
MDACT: A New Principle of Adjunctive Cancer Treatment Using Combinations of Multiple Repurposed Drugs, with an Example Regimen

 , , , , ,
, , , , ,  , , and
, , and
Abstract
:Simple Summary
Abstract
- Preface: gutta cavat lapidem, non vi, sed sæpe cadendo.
- Part One
1. Introduction
2. Attributes of Cancer Mandating an MDACT Type of Approach
- Spatial and temporal heterogeneity of growth-driving dependencies;
- Existence of mutually supporting, bilaterally communicating cell communities;
- Compensatory tumor responses to treatments;
- Existence of multiple cross-covering, growth-driving signaling pathways functioning in parallel;
- Metabolic flexibility reliance shifted to another energy source if one becomes inhibited;
- Pathological engagement of multiple normally functioning body systems to facilitate growth (e.g., cytokines, trophic factors, innervation, interacting stroma, angiogenesis);
- A subset of tumor stem cells with the potential to enter dormancy.
- An inverse relationship often seen between growth and invasion, where inhibiting one enhances the other.
3. Eight Overarching Principles Driving the Construction of MDACT-Type Regimens
3.1. The Principle of Breaking More than One Link in a Chain
3.2. The Principle of Palmer et al.
3.3. The Chess Principle of Shaping Versus Decisive Operations
3.4. The Principle of Chow et al.
3.5. The Principle of CUSP9v3
3.6. The Chess Aphorism—All Moves Create Strengths and Weaknesses
3.7. The Principle of Mass
3.8. The Principle of Blocking Parallel Pathways—The Nile Distributary Problem
- Part Two
4. The Drugs of gMDACT
4.1. Celecoxib
- A.
- Celecoxib in cholangiocarcinoma: High COX-2 expression is associated with shorter cholangiocarcinoma survival [41]. Preclinical studies have shown inhibition of cholangiocarcinoma growth by celecoxib [42,43,44]. Around 92% of cholangiocarcinoma tumors have strong CA-IX immunohistochemistry staining [45].
- B.
- Colon adenocarcinoma: COX-2-driven overproduction of prostaglandin E is an element of dysregulated excess growth across cancers, including colon adenocarcinoma [46,47,48,49]. CA-IX: CA-IX inhibitors increase colon adenocarcinoma cells’ sensitivity to temozolomide and other genotoxic chemotherapies [50]. CA-IX generally tends to be upregulated in hypoxic areas of cancers, and is found specifically in colon adenocarcinoma [51,52,53]. H+ export function by CA-IX has been shown to be crucial for keeping intracellular pH high enough to be compatible with growth in colon adenocarcinoma [54].
- C.
- Celecoxib in glioblastoma: COX-2 and CA-IX are elevated in glioblastoma, and are growth-facilitating elements. The potential usefulness of celecoxib in glioblastoma by inhibiting the function of both of these enzymes was recently reviewed in detail [36]. CA-IX upregulation characteristic of glioblastoma is crucial for this cancer’s adaptation to the hypoxic conditions in which it grows [55].
- D.
- Celecoxib in NSCLC: The roles of elevated COX-2 and CA-IX in NSCLC growth promotion, along with the potential benefit of celecoxib in NSCLC treatment, were recently reviewed [56,57,58]. Several studies show celecoxib’s potential for increasing immune response to NSCLC [59,60]. A recent human clinical immunization study in NSCLC showed that celecoxib enhanced immune responses to a lung cancer lysate vaccine [61]. COX-2 mediates aspects of NSCLC resistance to common traditional cytotoxic drugs, and celecoxib reduces that resistance in experimental models [62,63,64,65,66].
4.2. Dapsone
- A.
- Dapsone in cholangiocarcinoma: IL-8-driven chemotaxis of neutrophils infiltrating cholangiocarcinoma constitutes a trophic function in the growing tumor [91,92,93,94,95,96,97,98,99,100,101]. IL-8 drives angiogenesis, and is elevated in cholangiocarcinoma, where higher levels are associated with shorter survival [102,103,104,105,106,107,108,109,110]. As with other cancers, a higher NLR is strongly associated with shorter survival in cholangiocarcinoma [111].
- B.
- Dapsone in colon adenocarcinoma: Specifically in colon adenocarcinoma, a higher NLR is associated with shorter survival, while a low NLR is associated with longer survival [112,113,114]. Bevacizumab, a pharmaceutical monoclonal antibody to vascular endothelial growth factor (VEGF), is often used in the treatment of colon adenocarcinoma and NSCLC. The benefit of bevacizumab diminishes as the circulating absolute neutrophil count or NLR increases [75,115,116,117,118]. This inverse relationship is due to neutrophils’ delivery of intracellular VEGF, protected from circulating bevacizumab [119,120,121]. IL-8 is actively synthesized by both the malignant cells and their supporting nonmalignant stromata to support growth and angiogenesis in colon adenocarcinoma [122,123,124,125,126,127].
- C.
- Dapsone in glioblastoma: Dapsone’s suppression of IL-8-directed neutrophil chemotaxis and its consequent contributions to glioblastoma growth and angiogenesis were recently reviewed in detail [75]. IL-8 signaling at CXCR2 is a prominent member of the flood of cytokines driving glioblastoma growth [128]. Circulating neutrophils chemotactic to glioblastoma due to IL-8 are an element driving glioblastoma growth and related angiogenesis [129].
- D.
- Dapsone in NSCLC: IL-8 levels are elevated in NSCLC, and a degree of pretreatment elevation is associated with shorter OS [130]. IL-8 elevation in NSCLC is also associated with—and partially drives—increased myeloid-derived suppressor cells [131]. NSCLC tissue, sera, and pleural effusions have increased levels of IL-8 and its receptors, where the degree of elevation is correlated with shorter survival [132,133,134,135,136,137]. As seen commonly in other cancers, an NLR > 4 strongly predicts shorter OS in NSCLC [138,139,140,141,142]. Neutrophil extracellular traps, the presence of which shortens survival in NSCLC, are driven in part by excess IL-8 in NSCLC [68,71].
4.3. Disulfiram
- A.
- B.
- C.
- Disulfiram in glioblastoma: With potential utility in treating glioblastoma with temozolomide, disulfiram irreversibly inactivates P-gp [171,172,173]. As with other cancers, the ALDH-positive glioblastoma subpopulation has other stem attributes, and is more chemotherapy resistant than the ALDH-negative subpopulation [158,174,175,176].
- D.
4.4. Itraconazole
- A.
- Itraconazole in cholangiocarcinoma: 5-LO and its leukotriene products contribute to the growth of cholangiocarcinoma [207,208,209]. Hh signaling is a core growth-driving element in cholangiocarcinoma, and prominently so in the stem subset [210,211,212,213,214,215]. 5-LO is one of the drivers of both myeloid-derived suppressor cell immunosuppression and stemness in cholangiocarcinoma [216].
- B.
- Itraconazole in colon adenocarcinoma: Colon adenocarcinomas overexpress 5-LO, as well as COX-2 [217,218,219]. As found in other cancers, breast cancer’s overexpression of both COX-2 and 5-LO is associated with enhanced aggressiveness [220]. Dual inhibition of COX-2/5-LO inhibits colon cancer proliferation, migration, and invasion to a greater degree than either alone. Specifically, inhibition of 5-LO increases celecoxib’s cytotoxicity to colon cells [221,222,223]. Coordinated participation of COX-2 and 5-LO in carcinogenesis and cancer growth is recognized in several common cancers [220,221,224,225,226,227]. Hh signaling is a major growth-driving element in a variety of cancers, including colon adenocarcinoma [228]. Itraconazole interferes with colon cancer’s cytotoxicity resistance and growth by inhibiting Hh [229,230,231]. Hh signaling is a major driver of colon adenocarcinoma growth [232]. Itraconazole’s inhibition of Hh inhibits colon adenocarcinoma growth [229].
- C.
- Itraconazole in glioblastoma: The rationale for the use of itraconazole during glioblastoma treatment is based on its attributes of Hh inhibition, leukotriene signaling reduction, and reduction in P-gp-mediated cell export of temozolomide, as outlined in the three preceding CUSP9 papers [1,2,3,6]. 5-LO-generated leukotrienes promote glioblastoma migration, growth, and stem attributes [233].
- D.
- Itraconazole in NSCLC: A 2013 study showed that itraconazole plus pemetrexed in NSCLC doubled progression-free survival (PFS) and gave a fourfold increase in overall survival (OS) [234]. In 2017, three reviews of itraconazole’s attributes were published, suggesting its usefulness in interfering with cancer cells’ growth—two in general, and one specifically in NSCLC [235,236,237]. In 2018, Lee et al. outlined the potential of inhaled itraconazole to inhibit NSCLC growth [238]. In 2019, itraconazole was reformulated for superior pharmacokinetics in NSCLC treatment [239]. NSCLC patients given 300 mg of itraconazole orally, twice daily, for two weeks prior to surgery, had decreased tumor volume and reduced vascularity [240]. 5-LO-generated leukotrienes promote NSCLC migration and growth [233,241].
4.5. Pyrimethamine
- A.
- Pyrimethamine in cholangiocarcinoma: In cholangiocarcinoma, upregulated thymidine phosphorylase also contributes to chemotherapy resistance, and furthers survival [109,277,278,279]. Thymidine phosphorylase overexpression enhances growth and suppresses apoptosis in human umbilical vein endothelial cells, as well as increasing VEGF, IL-8, and the growth of cholangiocarcinoma cells [109]. STAT3 activation is an identified growth driver in cholangiocarcinoma [280,281,282].
- B.
- Pyrimethamine in colon adenocarcinoma: As commonly found in other cancers, STAT3 overactivation also constitutes a driving force in colon adenocarcinoma, and particularly so in the stem subpopulation [282,283,284,285,286,287,288]. Multiple experimental, non-marketed inhibitors of STAT3 reduced colon cancer growth in preclinical models [289,290,291]. Growth of colon cancer cells is suppressed when DNA binding of activated STAT3 is prevented [292].
- C.
- Pyrimethamine in glioblastoma: Of great interest for potential use in treating glioblastoma or brain metastases from breast or lung cancer is the unusual property of pyrimethamine in being concentrated in the brain at several times greater levels than in plasma [293,294]. Pyrimethamine is synergistically cytotoxic with temozolomide—the mainstay in current glioblastoma treatment—in melanoma and pituitary adenoma cell lines [295,296]. Glioblastomas have a greatly upregulated thymidine phosphorylase content and activity [297]. Experimental (non-marketed) thymidine phosphorylase inhibitors have no cytotoxicity alone, but are synergistic with temozolomide against glioblastoma cell lines [297].
- D.
4.6. Telmisartan
- A.
- Telmisartan in cholangiocarcinoma: Telmisartan triggers cholangiocarcinoma G0/G1 cell-cycle arrest in vitro [307]. Telmisartan also triggers cell-cycle arrest in a wide variety of gastrointestinal and other common cancers [308,309,310,311,312,313,314,315,316,317,318]. ACE and ACE-related signaling are active specifically as elements driving cholangiocarcinoma growth [319,320,321].
- B.
- Telmisartan in colon adenocarcinoma: Telmisartan blocks angiotensin II receptor type 1. Marketed to treat hypertension, it has several other attributes and uses. Colon adenocarcinoma cells express angiotensin II receptor 1. Telmisartan’s IC50 to several colon cancer cell lines in vitro is between 1 and 5 µM [322]. Irbesartan, a marketed pharmaceutical ARB similar to telmisartan, inhibits colitis-associated colon cancer development [323]. Candesartan, another pharmaceutical ARB similar to telmisartan, inhibits colon adenocarcinoma xenograft growth and tumor-related fibrosis [324]. Other studies have found that colon cancer cell growth inhibition is greater with telmisartan compared to candesartan [325]. Candesartan decreased the immune suppression function of tumor-associated CD11b+ T cells and decreased their production of VEGF and arginase, and increased interferon-γ synthesis in the lymph nodes of colon-cancer-bearing mice, without having effect on in vivo tumor growth [326].
- C.
- Telmisartan in glioblastoma: Telmisartan was cytotoxic via peroxisome proliferator-activated receptor gamma (PPAR-γ) agonism in glioblastoma cells in vitro, at low concentrations [327]. Telmisartan-induced inhibition of glioblastoma growth via angiotensin receptor inhibition has been extensively reviewed previously [328]. PPAR-γ is upregulated in mesenchymal glioblastoma stem cells, with agonism suppressing growth [329].
- D.
- Telmisartan in NSCLC: Several studies show longer survival in NSCLC in patients receiving an ARB [330,331,332,333]. This effect, although slight, has been consistently found across studies. Empirically, telmisartan inhibits experimental NSCLC growth [334]. Various putative MOAs for telmisartan’s inhibition of NSCLC have been identified [334,335,336,337,338,339].
