
Gene Expression Monotonicity across Bladder Cancer Stages Informs on the Molecular Pathogenesis and Identifies a Prognostic Eight-Gene Signature

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
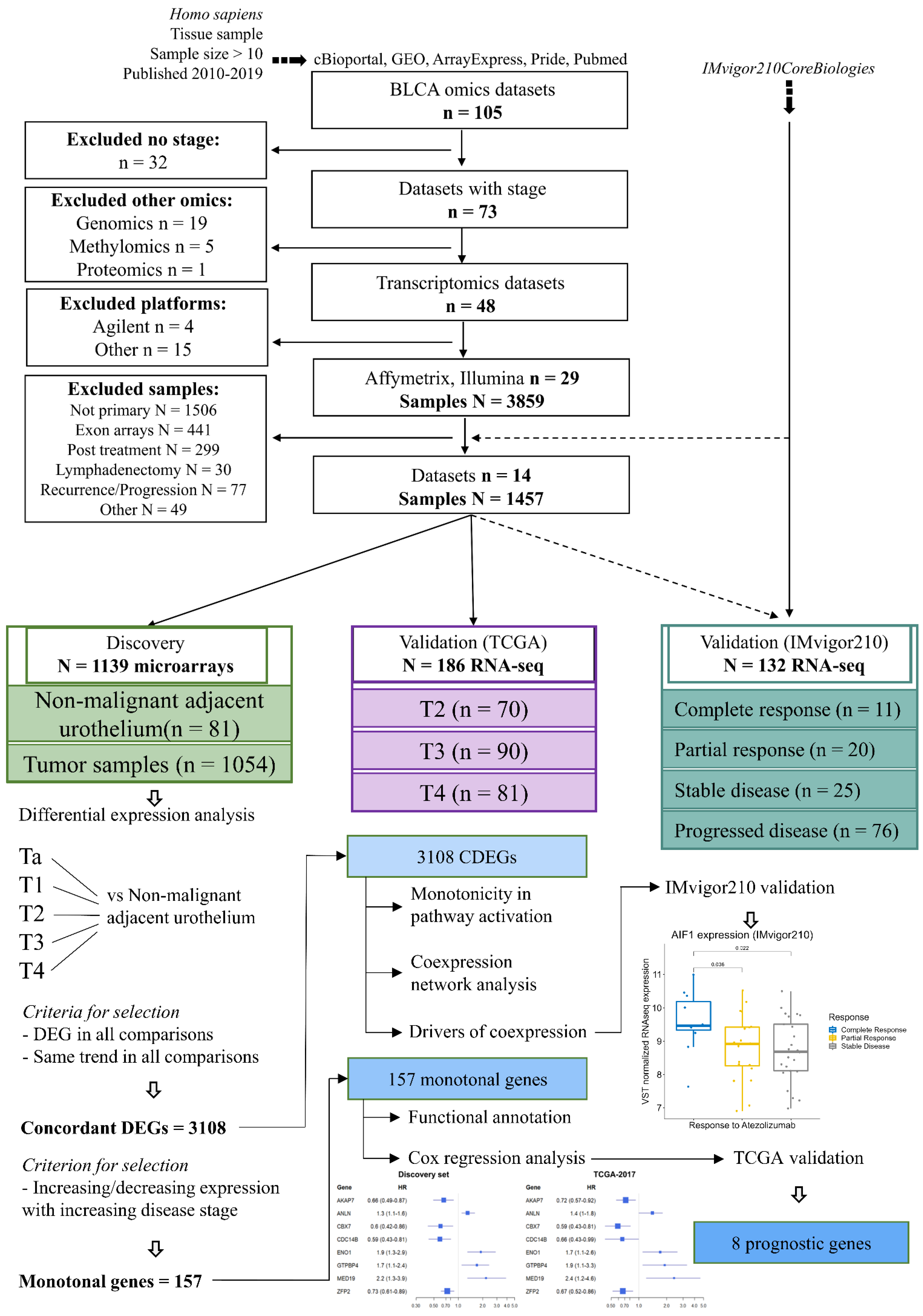
2. Materials and Methods
3. Results
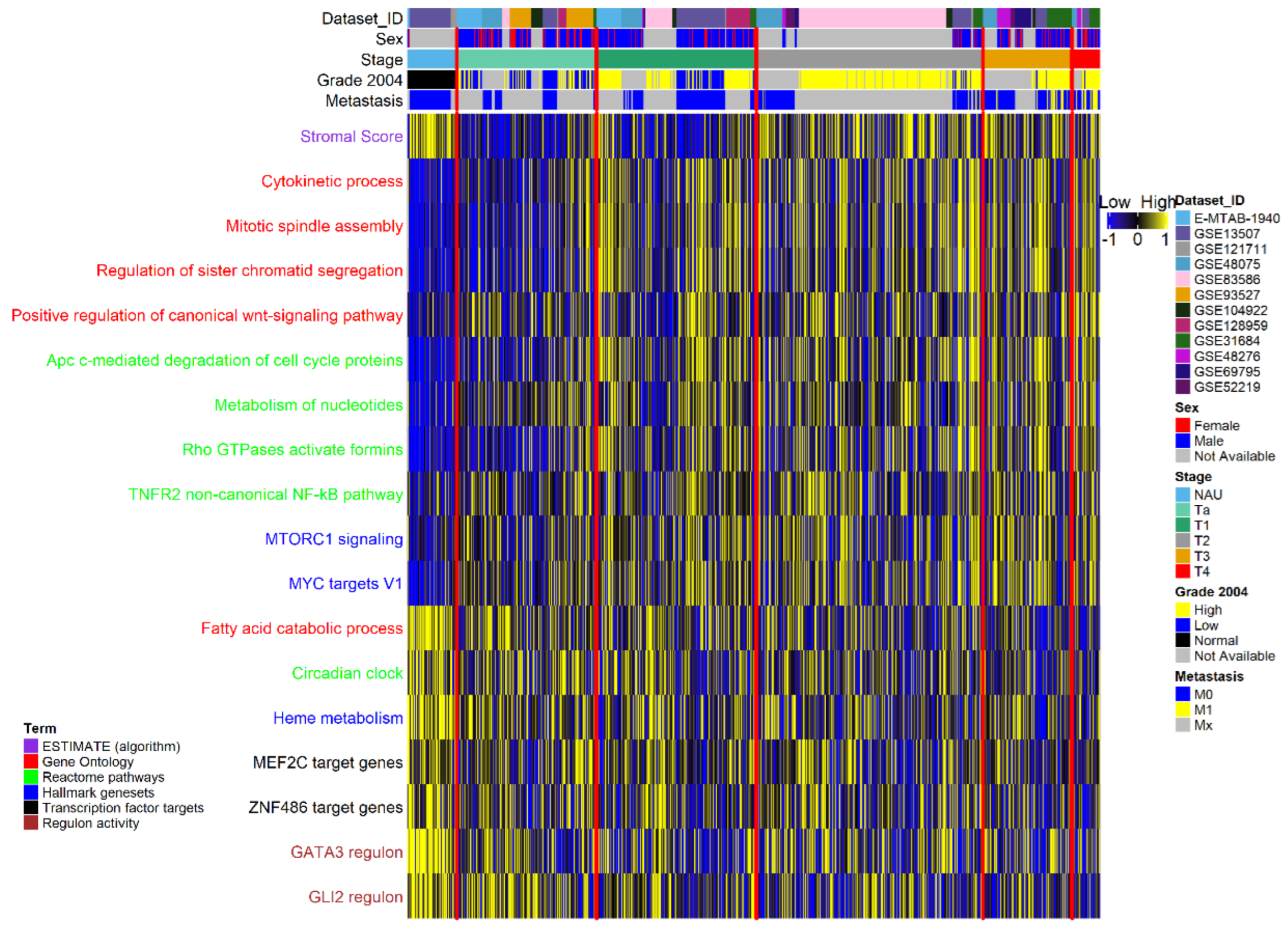
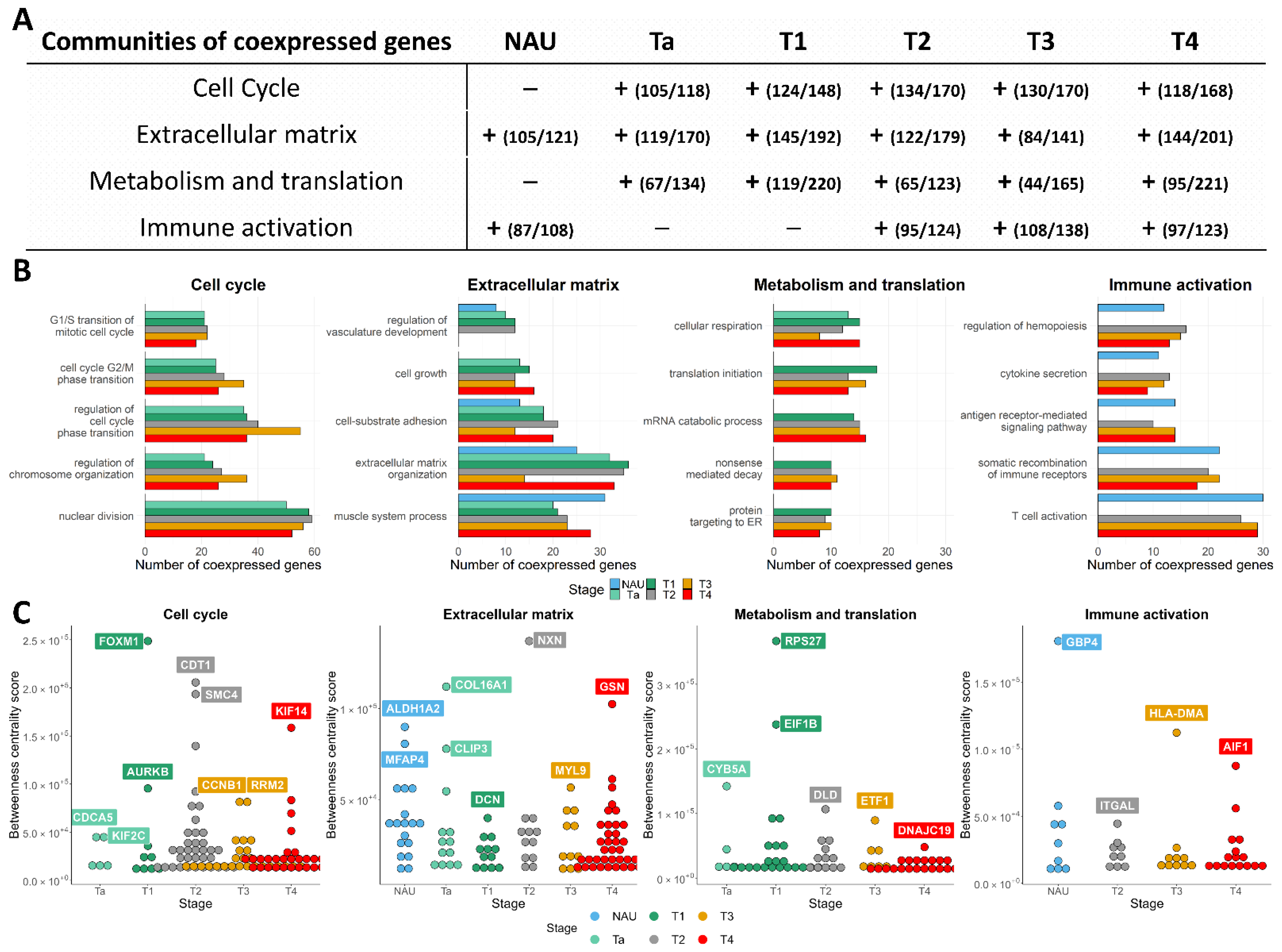
3.1. Pathway Activation and Gene Co-Expression Link the Cell Cycle to ECM and Metabolic Alterations
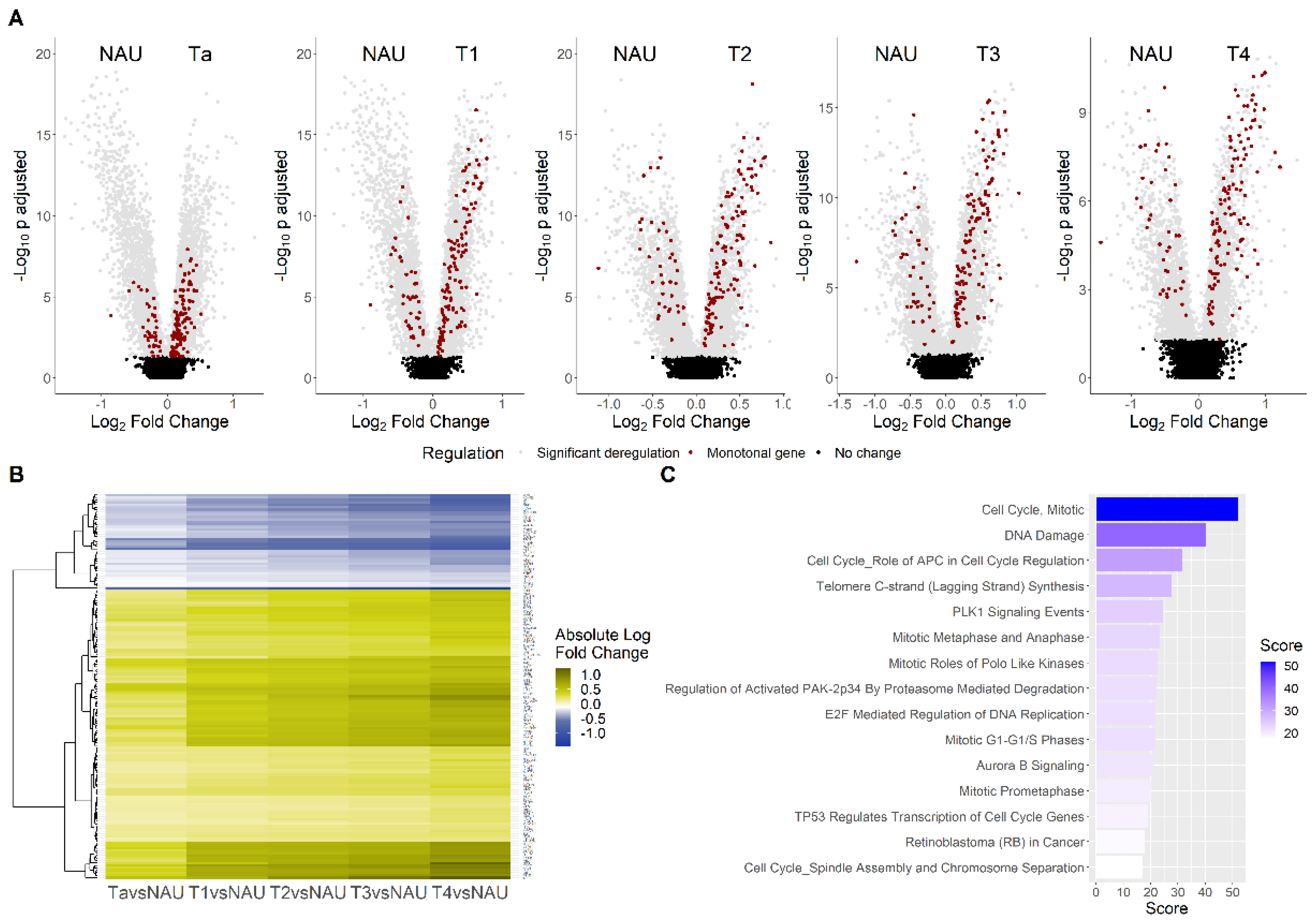
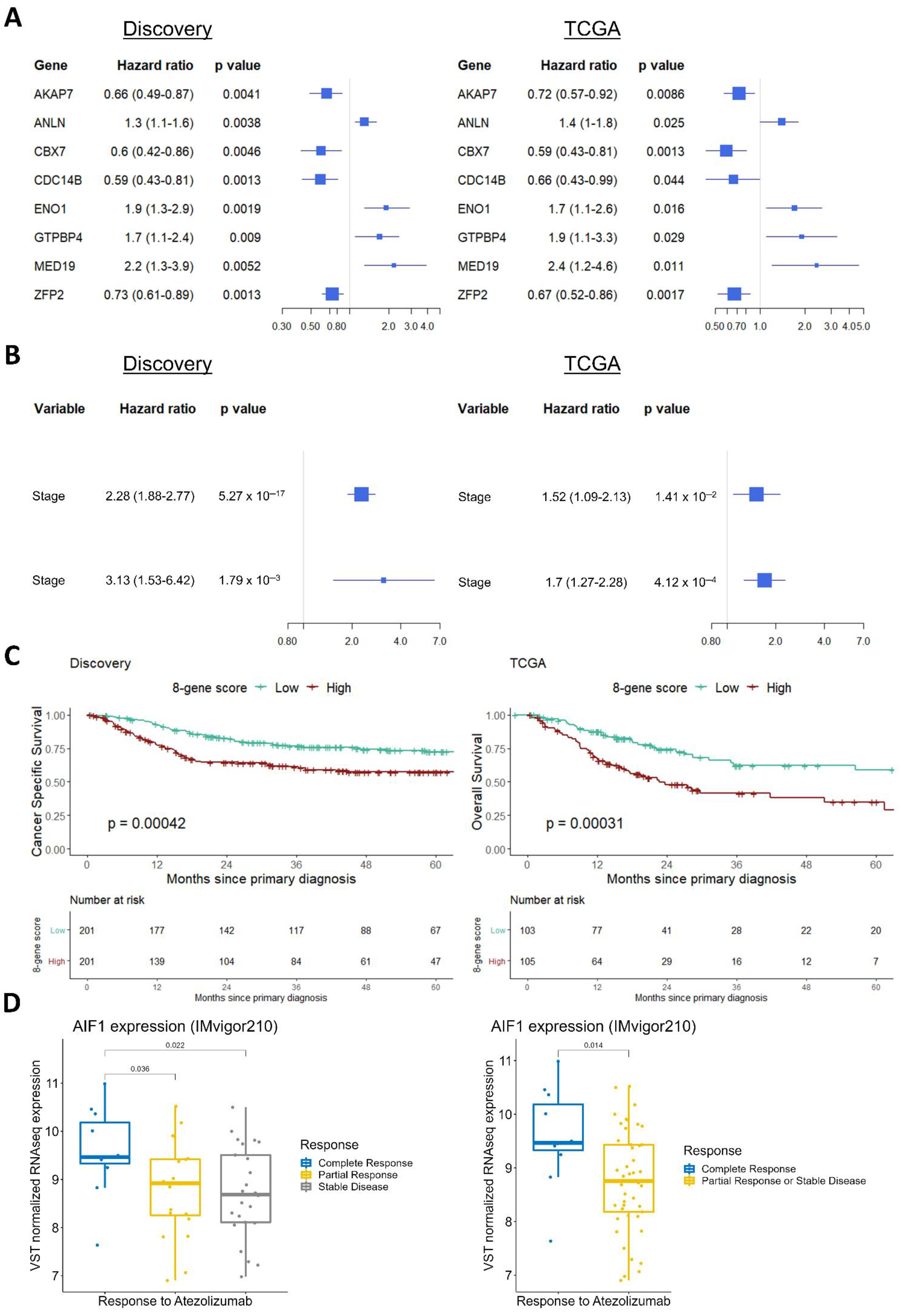
3.2. Monotonicity in Individual Genes, Prognostic Signatures, and Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Database Search Strategy, Cohorts, and Processing
Appendix A.2. Inclusion and Exclusion Criteria
Appendix A.3. Cohorts
- GSE121711 [64]: Loras A, et al., 2019. Cancers (Basel): Study of the metabolome and transcriptome of bladder cancer patients, aiming to identify tissue and urinary metabolic signatures as biomarkers of BC. Contains 8 tumoral and 10 normal urothelium samples taken from regions adjacent to the tumor (NAU).
- GSE93527 [65]: Hurst CD, et al., 2017. Cancer Cell: Subtyping of Ta-staged non-muscle-invasive bladder cancer patients based on copy number alterations, and subsequent expression profiling of 79 samples.
