makes no explicit assumption on the initial tumor cell distribution. This latter requirement of our approach implies that the tumor diffusivity and proliferation rate remain constant between the scans—which is in any case also implicitly assumed between the tumor emergence and the single scan time for the forward problem described in [
27]. As a consequence, our method is expected to be sensitive to any treatment administered between the scans that would significantly impact the tumor model parameters (chemotherapy, radiotherapy) or solving domain (surgery). Finally, as opposed to [
27], our method considers a spatially variable tumor cell diffusion tensor accounting for the preferential migration of tumor cells along white matter tracts, but is restricted to monofocal tumors in the present form.
As a future work, tumor-induced mass effect should be further integrated into the reaction-diffusion model since it is known to cause substantial deformations of the brain parenchyma and distortions of the white matter tracts as the tumor grows, which should also be taken into account for accurate treatment planning. Such effects have been previously considered [
6,
7], but would introduce additional parameters to be estimated. In addition, transient brain deformations would hardly be integrated into a regular grid-based approach such as the finite difference method used in this work without loss of precision. A finite element formulation over an unstructured mesh could be used instead but would be much more computationally expensive—hence less suited for the generation of large high-resolution datasets such as the one described herein. Tumor-induced destruction of the white matter tracts should also be further considered, as an accurate capture of the original orientation of the brain fibers within the tumor region is required for the evaluation of the cell-density distribution and model parameter values using both DCNNs. A solution to this problem has been previously proposed in [
25]—though subject to limitations—in which symmetry of the brain is exploited to artificially reconstruct the missing brain fibers. More advanced methods should be investigated in this sense. Necrosis could also be integrated into the model as proposed in [
15,
16], which would have avoided the counter-intuitive correspondence between the hyper-dense (
) region of the estimated tumor cell-density distribution and the necrotic area visible on MRI in
Figure 12. Furthermore, the deep neural networks presented herein remain little flexible as they would need to be retrained if different imaging threshold values were considered, although transfer learning could be used to benefit from the lower-level features learned herein and avoid retraining the networks from scratch [
54]. Ultimately, the threshold values could be fed to the networks along with the binary contours, but this would make the problem even more complex and would therefore require an even larger training dataset. Although real medical imaging data were used in this work, the verification of our approach still relied on healthy subject data. Therefore, the underlying hypothesis was made that the reaction-diffusion model defined by Equation (4)–(6) and used for tumor synthesis is indeed able to accurately capture the growth of real gliomas, which has never been extensively demonstrated so far to the best of our knowledge. Validation of our approach on actual glioma patient data should be further performed, but longitudinal imaging data with stereotactic biopsies of untreated glioma patients remain scarce. Including the effects of treatments into reaction-diffusion models has also been proposed previously [
4,
5,
17,
18,
19], but again introduces additional parameters, increasing the complexity of the problem. Alternately, the method could be applied to large publicly available datasets such as the BRaTS dataset [
28]. Whereas ground-truth cell-density distributions and model parameter values are unknown for such datasets, indirect validation by investigation of the predictive performance of the estimated model parameters for tumor bio-markers could still be performed, as attempted in [
29].
This work also highlights the added value of DCNNs for the resolution of ill-posed problems that are hardly solved by classical optimization methods, and provides encouraging results towards the full personalization of reaction-diffusion glioma growth models from medical imaging data, which has remained unsolved for decades.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}