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| ALDH | Aldehyde dehydrogenase |
| ARB | Angiotensin receptor-blocking drug |
| CA | Carbonic anhydrase |
| COX-2 | Cyclooxygenase-2 |
| DHFR | Dihydrofolate reductase |
| Hh | Hedgehog |
| 5-LO | 5-Lipoxygenase |
| MTX | Methotrexate |
| MOA | Mechanism of action |
| MDACT | Multidrug adjuvant for cancer treatment |
| NLR | Neutrophil-to-lymphocyte ratio |
| VEGF | Vascular endothelial growth factor |
References
- Halatsch, M.-E.; Kast, R.E.; Karpel-Massler, G.; Mayer, B.; Zolk, O.; Schmitz, B.; Scheuerle, A.; Maier, L.; Bullinger, L.; Mayer-Steinacker, R.; et al. A phase Ib/IIa trial of 9 repurposed drugs combined with temozolomide for the treatment of recurrent glioblastoma: CUSP9v3. Neurooncol. Adv. 2021, 3, vdab075. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Skaga, I.Ø.; Grieg, Z.; Sandberg, C.J.; Langmoen, I.A.; Vik-Mo, E.O. The efficacy of a coordinated pharmacological blockade in glioblastoma stem cells with nine repurposed drugs using the CUSP9 strategy. J. Cancer Res. Clin. Oncol. 2019, 145, 1495–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halatsch, M.-E.; Dwucet, A.; Schmidt, C.J.; Mühlnickel, J.; Heiland, T.; Zeiler, K.; Siegelin, M.D.; Kast, R.E.; Karpel-Massler, G. In Vitro and Clinical Compassionate Use Experiences with the Drug Repurposing Approach CUSP9v3 in Glioblastoma. Pharmaceuticals 2021, 14, 1241. [Google Scholar] [CrossRef] [PubMed]
- Mettang, M.; Meyer-Pannwitt, V.; Karpel-Massler, G.; Zhou, S.; Carragher, N.O.; Föhr, K.J.; Baumann, B.; Nonnenmacher, L.; Enzenmüller, S.; Dahlhaus, M.; et al. Blocking distinct interactions between Glioblastoma cells and their tissue microenvironment: A novel multi-targeted therapeutic approach. Sci. Rep. 2018, 8, 5527. [Google Scholar] [CrossRef]
- Kast, R.E.; Halatsch, M.-E. Matrix metalloproteinase-2 and -9 in glioblastoma: A trio of old drugs-captopril, disulfiram and nelfinavir-are inhibitors with potential as adjunctive treatments in glioblastoma. Arch. Med. Res. 2012, 43, 243–247. [Google Scholar] [CrossRef]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.-E. CUSP9* treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E.; Boockvar, J.A.; Brüning, A.; Cappello, F.; Chang, W.-W.; Cvek, B.; Dou, Q.P.; Duenas-Gonzalez, A.; Efferth, T.; Focosi, D.; et al. A conceptually new treatment approach for relapsed glioblastoma: Coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 2013, 4, 502–530. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.S.; Shabani, S.; Awad, A.J.; Kaushal, M.; Doan, N. Molecular Markers of Therapy Resistant Glioblastoma and Potential Strategy to Combat Resistance. Int. J. Mol. Sci. 2018, 19, 1765. [Google Scholar] [CrossRef] [Green Version]
- Collen, M.F.; Sellers, A.L.; Kast, E.C. Combined penicillin and sulfadiazine therapy in pneumococcic pneumonia. Am. J. Med. Sci. 1946, 211, 299–306. [Google Scholar] [CrossRef]
- Zapletalova, D.; André, N.; Deak, L.; Kyr, M.; Bajciova, V.; Mudry, P.; Dubska, L.; Demlova, R.; Pavelka, Z.; Zitterbart, K.; et al. Metronomic chemotherapy with the COMBAT regimen in advanced pediatric malignancies: A multicenter experience. Oncology 2012, 82, 249–260. [Google Scholar] [CrossRef]
- Peyrl, A.; Chocholous, M.; Kieran, M.W.; Azizi, A.A.; Prucker, C.; Czech, T.; Dieckmann, K.; Schmook, M.-T.; Haberler, C.; Leiss, U.; et al. Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr. Blood Cancer 2012, 59, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.; Hsu, S.H.; Blanco, A.; Buryanek, J.; Day, A.L.; McGuire, M.F.; Brown, R.E. Durable response of glioblastoma to adjuvant therapy consisting of temozolomide and a weekly dose of AMD3100 (plerixafor), a CXCR4 inhibitor, together with lapatinib, metformin and niacinamide. Oncoscience 2016, 3, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, T.; Sabit, H.; Dong, Y.; Miyashita, K.; Kinoshita, M.; Uchiyama, N.; Hayashi, Y.; Hayashi, Y.; Minamoto, T.; Nakada, M. Biological basis and clinical study of glycogen synthase kinase- 3β-targeted therapy by drug repositioning for glioblastoma. Oncotarget 2017, 8, 22811–22824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatokoro, M.; Fujii, Y.; Kawakami, S.; Saito, K.; Koga, F.; Matsuoka, Y.; Iimura, Y.; Masuda, H.; Kihara, K. Phase-II trial of combination treatment of interferon-α, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor (I-CCA therapy) for advanced renal cell carcinoma. Cancer Sci. 2011, 102, 137–143. [Google Scholar] [CrossRef]
- Maraka, S.; Groves, M.D.; Mammoser, A.G.; Melguizo-Gavilanes, I.; Conrad, C.A.; Tremont-Lukats, I.W.; Loghin, M.E.; O’Brien, B.J.; Puduvalli, V.K.; Sulman, E.P.; et al. Phase 1 lead-in to a phase 2 factorial study of temozolomide plus memantine, mefloquine, and metformin as postradiation adjuvant therapy for newly diagnosed glioblastoma. Cancer 2019, 125, 424–433, Erratum in Cancer 2019, 125, 1387. [Google Scholar] [CrossRef] [Green Version]
- Nogales, C.; Mamdouh, Z.M.; List, M.; Kiel, C.; Casas, A.I.; Schmidt, H.H. Network pharmacology: Curing causal mechanisms instead of treating symptoms. Trends Pharmacol. Sci. 2021, 43, 136–150. [Google Scholar] [CrossRef]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. Elife 2019, 8, e50036. [Google Scholar] [CrossRef]
- Candelaria, M.; Dueñas-Gonzalez, A. Rituximab in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in diffuse large B-cell lymphoma. Ther. Adv. Hematol. 2021, 12, 2040620721989579. [Google Scholar] [CrossRef]
- Czuczman, M.S.; Weaver, R.; Alkuzweny, B.; Berlfein, J.; Grillo-López, A.J. Prolonged clinical and molecular remission in patients with low-grade or follicular non-Hodgkin’s lymphoma treated with rituximab plus CHOP chemotherapy: 9-year follow-up. J. Clin. Oncol. 2004, 22, 4711–4716. [Google Scholar] [CrossRef]
- Moore, D.R. Decisive, Shaping, Sustaining Operations: An Operational Organization for the Contemporary Mission Environment. School of Advanced Military Studies United States Army Command and General Staff College Fort Leavenworth, Kansas, USA. Available online: https://apps.dtic.mil (accessed on 28 February 2022).
- Chow, C.K.; Atkins, E.R.; Hillis, G.S.; Nelson, M.R.; Reid, C.M.; Schlaich, M.P.; Hay, P.; Rogers, K.; Billot, L.; Burke, M.; et al. QUARTET Investigators. Initial treatment with a single pill containing quadruple combination of quarter doses of blood pressure medicines versus standard dose monotherapy in patients with hypertension (QUARTET): A phase 3, randomised, double-blind, active-controlled trial. Lancet 2021, 398, 1043–1052. [Google Scholar] [CrossRef]
- Gürgen, D.; Conrad, T.; Becker, M.; Sebens, S.; Röcken, C.; Hoffmann, J.; Langhammer, S. Breaking the crosstalk of the Cellular Tumorigenic Network by low-dose combination therapy in lung cancer patient-derived xenografts. Commun. Biol. 2022, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- De Monte, C.; Carradori, S.; Gentili, A.; Mollica, A.; Trisciuoglio, D.; Supuran, C.T. Dual Cyclooxygenase and Carbonic Anhydrase Inhibition by Nonsteroidal Anti-Inflammatory Drugs for the Treatment of Cancer. Curr. Med. Chem. 2015, 22, 2812–2818. [Google Scholar] [CrossRef] [PubMed]
- De Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved Brain Penetration and Antitumor Efficacy of Temozolomide by Inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; De Gooijer, M.C.; Roig, E.M.; Buil, L.C.; Christner, S.M.; Beumer, J.; Würdinger, T.; Beijnen, J.H.; Van Tellingen, O. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin. Cancer Res. 2014, 20, 2703–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, J.L.; Walker, N.D.; Scotto, K.W.; Rameshwar, P. Temozolomide competes for P-glycoprotein and contributes to chemoresistance in glioblastoma cells. Cancer Lett. 2015, 367, 69–75. [Google Scholar] [CrossRef]
- Mirzaei, S.; Gholami, M.H.; Hashemi, F.; Zabolian, A.; Farahani, M.V.; Hushmandi, K.; Zarrabi, A.; Goldman, A.; Ashrafizadeh, M.; Orive, G. Advances in understanding the role of P-gp in doxorubicin resistance: Molecular pathways, therapeutic strategies, and prospects. Drug Discov. Today 2021, 27, 436–455. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Ashby, C.R., Jr.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical molecular-based approach to overcome multidrug resistance in cancer by targeting P-glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef]
- Robinson, K.; Tiriveedhi, V. Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 2020, 10, 265. [Google Scholar] [CrossRef]
- Lim, J.S.; Park, Y.; Lee, B.M.; Kim, H.S.; Yoon, S. Co-treatment with Celecoxib or NS398 Strongly Sensitizes Resistant Cancer Cells to Antimitotic Drugs Independent of P-gp Inhibition. Anticancer Res. 2016, 36, 5063–5070. [Google Scholar] [CrossRef] [Green Version]
- Dharmapuri, G.; Doneti, R.; Philip, G.H.; Kalle, A.M. Celecoxib sensitizes imatinib-resistant K562 cells to imatinib by inhibiting MRP1-5, ABCA2 and ABCG2 transporters via Wnt and Ras signaling pathways. Leuk. Res. 2015, 39, 696–701. [Google Scholar] [CrossRef]
- Pagliarulo, V.; Ancona, P.; Niso, M.; Colabufo, N.A.; Contino, M.; Cormio, L.; Azzariti, A.; Pagliarulo, A. The interaction of celecoxib with MDR transporters enhances the activity of mitomycin C in a bladder cancer cell line. Mol. Cancer 2013, 12, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecil, D.L.; Gad, E.A.; Corulli, L.R.; Drovetto, N.; Lubet, R.A.; Disis, M.L. COX-2 Inhibitors Decrease Expression of PD-L1 in Colon Tumors and Increase the Influx of Type I Tumor-infiltrating Lymphocytes. Cancer Prev. Res. 2022, 15, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Tudor, D.V.; Bâldea, I.; Lupu, E.A.M.; Kacso, T.; Kutasi, E.; Hopârtean, A.; Stretea, R.; Filip, A.G. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol. Med. 2020, 17, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. Adding high-dose celecoxib to increase effectiveness of standard glioblastoma chemoirradiation. Ann. Pharm. Fr. 2021, 79, 481–488. [Google Scholar] [CrossRef]
- Mostafa, T.M.; Alm El-Din, M.A.; Rashdan, A.R. Celecoxib as an adjuvant to chemotherapy for patients with metastatic colorectal cancer: A randomized controlled clinical study. Saudi Med. J. 2022, 43, 37–44. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Krysan, K.; Morrow, J.D.; Milne, G.L.; Newman, R.A.; Tucker, C.; Elashoff, R.M.; Dubinett, S.M.; Figlin, R.A. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clin. Cancer Res. 2006, 12, 3381–3388. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.R.; Chen, J.Q.; Zhang, X.W.; Gao, Q.Y.; Li, W.H.; Yan, L.J.; Zhang, Y.Q.; Wu, C.J.; Xing, J.L.; Liu, J.P. Cardiovascular safety of celecoxib in rheumatoid arthritis and osteoarthritis patients: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0261239. [Google Scholar] [CrossRef]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Aldera, A.P.; Govender, D. Carbonic anhydrase IX: A regulator of pH and participant in carcinogenesis. J. Clin. Pathol. 2021, 74, 350–354. [Google Scholar] [CrossRef]
- Yeh, C.N.; Chiang, K.C.; Juang, H.H.; Pang, J.H.S.; Yu, C.S.; Lin, K.J.; Yeh, T.S.; Jan, Y.Y. Reappraisal of the therapeutic role of celecoxib in cholangiocarcinoma. PLoS ONE 2013, 8, e69928. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Chung, C.W.; Lee, H.M.; Kim, D.H.; Kwak, T.W.; Jeong, Y.I.; Kang, D.H. Synergistic effects of 5-aminolevulinic acid based photodynamic therapy and celecoxib via oxidative stress in human cholangiocarcinoma cells. Int. J. Nanomed. 2013, 8, 2173–2186. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Leng, J.; Han, C.; Demetris, A.J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 2004, 3, 299–307. [Google Scholar] [PubMed]