- E-MTAB-1940 [66]: Biton A, et al., 2014. Cell Reports: Characterization of the luminal and basal muscle-invasive bladder cancer subtypes based on an independent component analysis of 86 transcriptomes.
- GSE31684 [67]: Riester M, et al., 2012. Clinical Cancer Research: A study for the identification and validation of high-risk bladder cancer. It included 93 samples, taken from patients undergoing radical cystectomy (RC) at the Memorial Sloan-Kettering Cancer Center (MSKCC), of which 3 were collected post-treatment and were excluded from the analysis.
- GSE104922 [68]: Therkildsen C, et al., 2018. Molecular Oncology: Identification of molecular profiles in 41 bladder cancer patients with Lynch syndrome.
- GSE128959 [69]: Sjödahl G, et al., 2020. International Journal of Cancer: An analysis of multiple samples taken from 73 patients upon recurrence/progression, investigating transcriptional alterations and subtype shifts during disease development. Expression profiles are available for 55 bladder tumors, of which 2 were excluded due to missing stage information.
- GSE83586 [70]: Sjödahl G, et al., 2017. The Journal of Pathology: This study utilizes tumor samples from 307 advanced bladder cancer patients in order to compare molecular classification to tumor cell phenotype (the latter assessed via immunohistochemistry). We excluded six samples due to the absence of stage information.
- GSE48276 [71]: Choi W, et al., 2014. Cancer Cell: The MDA molecular classification of muscle-invasive bladder cancer. The GEO dataset contains 116 expression profiles, 16 of which were collected post-treatment and, thus, excluded. An additional set of 20 profiles had no information on the timing of sample collection, and were also excluded.
- GSE52219 [71]: Choi W, et al., 2014. Cancer Cell: The MDA molecular classification of muscle-invasive bladder cancer. The GEO dataset contains 23 FFPE pretreatment tumors from a phase III clinical trial, which were used to validate the MDA classification.
- GSE69795 [72]: McConkey, et al., 2016. European Urology: Investigation of gene expression signatures predicting response to methotrexate, vinblastine, doxorubicin, and cisplatin with bevacizumab. The GEO dataset contains 61 profiles, of which 23 and 1 were excluded due to post-treatment collection and the absence of stage information, respectively.
- GSE13507 [73]: Kim WJ, et al., 2010. Journal of Clinical Oncology: Analysis of primary bladder cancer for the identification of a prognostic gene expression classifier. The GEO dataset contains 256 profiles, of which 9 are normal bladders, 58 are NAU, 1 profile is taken from a C57BL/6 mouse sub-strain, 165 are primary tumor samples, and 23 correspond to recurrences. Due to the low number of normal bladder samples represented in the compiled discovery cohort, we grouped them together with the NAU samples. Samples collected after recurrence (n = 23), as well as the expression profile of the murine strain, were excluded from analysis.
- GSE48075 [71]: Choi W, et al., 2014. Cancer Cell: The MDA molecular classification of muscle-invasive bladder cancer. It includes 142 primary tumors of both non-muscle-invasive and muscle-invasive bladder cancer.
- TCGA-BLCA-2017 [42]: Robertson AG, et al., 2017. Cell: The TCGA molecular subtyping of muscle-invasive bladder cancer. Molecular and clinical data were downloaded from cBioPortal, containing 412 tumor samples. Of these, 109 had history of other malignancies, and were excluded as potentially being secondary bladder cancers. Additionally, 105 samples had unknown history of prior treatment for non-muscle-invasive bladder cancer, 2 samples were collected post-NAC, and 8 more samples had no stage information, so they were all excluded from further analysis (n analyzed = 186 samples).