- Zhang, Z.; Lai, G.H.; Sirica, A.E. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by Akt inactivation and Bax translocation. Hepatology 2004, 39, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Sadot, E.; Simpson, A.L.; Do, R.K.; Gonen, M.; Shia, J.; Allen, P.J.; D’Angelica, M.I.; DeMatteo, R.P.; Kingham, T.P.; Jarnagin, W.R. Cholangiocarcinoma: Correlation between Molecular Profiling and Imaging Phenotypes. PLoS ONE 2015, 10, e0132953. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.-B.; Li, B.; Zhang, Y.; Zhu, Y.-T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef] [PubMed]
- Tolloczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Aoki, T.; Narumiya, S. Prostaglandin E2-EP2 signaling as a node of chronic inflammation in the colon tumor microenvironment. Inflamm. Regen. 2017, 37, 4. [Google Scholar] [CrossRef] [Green Version]
- Andreucci, E.; Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Laurenzana, A.; Carta, F.; Supuran, C.T.; Calorini, L. The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells to conventional chemotherapy. J. Enzym. Inhib. Med. Chem. 2019, 34, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Kopecka, J.; Campia, I.; Jacobs, A.; Frei, A.P.; Ghigo, D.; Wollscheid, B.; Riganti, C. Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget 2015, 6, 6776–6793. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Kojima, M.; Aokage, K.; Sakai, T.; Nakamura, H.; Ohara, Y.; Tane, K.; Miyoshi, T.; Sugano, M.; Fujii, S.; et al. Clinicopathological characteristics associated with necrosis in pulmonary metastases from colorectal cancer. Virchows Arch. 2019, 474, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Nakada, N.; Mikami, T.; Horie, K.; Nagashio, R.; Sakurai, Y.; Sanoyama, I.; Yoshida, T.; Sada, M.; Kobayashi, K.; Sato, Y.; et al. Expression of CA2 and CA9 carbonic anhydrases in ulcerative colitis and ulcerative colitis-associated colorectal cancer. Pathol. Int. 2020, 70, 523–532. [Google Scholar] [CrossRef]
- Parks, S.K.; Cormerais, Y.; Durivault, J.; Pouyssegur, J. Genetic disruption of the pHi-regulating proteins Na+/H+ exchanger 1 (SLC9A1) and carbonic anhydrase 9 severely reduces growth of colon cancer cells. Oncotarget 2017, 8, 10225–10237. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.-R.; Liu, Y.-S.; Lai, S.-W.; Lin, H.-J.; Shen, C.-K.; Yang, L.-Y.; Lu, D.-Y. CAIX Regulates GBM Motility and TAM Adhesion and Polarization through EGFR/STAT3 under Hypoxic Conditions. Int. J. Mol. Sci. 2020, 21, 5838. [Google Scholar] [CrossRef] [PubMed]
- Kiran, A.V.V.V.R.; Kumari, G.K.; Krishnamurthy, P.T. Preliminary evaluation of anticancer efficacy of pioglitazone combined with celecoxib for the treatment of non-small cell lung cancer. Investig. New Drugs 2022, 40, 1–9. [Google Scholar] [CrossRef]
- Niu, K.; Chen, X.-W.; Qin, Y.; Zhang, L.-P.; Liao, R.-X.; Sun, J.-G. Celecoxib Blocks Vasculogenic Mimicry via an Off-Target Effect to Radiosensitize Lung Cancer Cells: An Experimental Study. Front. Oncol. 2021, 11, 697227. [Google Scholar] [CrossRef]
- Xu, Y.-Q.; Long, X.; Han, M.; Huang, M.-Q.; Lu, J.-F.; Sun, X.-D.; Han, W. Clinical benefit of COX-2 inhibitors in the adjuvant chemotherapy of advanced non-small cell lung cancer: A systematic review and meta-analysis. World J. Clin. Cases 2021, 9, 581–601. [Google Scholar] [CrossRef]
- Kim, J.; Noh, M.H.; Hur, D.Y.; Kim, B.; Kim, Y.S.; Lee, H.-K. Celecoxib upregulates ULBP-1 expression in lung cancer cells via the JNK/PI3K signaling pathway and increases susceptibility to natural killer cell cytotoxicity. Oncol. Lett. 2020, 20, 279. [Google Scholar] [CrossRef]
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy With Immunotherapy in Cancer. Front. Oncol. 2021, 11, 637504. [Google Scholar] [CrossRef]
- Zhang, M.; Hong, J.A.; Kunst, T.F.; Bond, C.D.; Kenney, C.M.; Warga, C.L.; Yeray, J.; Lee, M.-J.; Yuno, A.; Lee, S.; et al. Randomized phase II trial of a first-in-human cancer cell lysate vaccine in patients with thoracic malignancies. Transl. Lung Cancer Res. 2021, 10, 3079–3092. [Google Scholar] [CrossRef]
- Pan, C.; Zhang, Y.; Meng, Q.; Dai, G.; Jiang, Z.; Bao, H. Down Regulation of the Expression of ELMO3 by COX2 Inhibitor Suppresses Tumor Growth and Metastasis in Non-Small-Cell Lung Cancer. Front. Oncol. 2019, 9, 363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Kim, J.; Kim, Y.S. Celecoxib induces cell death on non-small cell lung cancer cells through endoplasmic reticulum stress. Anat. Cell Biol. 2017, 50, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.-F.; Fang, Q.-Y.; Ji, X.-X.; Zhou, S.-W. Cyclooxygenase-2 mediates gefitinib resistance in non-small cell lung cancer through the EGFR/PI3K/AKT axis. J. Cancer. 2020, 11, 3667–3674. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.-B.; Fang, H.-Y.; Tao, D.-Y.; Chen, X.-P.; Cao, F.-L. COX-2 potentiates cisplatin resistance of non-small cell lung cancer cells by promoting EMT in an AKT signaling pathway-dependent manner. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3838–3846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; He, D.; Song, E.; Jiang, M.; Song, Y. Celecoxib enhances the sensitivity of non-small-cell lung cancer cells to radiation-induced apoptosis through downregulation of the Akt/mTOR signaling pathway and COX-2 expression. PLoS ONE 2019, 14, e0223760, Erratum in PLoS ONE 2019, 14, e0224843. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Karalis, J.D.; Liu, C.; Murimwa, G.Z.; Park, J.V.; Heid, C.A.; Reznik, S.I.; Huang, E.; Minna, J.D.; Brekken, R.A. The Colorectal Cancer Tumor Microenvironment and Its Impact on Liver and Lung Metastasis. Cancers 2021, 13, 6206. [Google Scholar] [CrossRef]
- Zhou, Y.; Tao, W.; Shen, F.; Du, W.; Xu, Z.; Liu, Z. The Emerging Role of Neutrophil Extracellular Traps in Arterial, Venous and Cancer Associated Thrombosis. Front. Cardiovasc. Med. 2021, 8, 786387. [Google Scholar] [CrossRef]
- Valadez-Cosmes, P.; Raftopoulou, S.; Mihalic, Z.N.; Marsche, G.; Kargl, J. Myeloperoxidase: Growing importance in cancer pathogenesis and potential drug target. Pharmacol. Ther. 2021, 236, 108052. [Google Scholar] [CrossRef]
- Duits, D.E.; de Visser, K.E. Impact of cancer cell-intrinsic features on neutrophil behavior. Semin. Immunol. 2021, 101546, in press. [Google Scholar] [CrossRef]
- Kaltenmeier, C.; Simmons, R.L.; Tohme, S.; Yazdani, H.O. Neutrophil Extracellular Traps (NETs) in Cancer Metastasis. Cancers 2021, 13, 6131. [Google Scholar] [CrossRef]
- Taucher, E.; Taucher, V.; Fink-Neuboeck, N.; Lindenmann, J.; Smolle-Juettner, F.-M. Role of Tumor-Associated Neutrophils in the Molecular Carcinogenesis of the Lung. Cancers 2021, 13, 5972. [Google Scholar] [CrossRef] [PubMed]
- Mangrolia, U.; Osborne, J.W. Probiotics in Counteracting the Role of Neutrophils in Cancer Metastasis. Vaccines 2021, 9, 1306. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Dong, L.; Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. J. Hematol. Oncol. 2021, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. Research Supporting a Pilot Study of Metronomic Dapsone during Glioblastoma Chemoirradiation. Med. Sci. 2021, 9, 12. [Google Scholar] [CrossRef]
- Kast, R.E. Dapsone as treatment adjunct in ARDS. Exp. Lung Res. 2020, 46, 157–161. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E.; Hill, Q.; Wion, D.; Mellstedt, H.; Focosi, D.; Karpel-Massler, G.; Heiland, T.; Halatsch, M.-E. Glioblastoma synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: Potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumour Biol. 2017, 39, 1010428317699797. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E. Erlotinib augmentation with dapsone for rash mitigation and increased anti-cancer effectiveness. Springerplus 2015, 4, 638. [Google Scholar] [CrossRef] [PubMed]
- Boccellino, M.; Quagliuolo, L.; Alaia, C.; Grimaldi, A.; Addeo, R.; Nicoletti, G.F.; Kast, R.E.; Caraglia, M. The strange connection between epidermal growth factor receptor tyrosine kinase inhibitors and dapsone: From rash mitigation to the increase in anti-tumor activity. Curr. Med. Res. Opin. 2016, 32, 1839–1848. [Google Scholar] [CrossRef]
- Asokan, S.; Bandapalli, O.R. CXCL8 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1302, 25–39. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Ambati, R.; Gundamaraju, R. Exploring the Crosstalk between Inflammation and Epithelial-Mesenchymal Transition in Cancer. Mediat. Inflamm. 2021, 2021, 9918379. [Google Scholar] [CrossRef]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef] [PubMed]
- Wozel, G.; Blasum, C. Dapsone in dermatology and beyond. Arch. Dermatol. Res. 2014, 306, 103–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyfman, M.; Debabov, D.; Poloso, N.; Alvandi, N. Mechanistic insight into the activity of a sulfone compound dapsone on Propionibacterium (Newly Reclassified as Cutibacterium) Acnes-mediated cytokine production. Exp. Dermatol. 2019, 28, 190–197. [Google Scholar] [CrossRef]
- Lan, C.-C.E.; Wu, C.-S.; Huang, S.-M.; Wu, I.-H.; Chen, G.-S. High-glucose environment enhanced oxidative stress and increased interleukin-8 secretion from keratinocytes: New insights into impaired diabetic wound healing. Diabetes 2013, 62, 2530–2538. [Google Scholar] [CrossRef] [Green Version]
- Ghaoui, N.; Hanna, E.; Abbas, O.; Kibbi, A.-G.; Kurban, M. Update on the use of dapsone in dermatology. Int. J. Dermatol. 2020, 59, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Kast, R.E.; Siegelin, M.D.; Dwucet, A.; Schneider, E.; Westhoff, M.-A.; Wirtz, C.R.; Chen, X.Y.; Halatsch, M.-E.; Bolm, C. Anti-glioma Activity of Dapsone and Its Enhancement by Synthetic Chemical Modification. Neurochem. Res. 2017, 42, 3382–3389. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Liu, R.C.; Consuegra, G.; Hui, R.; Fernandez-Penas, P. Epidermal growth factor receptor inhibitor-induced papulopustular eruption successfully treated with low-dose oral dapsone. Australas. J. Dermatol. 2018, 59, e219–e220. [Google Scholar] [CrossRef]
- Yan, P.; Zhu, H.; Yin, L.; Wang, L.; Xie, P.; Ye, J.; Jiang, X.; He, X. Integrin αvβ6 Promotes Lung Cancer Proliferation and Metastasis through Upregulation of IL-8-Mediated MAPK/ERK Signaling. Transl. Oncol. 2018, 11, 619–627. [Google Scholar] [CrossRef]
- Kanwar, B.A.; Khattak, A.; Balentine, J.; Lee, J.H.; Kast, R.E. Benefits of Using Dapsone in Patients Hospitalized with COVID-19. Vaccines 2022, 10, 195. [Google Scholar] [CrossRef]
- Kaltenmeier, C.T.; Yazdani, H.; van der Windt, D.; Molinari, M.; Geller, D.; Tsung, A.; Tohme, S. Neutrophil extracellular traps as a novel biomarker to predict recurrence-free and overall survival in patients with primary hepatic malignancies. HPB 2021, 23, 309–320. [Google Scholar] [CrossRef]
- Huh, G.; Ryu, J.K.; Chun, J.W.; Kim, J.S.; Park, N.; Cho, I.R.; Paik, W.H.; Lee, S.H.; Kim, Y.-T. High platelet-to-lymphocyte ratio is associated with poor prognosis in patients with unresectable intrahepatic cholangiocarcinoma receiving gemcitabine plus cisplatin. BMC Cancer 2020, 20, 907. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Kim, S.J.; Yu, M.H.; Lee, K.J.; Cha, Y.S. Uses of Inflammatory Markers for Differentiation of Intrahepatic Mass-Forming Cholangiocarcinoma from Liver Abscess: Case-Control Study. J. Clin. Med. 2020, 9, 3194. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Kang, Q.; Wang, L.; Liu, L.; Ke, Y.; Zhu, Y.; Zhang, N.; Xiong, S.; Li, Y.; Zou, H. Prognostic significance of the neutrophil-to-lymphocyte ratio with distal cholangiocarcinoma patients. Medicine 2020, 99, e22827. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, C.C.; Portilho, A.L.C.; Pinheiro, L.V.; Vivas, R.A.; Britto, M.; Montenegro, M.; Rodrigues, L.F.D.F.; Arruda, S.; Lyra, A.C.; Cavalcante, L.N. Sweet syndrome as a paraneoplastic manifestation of cholangiocarcinoma: A case report. World J. Clin. Cases 2020, 8, 4122–4127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Y.; Hu, K.; Huang, Y. Investigating effects of preoperative inflammatory biomarkers on predicting survival outcomes of intrahepatic cholangiocarcinoma after curative resection. World J. Surg. Oncol. 2020, 18, 272. [Google Scholar] [CrossRef]
- Chiu, T.-J.; Chen, Y.-J.; Kuo, F.-Y.; Chen, Y.-Y. Elevated neutrophil-to-lymphocyte ratio and predominance of intrahepatic cholangiocarcinoma prediction of poor hepatectomy outcomes in patients with combined hepatocellular-cholangiocarcinoma. PLoS ONE 2020, 15, e0240791. [Google Scholar] [CrossRef]
- Ren, A.; Li, Z.; Cheng, P.; Zhang, X.; Deng, R.; Ma, Y. Systemic Immune Inflammation Index Is a Prognostic Predictor in Patients with Intrahepatic Cholangiocarcinoma Undergoing Liver Transplantation. Mediat. Inflamm. 2021, 2021, 6656996. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef]
- Branchi, V.; Jürgensen, B.; Esser, L.; Gonzalez-Carmona, M.; Weismüller, T.; Strassburg, C.; Henn, J.; Semaan, A.; Lingohr, P.; Manekeller, S.; et al. Tumor Infiltrating Neutrophils Are Frequently Found in Adenocarcinomas of the Biliary Tract and Their Precursor Lesions with Possible Impact on Prognosis. J. Pers Med. 2021, 11, 233. [Google Scholar] [CrossRef]
- Ma, B.; Meng, H.; Shen, A.; Ma, Y.; Zhao, D.; Liu, G.; Zheng, S.; Tian, Y.; Zhang, W.; Li, Q.; et al. Prognostic Value of Inflammatory and Tumour Markers in Small-Duct Subtype Intrahepatic Cholangiocarcinoma after Curative-Intent Resection. Gastroenterol. Res. Pract. 2021, 2021, 6616062. [Google Scholar] [CrossRef]
- Roy, S.; Kumaravel, S.; Banerjee, P.; White, T.K.; O’Brien, A.; Seelig, C.; Chauhan, R.; Ekser, B.; Bayless, K.J.; Alpini, G.; et al. Tumor Lymphatic Interactions Induce CXCR2-CXCL5 Axis and Alter Cellular Metabolism and Lymphangiogenic Pathways to Promote Cholangiocarcinoma. Cells 2021, 10, 3093. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Tsuneyama, K.; Ishikawa, A.; Nakanuma, Y. Intrahepatic cholangiocarcinoma in cirrhosis presents granulocyte and granulocyte-macrophage colony-stimulating factor. Hum. Pathol. 2003, 34, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Boonyanugomol, W.; Chomvarin, C.; Hahnvajanawong, C.; Sripa, B.; Kaparakis-Liaskos, M.; Ferrero, R.L. Helicobacter pylori cag pathogenicity island (cagPAI) involved in bacterial internalization and IL-8 induced responses via NOD1- and MyD88-dependent mechanisms in human biliary epithelial cells. PLoS ONE 2013, 8, e77358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueoka, H.; Hirano, T.; Uda, Y.; Iimuro, Y.; Yamanaka, J.; Fujimoto, J. Blockage of CXCR2 suppresses tumor growth of intrahepatic cholangiocellular carcinoma. Surgery 2014, 155, 640–649. [Google Scholar] [CrossRef]
- Sun, Q.; Li, F.; Sun, F.; Niu, J. Interleukin-8 is a prognostic indicator in human hilar cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 8376–8384. [Google Scholar]
- Thongchot, S.; Ferraresi, A.; Vidoni, C.; Loilome, W.; Yongvanit, P.; Namwat, N.; Isidoro, C. Resveratrol interrupts the pro-invasive communication between cancer associated fibroblasts and cholangiocarcinoma cells. Cancer Lett. 2018, 430, 160–171. [Google Scholar] [CrossRef]
- Yamanaka, T.; Harimoto, N.; Yokobori, T.; Muranushi, R.; Hoshino, K.; Hagiwara, K.; Gantumur, D.; Handa, T.; Ishii, N.; Tsukagoshi, M.; et al. Nintedanib inhibits intrahepatic cholangiocarcinoma aggressiveness via suppression of cytokines extracted from activated cancer associated fibroblasts. Br. J. Cancer. 2020, 122, 986–994. [Google Scholar] [CrossRef]
- Li, S.; Yang, H.; Li, K.; Fan, G.; Deng, L.; Xu, C. Thymidine phosphorylase promotes angiogenesis and tumour growth in intrahepatic cholangiocarcinoma. Cell Biochem. Funct. 2020, 38, 743–752. [Google Scholar] [CrossRef]
- Dana, P.; Kariya, R.; Lert-Itthiporn, W.; Seubwai, W.; Saisomboon, S.; Wongkham, C.; Okada, S.; Wongkham, S.; Vaeteewoottacharn, K. Homophilic Interaction of CD147 Promotes IL-6-Mediated Cholangiocarcinoma Invasion via the NF-κB-Dependent Pathway. Int. J. Mol. Sci. 2021, 22, 13496. [Google Scholar] [CrossRef]
- Filippi, R.; Montagnani, F.; Lombardi, P.; Fornaro, L.; Aprile, G.; Casadei-Gardini, A.; Faloppi, L.; Palloni, A.; Satolli, M.A.; Scartozzi, M.; et al. A prognostic model in patients with advanced biliary tract cancer receiving first-line chemotherapy. Acta Oncol. 2021, 60, 1317–1324. [Google Scholar] [CrossRef]
- Biró, A.; Kolozsi, P.; Nagy, A.; Varga, Z.; Káposztás, Z.; Tóth, D. Significance of preoperative blood tests in the prognosis of colorectal cancer: A prospective, multicenter study from Hungary. J. Clin. Lab. Anal. 2022, 36, e24128. [Google Scholar] [CrossRef] [PubMed]
- Turhan, V.B.; Ünsal, A.; Gök, H.F.; Öztürk, B.; Öztürk, D.; Simsek, G.G.; Buluş, H. Predictive Value of Preoperative Neutrophil-Lymphocyte and Platelet-Lymphocyte Ratio in Determining the Stage of Colon Tumors. Cureus 2021, 13, e18381. [Google Scholar] [CrossRef] [PubMed]
- Mazaki, J.; Katsumata, K.; Sujino, H.; Udo, R.; Tago, T.; Kasahara, K.; Kuwabara, H.; Enomoto, M.; Ishizaki, T.; Nagakawa, Y.; et al. Neutrophil-to-lymphocyte Ratio as a Prognostic Factor for Colon Cancer in Elderly Patients: A Propensity Score Analysis. Anticancer Res. 2021, 41, 4471–4478. [Google Scholar] [CrossRef] [PubMed]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangère, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L.; et al. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.J.; Burge, M.; Feeney, K.; Gibbs, P.; Jones, K.; Marx, G.; Molloy, M.P.; Price, T.; Reece, W.H.H.; Segelov, E.; et al. The prognostic role of inflammatory markers in patients with metastatic colorectal cancer treated with bevacizumab: A translational study. PLoS ONE 2020, 15, e0229900. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, L.M.; Fritsch, M.; Gebauer, F.; Günther, S.D.; Stair, N.R.; Seeger, J.M.; Thangarajah, F.; Dieplinger, G.; Bludau, M.; Alakus, H.; et al. Tumour-infiltrating neutrophils counteract anti-VEGF therapy in metastatic colorectal cancer. Br. J. Cancer. 2019, 120, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Jiguet-Jiglaire, C.; Boissonneau, S.; Denicolai, E.; Hein, V.; Lasseur, R.; Garcia, J.; Romain, S.; Appay, R.; Graillon, T.; Mason, W.; et al. Plasmatic MMP9 released from tumor-infiltrating neutrophils is predictive for bevacizumab efficacy in glioblastoma patients: An AVAglio ancillary study. Acta Neuropathol. Commun. 2022, 1, 1–14. [Google Scholar] [CrossRef]
- Tan, K.W.; Chong, S.Z.; Wong, F.H.S.; Evrard, M.; Tan, S.M.-L.; Keeble, J.; Kemeny, D.M.; Ng, L.G.; Abastado, J.-P.; Angeli, V. Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D. Blood 2013, 122, 3666–3677. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [Green Version]
- Itatani, Y.; Yamamoto, T.; Zhong, C.; Molinolo, A.A.; Ruppel, J.; Hegde, P.; Taketo, M.M.; Ferrara, N. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 21598–21608. [Google Scholar] [CrossRef]
- Fisher, R.C.; Bellamkonda, K.; Molina, L.A.; Xiang, S.; Liska, D.; Sarvestani, S.K.; Chakrabarti, S.; Berg, A.; Jorgensen, M.L.; Hatala, D.; et al. Disrupting Inflammation-Associated CXCL8-CXCR1 Signaling Inhibits Tumorigenicity Initiated by Sporadic- and Colitis-Colon Cancer Stem Cells. Neoplasia 2019, 21, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, J.; Liu, J.; Xu, B.; Liang, X.; Yang, X.; Feng, Y.; Liu, J. IL-8/CXCR2 mediates tropism of human bone marrow-derived mesenchymal stem cells toward CD133+ /CD44+ Colon cancer stem cells. J. Cell Physiol. 2021, 236, 3114–3128. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Seo, Y.; Kwon, J.; Shin, Y.; Kim, S.; Park, S.J.; Park, J.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. IL-6 and IL-8, secreted by myofibroblasts in the tumor microenvironment, activate HES1 to expand the cancer stem cell population in early colorectal tumor. Mol. Carcinog. 2021, 60, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Bronikowska, J.; Kłósek, M.; Janeczko, T.; Kostrzewa-Susłow, E.; Czuba, Z.P. The modulating effect of methoxy-derivatives of 2′-hydroxychalcones on the release of IL-8, MIF, VCAM-1 and ICAM-1 by colon cancer cells. Biomed. Pharmacother. 2022, 145, 112428. [Google Scholar] [CrossRef]
- Watanabe, K.; Shiga, K.; Maeda, A.; Harata, S.; Yanagita, T.; Suzuki, T.; Ushigome, H.; Maeda, Y.; Hirokawa, T.; Ogawa, R.; et al. Chitinase 3-like 1 secreted from cancer-associated fibroblasts promotes tumor angiogenesis via interleukin-8 secretion in colorectal cancer. Int. J. Oncol. 2022, 60, 3. [Google Scholar] [CrossRef]
- Ning, Y.; Lenz, H.-J. Targeting IL-8 in colorectal cancer. Expert Opin. Ther. Targets. 2012, 16, 491–497. [Google Scholar] [CrossRef]
- Urbantat, R.; Vajkoczy, P.; Brandenburg, S. Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers 2021, 13, 2983. [Google Scholar] [CrossRef]
- Basheer, A.S.; Abas, F.; Othman, I.; Naidu, R. Role of Inflammatory Mediators, Macrophages, and Neutrophils in Glioma Maintenance and Progression: Mechanistic Understanding and Potential Therapeutic Applications. Cancers 2021, 13, 4226. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, X.; Du, J.; Zhang, D.; Liu, J.; Chen, M.; Zhao, J.; Zhong, W.; Xu, Y.; Wang, M. Circulating cytokines associated with clinical outcomes in advanced non-small cell lung cancer patients who received chemoimmunotherapy. Thorac. Cancer 2022, 13, 219–227. [Google Scholar] [CrossRef]
- Zadian, S.S.; Adcock, I.M.; Salimi, B.; Mortaz, E. Circulating Levels of Monocytic Myeloid-Derived Suppressor Cells (M-MDSC) and CXCL-8 in Non-Small Cell Lung Cancer (NSCLC). Tanaffos 2021, 20, 15–21. [Google Scholar]
- Yang, F.; Zhang, S.; Meng, Q.; Zhou, F.; Pan, B.; Liu, F.; Yu, Y. CXCR1 correlates to poor outcomes of EGFR-TKI against advanced non-small cell lung cancer by activating chemokine and JAK/STAT pathway. Pulm. Pharmacol. Ther. 2021, 67, 102001. [Google Scholar] [CrossRef] [PubMed]
- Hayama, N.; Hattori, S.; Takahashi, G.; Takahashi, F.; Takeuchi, T.; Tanaka, J.; Horio, Y.; Takiguchi, H.; Tomomatsu, K.; Kitahara, A.; et al. Cytokine/Chemokine/Growth Factor Levels in Malignant Pleural Effusion of Non-Small Cell Lung Cancer. Tokai J. Exp. Clin. Med. 2020, 45, 224–229. [Google Scholar] [PubMed]
- Qu, J.; Cheng, T.; Liu, L.; Heng, J.; Liu, X.; Sun, Z.; Wang, W.; Li, K.; Yang, N. Mast cells induce epithelial-to-mesenchymal transition and migration in non-small cell lung cancer through IL-8/Wnt/β-catenin pathway. J. Cancer 2019, 10, 5567. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Xu, Y.; Ding, R.; Qiu, K.; Zhang, R.; Wang, H.; Huang, L.; Xie, X.; Yan, H.; Deng, Y.; et al. Extensive serum biomarker analysis in patients with non-small-cell lung carcinoma. Cytokine 2020, 126, 154868. [Google Scholar] [CrossRef] [PubMed]
- Cury, S.S.; de Moraes, D.; Freire, P.P.; de Oliveira, G.; Marques, D.V.P.; Fernandez, G.J.; Dal-Pai-Silva, M.; Hasimoto, N.; dos Reis, P.P.; Rogatto, S.R.; et al. Tumor Transcriptome Reveals High Expression of IL-8 in Non-Small Cell Lung Cancer Patients with Low Pectoralis Muscle Area and Reduced Survival. Cancers 2019, 11, 1251. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Li, A.; Yu, S.; Qin, S.; Han, N.; Pestell, R.G.; Han, X.; Wu, K. DACH1 antagonizes CXCL8 to repress tumorigenesis of lung adenocarcinoma and improve prognosis. J. Hematol. Oncol. 2018, 11, 53. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Prognostic significance of baseline neutrophil-lymphocyte ratio in patients with non-small-cell lung cancer: A pooled analysis of open phase III clinical trial data. Future Oncol. 2022, 18. [Google Scholar] [CrossRef]
- Liu, N.; Mao, J.; Tao, P.; Chi, H.; Jia, W.; Dong, C. The relationship between NLR/PLR/LMR levels and survival prognosis in patients with non-small cell lung carcinoma treated with immune checkpoint inhibitors. Medicine 2022, 101, e28617. [Google Scholar] [CrossRef]
- Aduquaye, M.; Dube, S.; Bashir, B.; Chowdhury, A.; Ahmed, N.; Leylek, A.; Kim, J.; Lambert, P.; Bucher, O.; Hunter, W.; et al. Impact of Pre-Treatment NLR and Other Hematologic Biomarkers on the Outcomes of Early-Stage Non-Small-Cell Lung Cancer Treated with Stereotactic Body Radiation Therapy. Curr. Oncol. 2022, 29, 193–208. [Google Scholar] [CrossRef]