- IMvigor210 [34]: Mariathasan S, et al., 2018. Nature: A study investigating molecular determinants of tumor response to atezolizumab immunotherapy, administered to metastatic bladder cancer patients. Transcriptomic profiles are available on GitHub (R package “IMvigor210CoreBiologies”) for 298 patients, from which we analyzed only samples collected pre-NAC (n = 132 samples).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technology | Platform | Dataset ID | NAU | Ta | T1 | T2 | T3 | T4 | Study Size |
|---|---|---|---|---|---|---|---|---|---|
| Affymetrix | GPL17586 | GSE121711 | 10 | 3 | 2 | 3 | 18 | ||
| Affymetrix | GPL17586 | GSE93527 | 79 | 79 | |||||
| Affymetrix | GPL570 | E-MTAB-1940 | 4 | 41 | 41 | 86 | |||
| Affymetrix | GPL570 | GSE31684 | 5 | 10 | 16 | 41 | 18 | 90 | |
| Affymetrix | GPL6244 | GSE104922 | 19 | 7 | 10 | 5 | 0 | 41 | |
| Affymetrix | GPL6244 | GSE128959 | 13 | 39 | 53 | ||||
| Affymetrix | GPL6244 | GSE83586 | 13 | 44 | 241 | 1 | 1 | 301 | |
| Illumina | GPL14951 | GSE48276 | 2 | 6 | 23 | 6 | 37 | ||
| Illumina | GPL14951 | GSE52219 | 15 | 7 | 1 | 23 | |||
| Illumina | GPL14951 | GSE69795 | 3 | 7 | 27 | 37 | |||
| Illumina | GPL6102 | GSE13507 | 67 | 23 | 80 | 31 | 19 | 12 | 232 |
| Illumina | GPL6947 | GSE48075 | 33 | 34 | 42 | 23 | 8 | 142 | |
| Stage size | 81 | 229 | 262 | 371 | 146 | 46 | 1135 |
Appendix A.4. Integration and Processing of the Transcriptomes
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, P.A.; Moch, H.; Cubilla, A.L.; Ulbright, T.M.; Reuter, V.E. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part B: Prostate and Bladder Tumours. Eur. Urol. 2016, 70, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magers, M.J.; Lopez-Beltran, A.; Montironi, R.; Williamson, S.R.; Kaimakliotis, H.Z.; Cheng, L. Staging of bladder cancer. Histopathology 2019, 74, 112–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2014, 15, 25–41. [Google Scholar] [CrossRef]
- Messing, E.M. Financial Toxicity of Having Bladder Cancer. Bladder Cancer 2018, 4, 351–352. [Google Scholar] [CrossRef] [Green Version]
- Casilla-Lennon, M.M.; Choi, S.K.; Deal, A.M.; Bensen, J.T.; Narang, G.; Filippou, P.; McCormick, B.; Pruthi, R.; Wallen, E.; Tan, H.J.; et al. Financial Toxicity among Patients with Bladder Cancer: Reasons for Delay in Care and Effect on Quality of Life. J. Urol. 2018, 199, 1166–1173. [Google Scholar] [CrossRef]
- Batista, R.; Vinagre, N.; Meireles, S.; Vinagre, J.; Prazeres, H.; Leao, R.; Maximo, V.; Soares, P. Biomarkers for Bladder Cancer Diagnosis and Surveillance: A Comprehensive Review. Diagnostics 2020, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Dyrskjot, L.; Reinert, T.; Algaba, F.; Christensen, E.; Nieboer, D.; Hermann, G.G.; Mogensen, K.; Beukers, W.; Marquez, M.; Segersten, U.; et al. Prognostic Impact of a 12-gene Progression Score in Non-muscle-invasive Bladder Cancer: A Prospective Multicentre Validation Study. Eur. Urol. 2017, 72, 461–469. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Wang, J.; Li, L.; Bi, J. Identification of a Prognosis-Related Risk Signature for Bladder Cancer to Predict Survival and Immune Landscapes. J. Immunol. Res. 2021, 2021, 3236384. [Google Scholar] [CrossRef]
- Dong, B.; Liang, J.; Li, D.; Song, W.; Song, J.; Zhu, M.; Zhao, S.; Ma, Y.; Yang, T. Identification of a Prognostic Signature Associated With the Homeobox Gene Family for Bladder Cancer. Front. Mol. Biosci. 2021, 8, 688298. [Google Scholar] [CrossRef]
- Qu, G.; Liu, Z.; Yang, G.; Xu, Y.; Xiang, M.; Tang, C. Development of a prognostic index and screening of prognosis related genes based on an immunogenomic landscape analysis of bladder cancer. Aging 2021, 13, 12099–12112. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Hong, X.; Yu, C.; Li, H.; Wang, Q.; Zhang, Y.; Wang, T.; Wang, X. Preclinical analysis of novel prognostic transcription factors and immune-related gene signatures for bladder cancer via TCGA-based bioinformatic analysis. Oncol. Lett. 2021, 21, 344. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Yuan, L.; Ma, B.; Wang, G.; Qiu, W.; Tian, Y. An EMT-related gene signature for the prognosis of human bladder cancer. J. Cell. Mol. Med. 2019, 24, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.Q.; Zhou, X.G.; Yi, Q.Y.; Deng, C.W.; Gao, J.M.; Chen, G.; Wang, Q.Y. Prognostic Signature of Alternative Splicing Events in Bladder Urothelial Carcinoma Based on Spliceseq Data from 317 Cases. Cell. Physiol. Biochem. 2018, 48, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Cumberbatch, M.G.K.; Jubber, I.; Black, P.C.; Esperto, F.; Figueroa, J.D.; Kamat, A.M.; Kiemeney, L.; Lotan, Y.; Pang, K.; Silverman, D.T.; et al. Epidemiology of Bladder Cancer: A Systematic Review and Contemporary Update of Risk Factors in 2018. Eur. Urol. 2018, 74, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, J.M. The origins of bladder cancer. Lab. Investig. 2008, 88, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.; Diaz, E.; Reya, T. Stem cells in cancer initiation and progression. J. Cell Biol. 2019, 219, e201911053. [Google Scholar] [CrossRef]