- Park, J.Y.; Jang, S.H.; Lee, C.Y.; Kim, T.; Chung, S.J.; Lee, Y.J.; Kim, H.I.; Kim, J.-H.; Park, S.; Hwang, Y.I.; et al. Pretreatment neutrophil-to-lymphocyte ratio and cigarette smoking as prognostic factors in patients with advanced NSCLC treated with osimertinib. Tuberc. Respir. Dis. 2022, 85, 115. [Google Scholar] [CrossRef]
- Bryant, A.K.; Sankar, K.; Strohbehn, G.W.; Zhao, L.; Elliott, D.; Qin, A.; Yentz, S.; Ramnath, N.; Green, M.D. Prognostic and predictive value of neutrophil-to-lymphocyte ratio with adjuvant immunotherapy in stage III non-small-cell lung cancer. Lung Cancer 2022, 163, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Poturnajova, M.; Kozovska, Z.; Matuskova, M. Aldehyde dehydrogenase 1A1 and 1A3 isoforms—Mechanism of activation and regulation in cancer. Cell Signal. 2021, 87, 110120. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J.; Saya, H. Molecular pathology underlying the robustness of cancer stem cells. Regen. Ther 2021, 17, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Li, X.; Ren, Y.; Zhang, X. Disulfiram: A novel repurposed drug for cancer therapy. Cancer Chemother. Pharmacol. 2021, 87, 159–172. [Google Scholar] [CrossRef]
- Belayneh, R.; Weiss, K. The Role of ALDH in the Metastatic Potential of Osteosarcoma Cells and Potential ALDH Targets. Adv. Exp. Med. Biol. 2020, 1258, 157–166. [Google Scholar] [CrossRef]
- Püschel, J.; Dubrovska, A.; Gorodetska, I. The Multifaceted Role of Aldehyde Dehydrogenases in Prostate Cancer Stem Cells. Cancers 2021, 13, 4703. [Google Scholar] [CrossRef]
- Salem, M.L.; El-Badawy, A.; Li, Z. Immunobiology and signaling pathways of cancer stem cells: Implication for cancer therapy. Cytotechnology 2015, 67, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.; Iniesta, C.B. Suppressing glioblastoma stem cell function by aldehyde dehydrogenase inhibition with chloramphenicol or disulfiram as a new treatment adjunct: An hypothesis. Curr. Stem. Cell Res. Ther. 2009, 4, 314–317. [Google Scholar] [CrossRef] [Green Version]
- Triscott, J.; Lee, C.; Hu, K.; Fotovati, A.; Berns, R.; Pambid, M.; Luk, M.; Kast, R.E.; Kong, E.; Toyota, E.; et al. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and overrides resistance to temozolomide. Oncotarget 2012, 3, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Rappa, F.; Cappello, F.; Halatsch, M.-E.; Scheuerle, A.; Kast, R. Aldehyde dehydrogenase and HSP90 co-localize in human glioblastoma biopsy cells. Biochimie 2013, 95, 782–786. [Google Scholar] [CrossRef]
- Brüning, A.; Kast, R.E. Oxidizing to death: Disulfiram for cancer cell killing. Cell Cycle 2014, 13, 1513–1514. [Google Scholar] [CrossRef]
- Kast, R.E.; Skuli, N.; Karpel-Massler, G.; Frosina, G.; Ryken, T.; Halatsch, M.-E. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: The EIS regimen. Oncotarget 2017, 8, 60727–60749. [Google Scholar] [CrossRef]
- Kast, R.E. The role of interleukin-18 in glioblastoma pathology implies therapeutic potential of two old drugs, disulfiram and ritonavir. Chin. J. Cancer 2015, 34, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, M.; Mylonas, I.; Kast, R.E.; Bruning, A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin mediated thioredoxin inhibition. Oncoscience 2013, 1, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Huang, T. Overview of Antabuse® (Disulfiram) in Radiation and Cancer Biology. Cancer Manag. Res. 2021, 13, 4095–4101. [Google Scholar] [CrossRef]
- Lu, Y.; Pan, Q.; Gao, W.; Pu, Y.; Luo, K.; He, B.; Gu, Z. Leveraging disulfiram to treat cancer: Mechanisms of action, delivery strategies, and treatment regimens. Biomaterials 2022, 281, 121335. [Google Scholar] [CrossRef]
- Chen, M.-H.; Weng, J.-J.; Cheng, C.-T.; Wu, R.-C.; Huang, S.-C.; Wu, C.-E.; Chung, Y.-H.; Liu, C.-Y.; Chang, M.-H.; Chiang, K.-C.; et al. ALDH1A3, the Major Aldehyde Dehydrogenase Isoform in Human Cholangiocarcinoma Cells, Affects Prognosis and Gemcitabine Resistance in Cholangiocarcinoma Patients. Clin. Cancer Res. 2016, 22, 4225–4235. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.-Y.; Hung, Y.-P.; Pan, Y.-R.; Chang, Y.-C.; Wu, C.-E.; Hsu, D.; Chang, P.; Lu, M.-L.; Huang, C.-Y.; Su, Y.; et al. Ruxolitinib Combined with Gemcitabine against Cholangiocarcinoma Growth via the JAK2/STAT1/3/ALDH1A3 Pathway. Biomedicines 2021, 9, 885. [Google Scholar] [CrossRef]
- Shuang, Z.-Y.; Wu, W.-C.; Xu, J.; Lin, G.; Liu, Y.-C.; Lao, X.-M.; Zheng, L.; Li, S. Transforming growth factor-β1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 2014, 354, 320–328. [Google Scholar] [CrossRef]
- Wang, M.; Xiao, J.; Jiang, J.; Qin, R. CD133 and ALDH may be the molecular markers of cholangiocarcinoma stem cells. Int. J. Cancer 2011, 128, 1996–1997. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, K.; Zhao, W.; Liu, Z.; Liu, J.; Shi, A.; Chen, T.; Mu, W.; Xu, Y.; Pan, C.; et al. Aldehyde dehydrogenase 3B2 promotes the proliferation and invasion of cholangiocarcinoma by increasing Integrin Beta 1 expression. Cell Death Dis. 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- McGrath, N.; Fu, J.; Gu, S.Z.; Xie, C. Targeting cancer stem cells in cholangiocarcinoma. Int. J. Oncol. 2020, 57, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Modarai, S.R.; Gupta, A.; Opdenaker, L.M.; Kowash, R.; Masters, G.; Viswanathan, V.; Zhang, T.; Fields, J.Z.; Boman, B.M. The anti-cancer effect of retinoic acid signaling in CRC occurs via decreased growth of ALDH+ colon cancer stem cells and increased differentiation of stem cells. Oncotarget 2018, 9, 34658–34669. [Google Scholar] [CrossRef] [Green Version]
- Holah, N.S.; Aiad, H.A.; Asaad, N.Y.; Elkhouly, E.A.; Lasheen, A.G. Evaluation of the Role of ALDH1 as Cancer Stem Cell Marker in Colorectal Carcinoma: An Immunohistochemical Study. J. Clin. Diagn Res. 2017, 11, EC17–EC23. [Google Scholar] [CrossRef]
- Shenoy, A.; Butterworth, E.; Huang, E.H. ALDH as a marker for enriching tumorigenic human colonic stem cells. Methods Mol. Biol. 2012, 916, 373–385. [Google Scholar] [CrossRef]
- Tsochantaridis, I.; Roupas, A.; Voulgaridou, G.-P.; Giatromanolaki, A.; Koukourakis, M.I.; Panayiotidis, M.I.; Pappa, A. Aldehyde Dehydrogenase 1B1 Is Associated with Altered Cell Morphology, Proliferation, Migration and Chemosensitivity in Human Colorectal Adenocarcinoma Cells. Biomedicines 2021, 9, 44. [Google Scholar] [CrossRef]
- Chen, Y.; Orlicky, D.J.; Matsumoto, A.; Singh, S.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenase 1B1 (ALDH1B1) is a potential biomarker for human colon cancer. Biochem. Biophys. Res. Commun. 2011, 405, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Abu-Serie, M.; El-Rashidy, F. In Vitro Collapsing Colon Cancer Cells by Selectivity of Disulfiram-Loaded Charge Switchable Nanoparticles Against Cancer Stem Cells. Recent Pat. Anticancer Drug Discov. 2017, 12, 260–271. [Google Scholar] [CrossRef]
- Guo, X.; Xu, B.; Pandey, S.; Goessl, E.; Brown, J.; Armesilla, A.; Darling, J.L.; Wang, W. Disulfiram/copper complex inhibiting NFkappaB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. 2010, 290, 104–113. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Peng, X.-H.; Nandigama, K.; Tekle, S.; Ambudkar, S.V. The molecular basis of the action of disulfiram as a modulator of the multidrug resistance-linked ATP binding cassette transporters MDR1 (ABCB1) and MRP1 (ABCC1). Mol. Pharmacol. 2004, 65, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J. Natl. Cancer Inst. 2000, 92, 898–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Disulfiram metabolites permanently inactivate the human multidrug resistance P-glycoprotein. Mol. Pharm. 2004, 1, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mo, Y.; Tan, Y.; Wen, Z.; Dai, Z.; Zhang, H.; Zhang, X.; Feng, S.; Liang, X.; Song, T.; et al. The ALDH Family Contributes to Immunocyte Infiltration, Proliferation and Epithelial Mesenchymal Transformation in Glioma. Front. Immunol. 2022, 12, 756606. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Wu, C.-L.; Lin, M.-X.; Sze, C.-I.; Gean, P.-W. Disulfiram Sensitizes a Therapeutic-Resistant Glioblastoma to the TGF-β Receptor Inhibitor. Int. J. Mol. Sci. 2021, 22, 10496. [Google Scholar] [CrossRef] [PubMed]
- Gelardi, E.; Colombo, G.; Picarazzi, F.; Ferraris, D.; Mangione, A.; Petrarolo, G.; Aronica, E.; Rizzi, M.; Mori, M.; La Motta, C.; et al. A Selective Competitive Inhibitor of Aldehyde Dehydrogenase 1A3 Hinders Cancer Cell Growth, Invasiveness and Stemness In Vitro. Cancers 2021, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.; Sullivan, L.A.; et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef] [Green Version]
- Serrano, D.; Bleau, A.-M.; Fernandez-Garcia, I.; Fernandez-Marcelo, T.; Iniesta, P.; Ortiz-De-Solorzano, C.; Calvo, A. Inhibition of telomerase activity preferentially targets aldehyde dehydrogenase-positive cancer stem-like cells in lung cancer. Mol. Cancer. 2011, 10, 96. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Sullivan, J.P.; Girard, L.; Augustyn, A.; Yenerall, P.; Rodriguez-Canales, J.; Liu, H.; Behrens, C.; Shay, J.W.; Wistuba, I.I.; et al. Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway. Clin. Cancer Res. 2014, 20, 4154–4166. [Google Scholar] [CrossRef] [Green Version]
- Terzuoli, E.; Bellan, C.; Aversa, S.; Ciccone, V.; Morbidelli, L.; Giachetti, A.; Donnini, S.; Ziche, M. ALDH3A1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance through Enhanced PD-L1 Output. Cancers 2019, 11, 1963. [Google Scholar] [CrossRef] [Green Version]
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. CD44+/EPCAM+ cells detect a subpopulation of ALDHhigh cells in human non-small cell lung cancer: A chance for targeting cancer stem cells? Oncotarget 2020, 11, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Cui, W.; Yuan, X.; Lin, L.; Cao, Q.; Wang, N.; Li, Y.; Guo, W.; Zhang, X.; et al. Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells. Oncotarget 2016, 7, 58516–58530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Xue, X.; Wang, L.; Wang, W.; Han, J.; Sun, X.; Zhang, H.; Liu, Y.; Che, X.; Yang, J.; et al. Suppressing autophagy enhances disulfiram/copper-induced apoptosis in non-small cell lung cancer. Eur. J. Pharmacol. 2018, 827, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Jeon, Y.; Kang, J.H.; Jang, H.; Lee, H.; Kim, S.-Y. The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers 2020, 12, 1382. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.W.; Elbassiouny, M.; Elkhodary, D.A.; Shawki, M.A.; Saad, A.S. The effect of itraconazole on the clinical outcomes of patients with advanced non-small cell lung cancer receiving platinum based chemotherapy: A randomized controlled study. Med. Oncol. 2021, 38, 23. [Google Scholar] [CrossRef]
- Shen, P.-W.; Chou, Y.-M.; Li, C.-L.; Liao, E.-C.; Huang, H.-S.; Yin, C.-H.; Chen, C.-L.; Yu, S.-J. Itraconazole improves survival outcomes in patients with colon cancer by inducing autophagic cell death and inhibiting transketolase expression. Oncol. Lett. 2021, 22, 768. [Google Scholar] [CrossRef]
- Zhang, W.; Bhagwath, A.S.; Ramzan, Z.; Williams, T.A.; Subramaniyan, I.; Edpuganti, V.; Kallem, R.R.; Dunbar, K.B.; Ding, P.; Gong, K.; et al. Itraconazole Exerts Its Antitumor Effect in Esophageal Cancer By Suppressing the HER2/AKT Signaling Pathway. Mol. Cancer Ther. 2021, 20, 1904–1915. [Google Scholar] [CrossRef]
- El-Sheridy, N.A.; El-Moslemany, R.M.; Ramadan, A.A.; Helmy, M.W.; El-Khordagui, L.K. Enhancing the in vitro and in vivo activity of itraconazole against breast cancer using miltefosine modified lipid nanocapsules. Drug Deliv. 2021, 28, 906–919. [Google Scholar] [CrossRef]
- Ghadi, M.; Hosseinimehr, S.J.; Amiri, F.T.; Mardanshahi, A.; Noaparast, Z. Data on the in vitro and in vivo anti-tumor effects of itraconazole, paclitaxel, and the two in combination in HT-29 and YM-1 cancer cell line and HT-29 colon cancer xenograft models. Data Brief. 2021, 35, 106862. [Google Scholar] [CrossRef]
- Li, K.; Fang, D.; Xiong, Z.; Luo, R. Inhibition of the hedgehog pathway for the treatment of cancer using Itraconazole. Onco Targets Ther. 2019, 12, 6875–6886. [Google Scholar] [CrossRef] [Green Version]
- Ban, L.; Mei, T.; Su, Q.; Li, W.; Huang, Z.; Liu, L.; Wu, Y.; Lv, S.; Wang, A.; Li, S. Anti-fungal drug itraconazole exerts anti-cancer effects in oral squamous cell carcinoma via suppressing Hedgehog pathway. Life Sci. 2020, 254, 117695. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Liu, W.; Wang, J.Q.; Tang, Z. “Hedgehog pathway”: A potential target of itraconazole in the treatment of cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, W. Itraconazole Alters the Stem Cell Characteristics of A549 and NCI-H460 Human Lung Cancer Cells by Suppressing Wnt Signaling. Med. Sci Monit. 2019, 25, 9509–9516. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.D.; Dias, R.B.; Vidal, M.T.A.; Valverde, L.D.F.; Costa, R.G.A.; Damasceno, A.K.A.; Sales, C.B.S.; Rocha, L.D.O.S.D.; Dos Reis, M.G.; Soares, M.B.P.; et al. Inhibition of CAL27 Oral Squamous Carcinoma Cell by Targeting Hedgehog Pathway With Vismodegib or Itraconazole. Front. Oncol. 2020, 10, 563838. [Google Scholar] [CrossRef]
- Lin, Y.; Cai, Q.; Chen, Y.; Shi, T.; Liu, W.; Mao, L.; Deng, B.; Ying, Z.; Gao, Y.; Luo, H.; et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5-lipoxygenase. Hepatology 2022, 75, 28–42. [Google Scholar] [CrossRef]
- Jaschonek, K.; Steinhilber, D.; Einsele, H.; Ehninger, G.; Roth, H.J. 5-Lipoxygenase inhibition by antifungal azole derivatives: New tools for immunosuppression? Eicosanoids 1989, 2, 189–190. [Google Scholar]
- Steel, H.C.; Tintinger, G.R.; Theron, A.J.; Anderson, R. Itraconazole mediated inhibition of calcium entry into platelet-activating factor-stimulated human neutrophils is due to interference with production of leukotriene B4. Clin. Exp. Immunol. 2007, 150, 144–150. [Google Scholar] [CrossRef]
- Steinhilber, D.; Jaschonek, K.; Knospe, J.; Morof, O.; Roth, H.J. Effects of novel antifungal azole derivatives on the 5-lipoxygenase and cyclooxygenase pathway. Arzneimittelforschung 1990, 40, 1260–1263. [Google Scholar]
- Lempers, V.J.C.; Heuvel, J.J.M.W.V.D.; Russel, F.G.M.; Aarnoutse, R.E.; Burger, D.M.; Brüggemann, R.J.; Koenderink, J.B. Inhibitory Potential of Antifungal Drugs on ATP-Binding Cassette Transporters P-Glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob. Agents Chemother. 2016, 60, 3372–3379. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Abe, Y.; Kawai, A.; Furihata, T.; Endo, T.; Takeda, H. Pharmacokinetic Drug Interactions of an Orally Available TRH Analog (Rovatirelin) with a CYP3A4/5 and P-Glycoprotein Inhibitor (Itraconazole). J. Clin. Pharmacol. 2020, 60, 1314–1323. [Google Scholar] [CrossRef]
- Ghadi, M.; Hosseinimehr, S.J.; Amiri, F.T.; Mardanshahi, A.; Noaparast, Z. Itraconazole synergistically increases therapeutic effect of paclitaxel and 99mTc-MIBI accumulation, as a probe of P-gp activity, in HT-29 tumor-bearing nude mice. Eur. J. Pharmacol. 2021, 895, 173892. [Google Scholar] [CrossRef] [PubMed]
- Quatannens, D.; Verhoeven, Y.; Van Dam, P.; Lardon, F.; Prenen, H.; Roeyen, G.; Peeters, M.; Smits, E.L.J.; Van Audenaerde, J. Targeting hedgehog signaling in pancreatic ductal adenocarcinoma. Pharmacol. Ther. 2022, 236, 108107. [Google Scholar] [CrossRef]
- Jain, R.; Dubey, S.K.; Singhvi, G. The Hedgehog pathway and its inhibitors: Emerging therapeutic approaches for basal cell carcinoma. Drug Discov. Today 2021, 27, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Gampala, S.; Yang, J.-Y. Hedgehog Pathway Inhibitors against Tumor Microenvironment. Cells 2021, 10, 3135. [Google Scholar] [CrossRef]
- Jeng, K.-S.; Chang, C.-F.; Lin, S.-S. Sonic Hedgehog Signaling in Organogenesis, Tumors, and Tumor Microenvironments. Int. J. Mol. Sci. 2020, 21, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wang, Y.; Xie, J. The Hedgehog pathway: Role in cell differentiation, polarity and proliferation. Arch. Toxicol. 2015, 89, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.-C.; Lin, J.-Y.; Hsu, S.-H.; Lan, W.-Y.; Kuo, C.-S.; Tian, Y.-F.; Sun, D.-P.; Huang, R.-F.S. Low folate metabolic stress reprograms DNA methylation-activated sonic hedgehog signaling to mediate cancer stem cell-like signatures and invasive tumour stage-specific malignancy of human colorectal cancers. Int. J. Cancer. 2017, 141, 2537–2550. [Google Scholar] [CrossRef]
- Cho, K.; Moon, H.; Seo, S.H.; Ro, S.W.; Kim, B.K. Pharmacological Inhibition of Sonic Hedgehog Signaling Suppresses Tumor Development in a Murine Model of Intrahepatic Cholangiocarcinoma. Int. J. Mol. Sci. 2021, 22, 13214. [Google Scholar] [CrossRef]
- Möbius, C.; Aust, G.; Wiedmann, M.; Wittekind, C.; Mössner, J.; Hauss, J.; Witzigmann, H. Prognostic value of eicosanoid pathways in extrahepatic cholangiocarcinoma. Anticancer Res. 2008, 28, 873–878. [Google Scholar]
- Khophai, S.; Thanee, M.; Techasen, A.; Namwat, N.; Klanrit, P.; Titapun, A.; Jarearnrat, A.; Sa-Ngiamwibool, P.; Loilome, W. Zileuton suppresses cholangiocarcinoma cell proliferation and migration through inhibition of the Akt signaling pathway. Onco Targets Ther. 2018, 11, 7019–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anichini, G.; Carrassa, L.; Stecca, B.; Marra, F.; Raggi, C. The Role of the Hedgehog Pathway in Cholangiocarcinoma. Cancers 2021, 13, 4774. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, H.-Y.; Zhang, L.; Zhou, Y.; Wu, J. Hedgehog Signaling, a Critical Pathway Governing the Development and Progression of Hepatocellular Carcinoma. Cells 2021, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Sirica, A.E. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 44–54. [Google Scholar] [CrossRef]
- Omenetti, A.; Diehl, A.M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 2011, 27, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Khamko, R.; Daduang, J.; Settasatian, C.; Limpaiboon, T. OPCML Exerts Antitumor Effects in Cholangiocarcinoma via AXL/STAT3 Inactivation and Rho GTPase Down-regulation. Cancer Genom. Proteomics 2021, 18, 771–780. [Google Scholar] [CrossRef]
- Wasilewicz, M.P.; Kołodziej, B.; Bojułko, T.; Kaczmarczyk, M.; Sulzyc-Bielicka, V.; Bielicki, D.; Ciepiela, K. Overexpression of 5-lipoxygenase in sporadic colonic adenomas and a possible new aspect of colon carcinogenesis. Int. J. Colorectal Dis. 2010, 25, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Che, X.H.; Chen, C.L.; Ye, X.L.; Weng, G.B.; Guo, X.Z.; Yu, W.Y.; Tao, J.; Chen, Y.C.; Chen, X. Dual inhibition of COX-2/5-LOX blocks colon cancer proliferation, migration and invasion in vitro. Oncol. Rep. 2016, 35, 1680–1688. [Google Scholar] [CrossRef] [Green Version]
- Barresi, V.; Grosso, M.; Vitarelli, E.; Tuccari, G.; Barresi, G. 5-Lipoxygenase is coexpressed with Cox-2 in sporadic colorectal cancer: A correlation with advanced stage. Dis. Colon Rectum 2007, 50, 1576–1584. [Google Scholar] [CrossRef]
- Shen, J.; Li, W.; Xiao, Z.; Zhang, L.; Li, M.; Li, L.; Hu, W.; Lu, L.; Boudreau, F.; Cho, C. The Co-regulatory Role of 5-Lipoxygenase and Cyclooxygenase-2 in the Carcinogenesis and their Promotion by Cigarette Smoking in Colons. Curr. Med. Chem. 2016, 23, 1131–1138. [Google Scholar] [CrossRef]
- Rao, C.V.; Janakiram, N.B.; Mohammed, A. Lipoxygenase and Cyclooxygenase Pathways and Colorectal Cancer Prevention. Curr. Colorectal Cancer Rep. 2012, 8, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, U.D.; Garona, J.; Ripoll, G.V.; Maloberti, P.M.; Solano, A.R.; Avagnina, A.; Gomez, D.E.; Alonso, D.F.; Podesta, E.J. The functional interaction between Acyl-CoA synthetase 4, 5-lipooxygenase and cyclooxygenase-2 controls tumor growth: A novel therapeutic target. PLoS ONE 2012, 7, e40794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Guan, L.; Zheng, J. Role of leukotriene B4 in celecoxib-mediated anticancer effect. Biochem. Biophys. Res. Commun. 2010, 402, 308–311. [Google Scholar] [CrossRef]
- Cianchi, F.; Cortesini, C.; Magnelli, L.; Fanti, E.; Papucci, L.; Schiavone, N.; Messerini, L.; Vannacci, A.; Capaccioli, S.; Perna, F.; et al. Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol. Cancer Ther. 2006, 5, 2716–2726. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.; Vyas, A.; Misra, S.; O’Brien, P.; Zambre, A.; Fresco, V.M.; Markwald, R.R.; Swamy, K.V.; Afrasiabi, Z.; Choudhury, A.; et al. Novel di-tertiary-butyl phenylhydrazones as dual cyclooxygenase-2/5-lipoxygenase inhibitors: Synthesis, COX/LOX inhibition, molecular modeling, and insights into their cytotoxicities. Bioorg. Med. Chem. Lett. 2014, 24, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Knab, L.M.; Grippo, P.J.; Bentrem, D.J. Involvement of eicosanoids in the pathogenesis of pancreatic cancer: The roles of cyclooxygenase-2 and 5-lipoxygenase. World J. Gastroenterol. 2014, 20, 10729–10739. [Google Scholar] [CrossRef]
- Costa, H.; Touma, J.; Davoudi, B.; Benard, M.; Sauer, T.; Geisler, J.; Vetvik, K.; Rahbar, A.; Söderberg-Naucler, C. Human cytomegalovirus infection is correlated with enhanced cyclooxygenase-2 and 5-lipoxygenase protein expression in breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2083–2095. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Huang, L.; Zhang, W.; Ning, X. Tepoxalin a dual 5-LOX-COX inhibitor and erlotinib an EGFR inhibitor halts progression of gastric cancer in tumor xenograft mice. Am. J. Transl Res. 2018, 10, 3847–3856. [Google Scholar]
- Cummings, M.; Massey, K.A.; Mappa, G.; Wilkinson, N.; Hutson, R.; Munot, S.; Saidi, S.; Nugent, D.; Broadhead, T.; Wright, A.I.; et al. Integrated eicosanoid lipidomics and gene expression reveal decreased prostaglandin catabolism and increased 5-lipoxygenase expression in aggressive subtypes of endometrial cancer. J. Pathol. 2019, 247, 21–34. [Google Scholar] [CrossRef]
- Douard, R.; Moutereau, S.; Pernet, P.; Chimingqi, M.; Allory, Y.; Manivet, P.; Conti, M.; Vaubourdolle, M.; Cugnenc, P.-H.; Loric, S. Sonic Hedgehog-dependent proliferation in a series of patients with colorectal cancer. Surgery 2006, 139, 665–670. [Google Scholar] [CrossRef]
- Deng, H.; Huang, L.; Liao, Z.; Liu, M.; Li, Q.; Xu, R. Itraconazole inhibits the Hedgehog signaling pathway thereby inducing autophagy-mediated apoptosis of colon cancer cells. Cell Death Dis. 2020, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.A.; Buczacki, S.J.A. Itraconazole perturbs colorectal cancer dormancy through SUFU-mediated WNT inhibition. Mol. Cell. Oncol. 2018, 5, e1494950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczacki, S.J.A.; Popova, S.; Biggs, E.; Koukorava, C.; Buzzelli, J.; Vermeulen, L.; Hazelwood, L.; Francies, H.; Garnett, M.J.; Winton, D.J. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J. Exp. Med. 2018, 215, 1891–1912. [Google Scholar] [CrossRef] [PubMed]
- Geyer, N.; Gerling, M. Hedgehog Signaling in Colorectal Cancer: All in the Stroma? Int. J. Mol. Sci. 2021, 22, 1025. [Google Scholar] [CrossRef]
- Lukic, A.; Wahlund, C.J.; Gómez, C.; Brodin, D.; Samuelsson, B.; Wheelock, C.E.; Gabrielsson, S.; Rådmark, O. Exosomes and cells from lung cancer pleural exudates transform LTC4 to LTD4, promoting cell migration and survival via CysLT1. Cancer Lett. 2019, 444, 1–8. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 619–623. [Google Scholar] [CrossRef] [Green Version]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Liu, M.; Wang, Q.; Shen, Y.; Mei, H.; Li, D.; Liu, W. Itraconazole exerts its anti-melanoma effect by suppressing Hedgehog, Wnt, and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 28510–28525. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Liu, Z.; Yang, K.; Kong, C.; Liu, C.; Chen, H.; Huang, J.; Qian, F. Tumor Progression of Non-Small Cell Lung Cancer Controlled by Albumin and Micellar Nanoparticles of Itraconazole, a Multitarget Angiogenesis Inhibitor. Mol. Pharm. 2017, 14, 4705–4713. [Google Scholar] [CrossRef]