- Czerniak, B.; Dinney, C.; McConkey, D. Origins of Bladder Cancer. Annu. Rev. Pathol. 2016, 11, 149–174. [Google Scholar] [CrossRef]
- Fong, M.H.Y.; Feng, M.; McConkey, D.J.; Choi, W. Update on bladder cancer molecular subtypes. Transl. Androl. Urol. 2020, 9, 2881–2889. [Google Scholar] [CrossRef]
- Warrick, J.I.; Sjodahl, G.; Kaag, M.; Raman, J.D.; Merrill, S.; Shuman, L.; Chen, G.; Walter, V.; DeGraff, D.J. Intratumoral Heterogeneity of Bladder Cancer by Molecular Subtypes and Histologic Variants. Eur. Urol. 2019, 75, 18–22. [Google Scholar] [CrossRef]
- Manimaran, S.; Selby, H.M.; Okrah, K.; Ruberman, C.; Leek, J.T.; Quackenbush, J.; Haibe-Kains, B.; Bravo, H.C.; Johnson, W.E. BatchQC: Interactive software for evaluating sample and batch effects in genomic data. Bioinformatics 2016, 32, 3836–3838. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Duron, C.; Pan, Y.; Gutmann, D.H.; Hardin, J.; Radunskaya, A. Variability of Betweenness Centrality and Its Effect on Identifying Essential Genes. Bull. Math. Biol. 2019, 81, 3655–3673. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Wullweber, A.; Strick, R.; Lange, F.; Sikic, D.; Taubert, H.; Wach, S.; Wullich, B.; Bertz, S.; Weyerer, V.; Stoehr, R.; et al. Bladder Tumor Subtype Commitment Occurs in Carcinoma In Situ Driven by Key Signaling Pathways Including ECM Remodeling. Cancer Res. 2021, 81, 1552–1566. [Google Scholar] [CrossRef]
- Yang, J.; Song, H.; Chen, L.; Cao, K.; Zhang, Y.; Li, Y.; Hao, X. Integrated analysis of microfibrillar-associated proteins reveals MFAP4 as a novel biomarker in human cancers. Epigenomics 2019, 11, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lapointe, J.; Kaygusuz, G.; Ong, D.E.; Li, C.; van de Rijn, M.; Brooks, J.D.; Pollack, J.R. The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer Res. 2005, 65, 8118–8124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Du, K. ClipR-59 interacts with Akt and regulates Akt cellular compartmentalization. Mol. Cell. Biol. 2009, 29, 1459–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Egana-Gorrono, L.; Chinnasamy, P.; Casimiro, I.; Almonte, V.M.; Parikh, D.; Oliveira-Paula, G.H.; Jayakumar, S.; Law, C.; Riascos-Bernal, D.F.; Sibinga, N.E.S. Allograft inflammatory factor-1 supports macrophage survival and efferocytosis and limits necrosis in atherosclerotic plaques. Atherosclerosis 2019, 289, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Roh, Y.G.; Kim, S.K.; Lee, J.S.; Seol, S.Y.; Lee, H.H.; Kim, W.T.; Kim, W.J.; Heo, J.; Cha, H.J.; et al. Activation of EZH2 and SUZ12 Regulated by E2F1 Predicts the Disease Progression and Aggressive Characteristics of Bladder Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5391–5403. [Google Scholar] [CrossRef] [Green Version]
- Kanehira, M.; Harada, Y.; Takata, R.; Shuin, T.; Miki, T.; Fujioka, T.; Nakamura, Y.; Katagiri, T. Involvement of upregulation of DEPDC1 (DEP domain containing 1) in bladder carcinogenesis. Oncogene 2007, 26, 6448–6455. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.L.; Liu, J.S.; Wu, P.L.; Guan, H.H.; Chen, Y.L.; Lin, A.C.; Ting, H.J.; Pang, S.T.; Yeh, S.D.; Ma, W.L.; et al. Identification of a new androgen receptor (AR) co-regulator BUD31 and related peptides to suppress wild-type and mutated AR-mediated prostate cancer growth via peptide screening and X-ray structure analysis. Mol. Oncol. 2014, 8, 1575–1587. [Google Scholar] [CrossRef]
- Kawahara, T.; Shareef, H.K.; Aljarah, A.K.; Ide, H.; Li, Y.; Kashiwagi, E.; Netto, G.J.; Zheng, Y.; Miyamoto, H. ELK1 is up-regulated by androgen in bladder cancer cells and promotes tumor progression. Oncotarget 2015, 6, 29860–29876. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.S.; Chen, X.; Guo, L.Y.; Li, X.D.; Deng, M.H.; Yuan, G.J.; He, L.Y.; Li, Y.H.; Zhang, Z.L.; Jiang, L.J.; et al. TRIM65 supports bladder urothelial carcinoma cell aggressiveness by promoting ANXA2 ubiquitination and degradation. Cancer Lett. 2018, 435, 10–22. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.W.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.Z.; Rouanne, M.; Tan, K.T.; Huang, R.Y.; Thiery, J.P. Molecular Subtypes of Urothelial Bladder Cancer: Results from a Meta-cohort Analysis of 2411 Tumors. Eur. Urol. 2019, 75, 423–432. [Google Scholar] [CrossRef]
- Chen, Z.; Zhou, L.; Liu, L.; Hou, Y.; Xiong, M.; Yang, Y.; Hu, J.; Chen, K. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 2020, 11, 5077. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, H.; He, D.; Cheng, Y.; Zhu, Y.; Xiao, M.; Lan, H.; Wang, Z.; Cao, K. Analysis of Tumor Microenvironment Characteristics in Bladder Cancer: Implications for Immune Checkpoint Inhibitor Therapy. Front. Immunol. 2021, 12, 672158. [Google Scholar] [CrossRef]
- Ching, C.B.; Hansel, D.E. Expanding therapeutic targets in bladder cancer: The PI3K/Akt/mTOR pathway. Lab. Investig. 2010, 90, 1406–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahe, M.; Dufour, F.; Neyret-Kahn, H.; Moreno-Vega, A.; Beraud, C.; Shi, M.; Hamaidi, I.; Sanchez-Quiles, V.; Krucker, C.; Dorland-Galliot, M.; et al. An FGFR3/MYC positive feedback loop provides new opportunities for targeted therapies in bladder cancers. EMBO Mol. Med. 2018, 10, e8163. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Kettner, N.M. The circadian clock in cancer development and therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar] [PubMed] [Green Version]
- Chen, Y.; Fan, Z.; Yang, Y.; Gu, C. Iron metabolism and its contribution to cancer (Review). Int. J. Oncol. 2019, 54, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Mechlin, C.W.; Tanner, M.J.; Chen, M.; Buttyan, R.; Levin, R.M.; Mian, B.M. Gli2 expression and human bladder transitional carcinoma cell invasiveness. J. Urol. 2010, 184, 344–351. [Google Scholar] [CrossRef]
- Ozgun, G.; Senturk, S.; Erkek-Ozhan, S. Retinoic acid signaling and bladder cancer: Epigenetic deregulation, therapy and beyond. Int. J. Cancer 2020, 148, 2364–2374. [Google Scholar] [CrossRef]
- Cai, H.; Zhu, X.D.; Ao, J.Y.; Ye, B.G.; Zhang, Y.Y.; Chai, Z.T.; Wang, C.H.; Shi, W.K.; Cao, M.Q.; Li, X.L.; et al. Colony-stimulating factor-1-induced AIF1 expression in tumor-associated macrophages enhances the progression of hepatocellular carcinoma. OncoImmunology 2017, 6, e1333213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanelli, G.; Romano, M.; Nova-Lamperti, E.; Werner Sunderland, M.; Nerviani, A.; Scotta, C.; Bombardieri, M.; Quezada, S.A.; Sacks, S.H.; Noelle, R.J.; et al. PD-L1 signaling on human memory CD4+ T cells induces a regulatory phenotype. PLoS Biol. 2021, 19, e3001199. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Wang, Z.; Chen, J.; Gu, L.; Chen, M.; Ding, Y.; Liu, T. Up-regulated ENO1 promotes the bladder cancer cell growth and proliferation via regulating beta-catenin. Biosci. Rep. 2019, 39, BSR20190503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Yan, Y.; Zhu, Z.; Liu, J.; He, X.; Dalangood, S.; Li, M.; Tan, M.; Cai, J.; Tang, P.; et al. CBX7 suppresses urinary bladder cancer progression via modulating AKR1B10-ERK signaling. Cell Death Dis. 2021, 12, 537. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, H.; Wang, W.; Gong, J.; Zhang, L.; Chen, Z.; Ding, Q. Expression of Med19 in bladder cancer tissues and its role on bladder cancer cell growth. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 920–927. [Google Scholar] [CrossRef]
- Liu, W.B.; Jia, W.D.; Ma, J.L.; Xu, G.L.; Zhou, H.C.; Peng, Y.; Wang, W. Knockdown of GTPBP4 inhibits cell growth and survival in human hepatocellular carcinoma and its prognostic significance. Oncotarget 2017, 8, 93984–93997. [Google Scholar] [CrossRef] [Green Version]
- da Silva, G.N.; Evangelista, A.F.; Magalhaes, D.A.; Macedo, C.; Bufalo, M.C.; Sakamoto-Hojo, E.T.; Passos, G.A.; Salvadori, D.M. Expression of genes related to apoptosis, cell cycle and signaling pathways are independent of TP53 status in urinary bladder cancer cells. Mol. Biol. Rep. 2011, 38, 4159–4170. [Google Scholar] [CrossRef]
- Li, L.; Ljungman, M.; Dixon, J.E. The human Cdc14 phosphatases interact with and dephosphorylate the tumor suppressor protein p53. J. Biol. Chem. 2000, 275, 2410–2414. [Google Scholar] [CrossRef] [Green Version]
- Dietachmayr, M.; Rathakrishnan, A.; Karpiuk, O.; von Zweydorf, F.; Engleitner, T.; Fernandez-Saiz, V.; Schenk, P.; Ueffing, M.; Rad, R.; Eilers, M.; et al. Antagonistic activities of CDC14B and CDK1 on USP9X regulate WT1-dependent mitotic transcription and survival. Nat. Commun. 2020, 11, 1268. [Google Scholar] [CrossRef] [Green Version]
- Moussa, H.F.; Bsteh, D.; Yelagandula, R.; Pribitzer, C.; Stecher, K.; Bartalska, K.; Michetti, L.; Wang, J.; Zepeda-Martinez, J.A.; Elling, U.; et al. Canonical PRC1 controls sequence-independent propagation of Polycomb-mediated gene silencing. Nat. Commun. 2019, 10, 1931. [Google Scholar] [CrossRef]
- Grossniklaus, U.; Paro, R. Transcriptional silencing by polycomb-group proteins. Cold Spring Harb. Perspect. Biol. 2014, 6, a019331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobocinska, J.; Molenda, S.; Machnik, M.; Oleksiewicz, U. KRAB-ZFP Transcriptional Regulators Acting as Oncogenes and Tumor Suppressors: An Overview. Int. J. Mol. Sci. 2021, 22, 2212. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Lu, X.; Bazai, S.K.; Comperat, E.; Mouawad, R.; Yao, H.; Roupret, M.; Spano, J.P.; Khayat, D.; Davidson, I.; et al. Comprehensive integrative profiling of upper tract urothelial carcinomas. Genome Biol. 2021, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Loras, A.; Suarez-Cabrera, C.; Martinez-Bisbal, M.C.; Quintas, G.; Paramio, J.M.; Martinez-Manez, R.; Gil, S.; Ruiz-Cerda, J.L. Integrative Metabolomic and Transcriptomic Analysis for the Study of Bladder Cancer. Cancers 2019, 11, 686. [Google Scholar] [CrossRef] [Green Version]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715.e7. [Google Scholar] [CrossRef]