- Lee, W.-H.; Loo, J.C.Y.; Ghadiri, M.; Leong, C.-R.; Young, P.; Traini, D. The potential to treat lung cancer via inhalation of repurposed drugs. Adv. Drug Deliv. Rev. 2018, 133, 107–130. [Google Scholar] [CrossRef]
- Alhakamy, N.A.; Md, S. Repurposing Itraconazole Loaded PLGA Nanoparticles for Improved Antitumor Efficacy in Non-Small Cell Lung Cancers. Pharmaceutics 2019, 11, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.E.; Putnam, W.C.; Fattah, F.J.; Kernstine, K.H.; Brekken, R.A.; Pedrosa, I.; Skelton, R.; Saltarski, J.M.; Lenkinski, R.E.; Leff, R.D.; et al. Concentration-dependent Early Antivascular and Antitumor Effects of Itraconazole in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 6017–6027. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Leyton, J.; Martinez, A.; Hong, S.; Malkinson, A.; Mulshine, J.L. Lipoxygenase inhibitors prevent lung carcinogenesis and inhibit non-small cell lung cancer growth. Exp. Lung Res. 1998, 24, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.; Halatsch, M.-E.; Rosell, R. OPALS: A New Osimertinib Adjunctive Treatment of Lung Adenocarcinoma or Glioblastoma Using Five Repurposed Drugs. Cells 2021, 10, 1148. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Walker, S.R.; Heppler, L.N.; Tyekucheva, S.; Nelson, E.A.; Klitgaard, J.; Nicolais, M.; Kroll, Y.; Xiang, M.; Yeh, J.E.; et al. Targeting constitutively active STAT3 in chronic lymphocytic leukemia: A clinical trial of the STAT3 inhibitor pyrimethamine with pharmacodynamic analyses. Am. J. Hematol. 2021, 96, E95–E98. [Google Scholar] [CrossRef] [PubMed]
- Ramchandani, S.; Mohan, C.D.; Mistry, J.R.; Su, Q.; Naz, I.; Rangappa, K.S.; Ahn, K.S. The multifaceted antineoplastic role of pyrimethamine against different human malignancies. IUBMB Life 2021, 74, 198–212. [Google Scholar] [CrossRef]
- Assaraf, Y.; Slotky, J. Characterization of a lipophilic antifolate resistance provoked by treatment of mammalian cells with the antiparasitic agent pyrimethamine. J. Biol. Chem. 1993, 268, 4556–4566. [Google Scholar] [CrossRef]
- Hannewald, P.; Maunit, B.; Muller, J.-F. Screening of DHFR-binding drugs by MALDI-TOFMS. Anal. Bioanal. Chem. 2008, 392, 1335–1344. [Google Scholar] [CrossRef]
- Reynolds, E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006, 5, 949–960. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Koźmiński, P.; Halik, P.K.; Chesori, R.; Gniazdowska, E. Overview of Dual-Acting Drug Methotrexate in Different Neurological Diseases, Autoimmune Pathologies and Cancers. Int. J. Mol. Sci. 2020, 21, 3483. [Google Scholar] [CrossRef]
- Sharma, A.; Jyotsana, N.; Lai, C.K.; Chaturvedi, A.; Gabdoulline, R.; Görlich, K.; Murphy, C.; Blanchard, J.E.; Ganser, A.; Brown, E.; et al. Pyrimethamine as a Potent and Selective Inhibitor of Acute Myeloid Leukemia Identified by High-throughput Drug Screening. Curr. Cancer Drug Targets 2016, 16, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M. Molecular and cellular effects of methotrexate. Curr. Opin. Rheumatol. 1999, 11, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Bedoui, Y.; Guillot, X.; Sélambarom, J.; Guiraud, P.; Giry, C.; Jaffar-Bandjee, M.C.; Ralandison, S.; Gasque, P. Methotrexate an Old Drug with New Tricks. Int. J. Mol. Sci. 2019, 20, 5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowcock, S.; Linch, D.; Machin, S.; Stewart, J. Pyrimethamine in the myeloproliferative disorders: A forgotten treatment? Clin. Lab. Haematol. 1987, 9, 129–136. [Google Scholar] [CrossRef]
- Hage, A.; Schoemaker, H.E.; Wever, R.; Zennaro, E.; Heipieper, H.J. Pyrimethamine in prevention of relapses of meningeal leukemia: Report of two cases. Cancer 1978, 42, 1216–1218. [Google Scholar] [CrossRef]
- Smyth, A.C.; Wiernik, P.H. Combination chemotherapy of adult acute lymphocytic leukemia. Clin. Pharmacol. Ther. 1976, 19, 240–245. [Google Scholar] [CrossRef]
- Liu, H.; Qin, Y.; Zhai, D.; Zhang, Q.; Gu, J.; Tang, Y.; Yang, J.; Li, K.; Yang, L.; Chen, S.; et al. Antimalarial Drug Pyrimethamine Plays a Dual Role in Antitumor Proliferation and Metastasis through Targeting DHFR and TP. Mol. Cancer Ther. 2019, 18, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Tertil, M.; Skrzypek, K.; Florczyk, U.; Węglarczyk, K.; Was, H.; Collet, G.; Guichard, A.; Gil, T.; Kuzdzal, J.; Jozkowicz, A.; et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: Crosstalk with Nrf2 and HO-1. PLoS ONE 2014, 9, e97070. [Google Scholar] [CrossRef]
- Furukawa, T.; Tabata, S.; Yamamoto, M.; Kawahara, K.; Shinsato, Y.; Minami, K.; Shimokawa, M.; Akiyama, S.-I. Thymidine phosphorylase in cancer aggressiveness and chemoresistance. Pharmacol. Res. 2018, 132, 15–20. [Google Scholar] [CrossRef]
- Tabata, S.; Yamamoto, M.; Goto, H.; Hirayama, A.; Ohishi, M.; Kuramoto, T.; Mitsuhashi, A.; Ikeda, R.; Haraguchi, M.; Kawahara, K.; et al. Thymidine Catabolism as a Metabolic Strategy for Cancer Survival. Cell Rep. 2017, 19, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, S.; Ikeda, R.; Yamamoto, M.; Shimaoka, S.; Mukaida, N.; Takeda, Y.; Yamada, K.; Soga, T.; Furukawa, T.; Akiyama, S.-I. Thymidine phosphorylase activates NFκB and stimulates the expression of angiogenic and metastatic factors in human cancer cells. Oncotarget 2014, 5, 10473–10485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, S.-I.; Tabata, S.; Ikeda, R.; Yamamoto, M.; Furukawa, T.; Kuramoto, T.; Takeda, Y.; Yamada, K.; Haraguchi, M.; Nishioka, Y.; et al. Thymidine phosphorylase enhances reactive oxygen species generation and interleukin-8 expression in human cancer cells. Oncol. Rep. 2012, 28, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.W.; Saadalla, A.; Ewida, A.H.; Al-Katranji, K.; Al-Saoudi, G.; Giaccone, Z.T.; Gounari, F.; Zhang, M.; Frank, D.A.; Khazaie, K. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol. Immunother. 2018, 67, 13–23. [Google Scholar] [CrossRef]
- Wu, B.; Fathi, S.; Mortley, S.; Mohiuddin, M.; Jang, Y.C.; Oyelere, A.K. Pyrimethamine conjugated histone deacetylase inhibitors: Design, synthesis and evidence for triple negative breast cancer selective cytotoxicity. Bioorg. Med. Chem. 2020, 28, 115345. [Google Scholar] [CrossRef] [PubMed]
- Egusquiaguirre, S.P.; Yeh, J.E.; Walker, S.R.; Liu, S.; Frank, D.A. The STAT3 Target Gene TNFRSF1A Modulates the NF-κB Pathway in Breast Cancer Cells. Neoplasia 2018, 20, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, M.; Case, A.E.; Larios, D.; Gandler, H.I.; Meng, C.; Tošić, I.; Weisberg, E.L.; Poitras, M.J.; Gokhale, P.C.; Paweletz, C.P.; et al. Inhibitors of the Transcription Factor STAT3 Decrease Growth and Induce Immune Response Genes in Models of Malignant Pleural Mesothelioma (MPM). Cancers 2020, 13, 7. [Google Scholar] [CrossRef]
- Heppler, L.N.; Attarha, S.; Persaud, R.; Brown, J.I.; Wang, P.; Petrova, B.; Tošić, I.; Burton, F.B.; Flamand, Y.; Walker, S.R.; et al. The antimicrobial drug pyrimethamine inhibits STAT3 transcriptional activity by targeting the enzyme dihydrofolate reductase. J. Biol. Chem. 2022, 298, 101531. [Google Scholar] [CrossRef]
- Wu, B.; Payero, B.; Taylor, S.; Oyelere, A.K. Discovery of novel STAT3 DNA binding domain inhibitors. Future Med. Chem. 2021, 13, 1253–1269. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z.; Han, J.; Ma, X.; Zheng, X.; Chen, J. Functional and Therapeutic Significance of Tumor-Associated Macrophages in Colorectal Cancer. Front. Oncol. 2022, 12, 781233. [Google Scholar] [CrossRef]
- El-Tanani, M.; Al Khatib, A.O.; Aladwan, S.M.; Abuelhana, A.; McCarron, P.A.; Tambuwala, M.M. Importance of STAT3 signalling in cancer, metastasis and therapeutic interventions. Cell Signal. 2022, 92, 110275. [Google Scholar] [CrossRef] [PubMed]
- Ayele, T.M.; Muche, Z.T.; Teklemariam, A.B.; Bogale, A.; Abebe, E.C. Role of JAK2/STAT3 Signaling Pathway in the Tumorigenesis, Chemotherapy Resistance, and Treatment of Solid Tumors: A Systemic Review. J. Inflamm. Res. 2022, 15, 1349–1364. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Mohammad, I.S.; Liu, Z. Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors. Oncol. Lett. 2020, 19, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.; Yakou, M.; Pang, L.; Ernst, M.; Huynh, J. Exploiting the STAT3 Nexus in Cancer-Associated Fibroblasts to Improve Cancer Therapy. Front. Immunol. 2021, 12, 767939. [Google Scholar] [CrossRef]
- Parakh, S.; Ernst, M.; Poh, A.R. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef]
- Tošić, I.; Frank, D.A. STAT3 as a mediator of oncogenic cellular metabolism: Pathogenic and therapeutic implications. Neoplasia 2021, 23, 1167–1178. [Google Scholar] [CrossRef]
- Su, Y.-L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int. J. Mol. Sci. 2018, 19, 1803. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, D.; Uhle, F.; Sahin, D.; Krauser, U.; Weigand, M.A.; Heeg, K.; Hildebrand, D.; Uhle, F.; Sahin, D.; Krauser, U.; et al. The Interplay of Notch Signaling and STAT3 in TLR-Activated Human Primary Monocytes. Front. Cell. Infect. Microbiol. 2018, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.H.; Lee, W.S.; Go, S.-I.; Kim, M.J.; Lee, U.S.; Choi, H.J.; Kim, D.C.; Lee, J.-H.; Kim, H.-G.; Bae, K.S.; et al. Can thymidine phosphorylase be a predictive marker for gemcitabine and doxifluridine combination chemotherapy in cholangiocarcinoma?: Case series. Medicine 2014, 93, e305. [Google Scholar] [CrossRef]
- Ge, X.; Wang, Y.; Li, Q.; Yu, H.; Ji, G.; Miao, L. NK4 regulates 5-fluorouracil sensitivity in cholangiocarcinoma cells by modulating the intrinsic apoptosis pathway. Oncol. Rep. 2013, 30, 448–454. [Google Scholar] [CrossRef]
- Thanasai, J.; Limpaiboon, T.; Jearanaikoon, P.; Sripa, B.; Pairojkul, C.; Tantimavanich, S.; Miwa, M. Effects of thymidine phosphorylase on tumor aggressiveness and 5-fluorouracil sensitivity in cholangiocarcinoma. World J. Gastroenterol. 2010, 16, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Farren, M.; Ahn, D.; Bekaii-Saab, T.; Lesinski, G.B. Signaling pathways as therapeutic targets in biliary tract cancer. Expert Opin. Ther. Targets 2017, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, H. Epigenetic alterations in cholangiocarcinoma sustained IL-6/STAT3 signaling in cholangio- carcinoma due to SOCS3 epigenetic silencing. Digestion 2009, 79 (Suppl. 1), 2–8. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: Pathogenesis and rationale for molecular therapies. Oncogene. 2013, 32, 4861–4870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Abdel-Aziz, M. STAT3 transcription factor as target for anti-cancer therapy. Pharmacol. Rep. 2020, 72, 1101–1124. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.-M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Shi, X.; Kaller, M.; Rokavec, M.; Kirchner, T.; Horst, D.; Hermeking, H. Characterization of a p53/miR-34a/CSF1R/STAT3 Feedback Loop in Colorectal Cancer. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 391–418. [Google Scholar] [CrossRef]
- Wei, N.; Li, J.; Fang, C.; Chang, J.; Xirou, V.; Syrigos, N.; Marks, B.J.; Chu, E.; Schmitz, J.C. Targeting colon cancer with the novel STAT3 inhibitor bruceantinol. Oncogene 2019, 38, 1676–1687. [Google Scholar] [CrossRef]
- De Simone, V.; Franzè, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; Macdonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-α synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Lin, L.; Liu, A.; Peng, Z.; Lin, H.-J.; Li, P.-K.; Li, C.; Lin, J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011, 71, 7226–7237. [Google Scholar] [CrossRef] [Green Version]