- Biton, A.; Bernard-Pierrot, I.; Lou, Y.; Krucker, C.; Chapeaublanc, E.; Rubio-Perez, C.; Lopez-Bigas, N.; Kamoun, A.; Neuzillet, Y.; Gestraud, P.; et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014, 9, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Riester, M.; Taylor, J.M.; Feifer, A.; Koppie, T.; Rosenberg, J.E.; Downey, R.J.; Bochner, B.H.; Michor, F. Combination of a novel gene expression signature with a clinical nomogram improves the prediction of survival in high-risk bladder cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Therkildsen, C.; Eriksson, P.; Hoglund, M.; Jonsson, M.; Sjodahl, G.; Nilbert, M.; Liedberg, F. Molecular subtype classification of urothelial carcinoma in Lynch syndrome. Mol. Oncol. 2018, 12, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Sjodahl, G.; Eriksson, P.; Patschan, O.; Marzouka, N.A.; Jakobsson, L.; Bernardo, C.; Lovgren, K.; Chebil, G.; Zwarthoff, E.; Liedberg, F.; et al. Molecular changes during progression from nonmuscle invasive to advanced urothelial carcinoma. Int. J. Cancer 2020, 146, 2636–2647. [Google Scholar] [CrossRef] [Green Version]
- Sjodahl, G.; Eriksson, P.; Liedberg, F.; Hoglund, M. Molecular classification of urothelial carcinoma: Global mRNA classification versus tumour-cell phenotype classification. J. Pathol. 2017, 242, 113–125. [Google Scholar] [CrossRef]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConkey, D.J.; Choi, W.; Shen, Y.; Lee, I.L.; Porten, S.; Matin, S.F.; Kamat, A.M.; Corn, P.; Millikan, R.E.; Dinney, C.; et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naive Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur. Urol. 2016, 69, 855–862. [Google Scholar] [PubMed] [Green Version]
- Kim, W.J.; Kim, E.J.; Kim, S.K.; Kim, Y.J.; Ha, Y.S.; Jeong, P.; Kim, M.J.; Yun, S.J.; Lee, K.M.; Moon, S.K.; et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol. Cancer 2010, 9, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Weishaupt, H.; Johansson, P.; Sundstrom, A.; Lubovac-Pilav, Z.; Olsson, B.; Nelander, S.; Swartling, F.J. Batch-normalization of cerebellar and medulloblastoma gene expression datasets utilizing empirically defined negative control genes. Bioinformatics 2019, 35, 3357–3364. [Google Scholar] [CrossRef] [Green Version]





| Kerrypnx | Discovery Set | TCGA-BLCA-2017 | IMvigor210 |
|---|---|---|---|
| No. of patients | 1135 | 188 | 132 |
| Median age (range) | 67 (24–95) | 69 (34–90) | |
| Sex | |||
| Female | 135 (11.9%) | 45 (23.9%) | 29 (22.0%) |
| Male | 459 (40.5%) | 143 (76.1%) | 103 (78.0%) |
| No info | 541 (47.6%) | 0 (0.0%) | 0 (0.0%) |
| Stage | |||
| NAU | 81 (7.1%) | 0 (0.0%) | |
| Ta | 229 (20.2%) | 0 (0.0%) | |
| T1 | 262 (23.1%) | 0 (0.0%) | |
| T2 | 371 (32.7%) | 70 (37.3%) | |
| T3 | 146 (12.9%) | 90 (47.9%) | |
| T4 | 46 (4.0%) | 28 (14.8%) | |
| Grade | |||
| G1 | 13 (1.1%) | 0 (0.0%) | |
| G2 | 164 (14.5%) | 0 (0.0%) | |
| G3 | 372 (32.8%) | 188 (100%) | |
| Gx | 13 (1.1%) | 0 (0.0%) | |
| No info | 573 (50.5%) | 0 (0.0%) | |
| Response to atezolizumab | |||
| Complete | 11 (8.3%) | ||
| Partial | 20 (15.2%) | ||
| Stable disease | 25 (18.9%) | ||
| Progressed disease | 76 (57.6%) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stroggilos, R.; Frantzi, M.; Zoidakis, J.; Mokou, M.; Moulavasilis, N.; Mavrogeorgis, E.; Melidi, A.; Makridakis, M.; Stravodimos, K.; Roubelakis, M.G.; et al. Gene Expression Monotonicity across Bladder Cancer Stages Informs on the Molecular Pathogenesis and Identifies a Prognostic Eight-Gene Signature. Cancers 2022, 14, 2542. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102542
Stroggilos R, Frantzi M, Zoidakis J, Mokou M, Moulavasilis N, Mavrogeorgis E, Melidi A, Makridakis M, Stravodimos K, Roubelakis MG, et al. Gene Expression Monotonicity across Bladder Cancer Stages Informs on the Molecular Pathogenesis and Identifies a Prognostic Eight-Gene Signature. Cancers. 2022; 14(10):2542. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102542
Chicago/Turabian StyleStroggilos, Rafael, Maria Frantzi, Jerome Zoidakis, Marika Mokou, Napoleon Moulavasilis, Emmanouil Mavrogeorgis, Anna Melidi, Manousos Makridakis, Konstantinos Stravodimos, Maria G. Roubelakis, and et al. 2022. "Gene Expression Monotonicity across Bladder Cancer Stages Informs on the Molecular Pathogenesis and Identifies a Prognostic Eight-Gene Signature" Cancers 14, no. 10: 2542. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102542