- Mi, C.; Cao, X.; Ma, K.; Wei, M.; Xu, W.; Lin, Y.; Zhang, J.; Wang, T.-Y. Digitoxin promotes apoptosis and inhibits proliferation and migration by reducing HIF-1α and STAT3 in KRAS mutant human colon cancer cells. Chem. Biol. Interact. 2022, 351, 109729. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Liu, S.; Ma, X.; Liu, J.; Liu, J.; Lei, M.; Cao, Y. The effect and mechanism of erianin on the reversal of oxaliplatin resistance in human colon cancer cells. Cell Biol. Int. 2021, 45, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.-W.; Xia, J.; Cheng, B.-H.; Gao, H.-C.; Fu, L.-W.; Luo, X.-L. Tea polyphenol EGCG inhibited colorectal-cancer-cell proliferation and migration via downregulation of STAT3. Gastroenterol. Rep. 2020, 9, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.-J.; Liu, D.-Y.; Shen, R.-R.; Xiong, Y. A short deletion in the DNA-binding domain of STAT3 suppresses growth and progression of colon cancer cells. Aging 2021, 13, 5185–5196. [Google Scholar] [CrossRef]
- Leport, C.; Meulemans, A.; Robine, D.; Dameron, G.; Vildé, J.L. Levels of pyrimethamine in serum and penetration into brain tissue in humans. AIDS 1992, 6, 1040–1041. [Google Scholar] [CrossRef]
- Schoondermark-van, d.V.E.; Galama, J.; Vree, T.; Camps, W.; Baars, I.; Eskes, T.; Meuwissen, J.; Melchers, W. Study of treatment of congenital Toxoplasma gondii infection in rhesus monkeys with pyrimethamine and sulfadiazine. Antimicrob. Agents Chemother. 1995, 39, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Osman, I.; Orlow, S.J. Antifolate activity of pyrimethamine enhances temozolomide induced cytotoxicity in melanoma cells. Mol. Cancer Res. 2009, 7, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Zhang, B.; Liu, X.; Guo, K.; Ma, S.; Cai, F.; Yang, Y.; Yao, Y.; Feng, M.; Bao, X.; et al. Pyrimethamine sensitizes pituitary adenomas cells to temozolomide through cathepsin B-dependent and caspase-dependent apoptotic pathways. Int. J. Cancer 2013, 133, 1982–1993. [Google Scholar] [CrossRef] [Green Version]
- Warfield, B.M.; Matheson, C.J.; McArthur, D.G.; Backos, D.S.; Reigan, P. Evaluation of Thymidine Phosphorylase Inhibitors in Glioblastoma and Their Capacity for Temozolomide Potentiation. ACS Chem. Neurosci. 2021, 12, 3477–3486. [Google Scholar] [CrossRef]
- Servat, C.C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; Gil, M.D.L.L.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in EGFR mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017, 8, 47305–47316. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-X.; Lin, S.-H.; Lin, C.-C.; Yang, C.-C.; Yuan, S.-Y. In Vitro and In Vivo Antitumor Effects of Pyrimethamine on Non-small Cell Lung Cancers. Anticancer Res. 2018, 38, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Menamin, M.C.; Murray, L.J.; Cantwell, M.M.; Hughes, C.M. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in cancer progression and survival: A systematic review. Cancer Causes Control. 2012, 23, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Meiners, J.; Jansen, K.; Gorbokon, N.; Büscheck, F.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Höflmayer, D.; Weidemann, S.; Fraune, C.; et al. Angiotensin Converting Enzyme 2 Protein Is Overexpressed in a Wide Range of Human Tumour Types: A Systematic Tissue Microarray Study on >15,000 Tumours. Biomedicines 2021, 9, 1831. [Google Scholar] [CrossRef]
- Kilmister, E.J.; Tan, S.T. The Role of the Renin-Angiotensin System in the Cancer Stem Cell Niche. J. Histochem. Cytochem. 2021, 69, 835–847. [Google Scholar] [CrossRef]
- Bernardo, A.; Malara, M.; Bertuccini, L.; De Nuccio, C.; Visentin, S.; Minghetti, L. The Antihypertensive Drug Telmisartan Protects Oligodendrocytes from Cholesterol Accumulation and Promotes Differentiation by a PPAR-γ-Mediated Mechanism. Int. J. Mol. Sci. 2021, 22, 9434. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.M.; Elsheakh, A.R.; Suddek, G.M.; Abdelaziz, R.R. Telmisartan alleviates alcohol induced liver injury by activation of PPAR-γ/ Nrf-2 crosstalk in mice. Int. Immunopharmacol. 2021, 99, 107963. [Google Scholar] [CrossRef] [PubMed]
- Devan, A.R.; Nair, B.; Kumar, A.R.; Nath, L.R. An insight into the role of telmisartan as PPAR-γ/α dual activator in the management of nonalcoholic fatty liver disease. Biotechnol. Appl. Biochem. 2021, 69, 461–648. [Google Scholar] [CrossRef] [PubMed]
- Kubik, M.; Chudek, J.; Adamczak, M.; Wiecek, A. Telmisartan improves cardiometabolic profile in obese patients with arterial hypertension. Kidney Blood Press. Res. 2012, 35, 281–289. [Google Scholar] [CrossRef]
- Samukawa, E.; Fujihara, S.; Oura, K.; Iwama, H.; Yamana, Y.; Tadokoro, T.; Chiyo, T.; Kobayashi, K.; Morishita, A.; Nakahara, M.; et al. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int. J. Oncol. 2017, 51, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Tsujiya, Y.; Yamamori, M.; Hasegawa, A.; Yamamoto, Y.; Yashiro, M.; Okamura, N. Telmisartan Exerts Cytotoxicity in Scirrhous Gastric Cancer Cells by Inducing G0/G1 Cell Cycle Arrest. Anticancer Res. 2021, 41, 5461–5468. [Google Scholar] [CrossRef]
- Fujita, N.; Fujita, K.; Iwama, H.; Kobara, H.; Fujihara, S.; Chiyo, T.; Namima, D.; Yamana, H.; Kono, T.; Takuma, K.; et al. Antihypertensive drug telmisartan suppresses the proliferation of gastric cancer cells in vitro and in vivo. Oncol. Rep. 2020, 44, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Kobara, H.; Fujihara, S.; Iwama, H.; Matsui, T.; Fujimori, A.; Chiyo, T.; Tingting, S.; Kobayashi, N.; Nishiyama, N.; Yachida, T.; et al. Antihypertensive drug telmisartan inhibits cell proliferation of gastrointestinal stromal tumor cells in vitro. Mol. Med. Rep. 2020, 22, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Saber, S.; Khodir, A.E.; Soliman, W.E.; Salama, M.M.; Abdo, W.S.; Elsaeed, B.; Nader, K.; Abdelnasser, A.; Megahed, N.; Basuony, M.; et al. Telmisartan attenuates N-nitrosodiethylamine induced hepatocellular carcinoma in mice by modulating the NF-κB-TAK1-ERK1/2 axis in the context of PPARγ agonistic activity. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Chiyo, T.; Kobara, H.; Fujihara, S.; Fujita, K.; Namima, D.; Nakahara, M.; Kobayashi, N.; Nishiyama, N.; Yachida, T.; et al. Telmisartan Inhibits Cell Proliferation and Tumor Growth of Esophageal Squamous Cell Carcinoma by Inducing S-Phase Arrest In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3197. [Google Scholar] [CrossRef] [Green Version]
- Oura, K.; Tadokoro, T.; Fujihara, S.; Morishita, A.; Chiyo, T.; Samukawa, E.; Yamana, Y.; Fujita, K.; Sakamoto, T.; Nomura, T.; et al. Telmisartan inhibits hepatocellular carcinoma cell proliferation in vitro by inducing cell cycle arrest. Oncol. Rep. 2017, 38, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, S.; Morishita, A.; Ogawa, K.; Tadokoro, T.; Chiyo, T.; Kato, K.; Kobara, H.; Mori, H.; Iwama, H.; Masaki, T. The angiotensin II type 1 receptor antagonist telmisartan inhibits cell proliferation and tumor growth of esophageal adenocarcinoma via the AMPKα/mTOR pathway in vitro and in vivo. Oncotarget 2017, 8, 8536–8549. [Google Scholar] [CrossRef] [Green Version]
- De Araújo, R.F., Jr.; Oliveira, A.L.C.L.; Silveira, R.F.D.M.; Rocha, H.A.D.O.; Cavalcanti, P.D.F.; Araújo, A. Telmisartan induces apoptosis and regulates Bcl-2 in human renal cancer cells. Exp. Biol. Med. 2015, 240, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Koyama, N.; Nishida, Y.; Ishii, T.; Yoshida, T.; Furukawa, Y.; Narahara, H. Telmisartan induces growth inhibition, DNA double-strand breaks and apoptosis in human endometrial cancer cells. PLoS ONE 2014, 9, e93050. [Google Scholar] [CrossRef]
- Nenicu, A.; Korbel, C.; Gu, Y.; Menger, M.D.; Laschke, M.W. Combined blockade of angiotensin II type 1 receptor and activation of peroxisome proliferator-activated receptor-γ by telmisartan effectively inhibits vascularization and growth of murine endometriosis-like lesions. Hum. Reprod. 2014, 29, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, R.; Funao, K.; Matsuyama, M.; Kawahito, Y.; Sano, H.; Chargui, J.; Touraine, J.-L.; Nakatani, T. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncol. Rep. 2008, 20, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Tajima, H.; Ohta, T.; Nakanuma, S.; Hayashi, H.; Nakagawara, H.; Onishi, I.; Takamura, H.; Ninomiya, I.; Kitagawa, H.; et al. Angiotensin II induces tumor progression and fibrosis in intrahepatic cholangiocarcinoma through an interaction with hepatic stellate cells. Int. J. Oncol. 2010, 37, 1251–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyazit, Y.; Purnak, T.; Suvak, B.; Kurt, M.; Sayilir, A.; Turhan, T.; Tas, A.; Torun, S.; Celik, T.; Ibis, M.; et al. Increased ACE in extrahepatic cholangiocarcinoma as a clue for activated RAS in biliary neoplasms. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Saikawa, S.; Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Nakanishi, K.; Kitagawa, K.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Angiotensin receptor blockade attenuates cholangiocarcinoma cell growth by inhibiting the oncogenic activity of Yes-associated protein. Cancer Lett. 2018, 434, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.D.; Mafura, B.; Lauscher, J.C.; Seeliger, H.; Kreis, M.E.; Gröne, J. Antiproliferative and apoptotic effects of telmisartan in human colon cancer cells. Oncol. Lett. 2014, 8, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Hachiya, K.; Masuya, M.; Kuroda, N.; Yoneda, M.; Tsuboi, J.; Nagaharu, K.; Nishimura, K.; Shiotani, T.; Ohishi, K.; Tawara, I.; et al. Irbesartan, an angiotensin II type 1 receptor blocker, inhibits colitis-associated tumourigenesis by blocking the MCP-1/CCR2 pathway. Sci. Rep. 2021, 11, 19943. [Google Scholar] [CrossRef]
- Tabatabai, E.; Khazaei, M.; Asgharzadeh, F.; Nazari, S.E.; Shakour, N.; Fiuji, H.; Ziaeemehr, A.; Mostafapour, A.; Parizadeh, M.R.; Nouri, M.; et al. Inhibition of angiotensin II type 1 receptor by candesartan reduces tumor growth and ameliorates fibrosis in colorectal cancer. EXCLI J. 2021, 20, 863–878. [Google Scholar] [CrossRef]
- Ozeki, K.; Tanida, S.; Morimoto, C.; Inoue, Y.; Mizoshita, T.; Tsukamoto, H.; Shimura, T.; Kataoka, H.; Kamiya, T.; Nishiwaki, E.; et al. Telmisartan inhibits cell proliferation by blocking nuclear translocation of ProHB-EGF C-terminal fragment in colon cancer cells. PLoS ONE 2013, 8, e56770. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Yaguchi, T.; Ohmura, G.; Kobayashi, A.; Kawamura, N.; Iwata, T.; Kiniwa, Y.; Okuyama, R.; Kawakami, Y. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. 2018, 109, 54–64. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Li, C.; Guo, J.; Xu, B.; Xue, L. Telmisartan attenuates human glioblastoma cells proliferation and oncogenicity by inducing the lipid oxidation. Asia-Pac. J. Clin. Oncol 2021. ahead of print. [Google Scholar] [CrossRef]
- Kast, R.E. Paths for Improving Bevacizumab Available in 2018: The ADZT Regimen for Better Glioblastoma Treatment. Med. Sci. 2018, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.N.M.; Oh, J.; Kim, S.; Antonio, J.M.; Vo, V.T.A.; Om, J.; Choi, J.-W.; Kim, J.-Y.; Jung, C.-W.; Park, M.-J.; et al. Peroxisome proliferator-activated receptor gamma as a theragnostic target for mesenchymal type glioblastoma patients. Exp. Mol. Med. 2020, 52, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Wilop, S.; Von Hobe, S.; Crysandt, M.; Esser, A.; Osieka, R.; Jost, E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. J. Cancer Res. Clin. Oncol. 2009, 135, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Arthur, P.; Patel, N.; Surapaneni, S.K.; Mondal, A.; Gebeyehu, A.; Bagde, A.; Kutlehria, S.; Nottingham, E.; Singh, M. Targeting lung cancer stem cells using combination of Tel and Docetaxel liposomes in 3D cultures and tumor xenografts. Toxicol. Appl. Pharmacol. 2020, 401, 115112. [Google Scholar] [CrossRef] [PubMed]
- Aydiner, A.; Ciftci, R.; Sen, F. Renin-Angiotensin system blockers may prolong survival of metastatic non-small cell lung cancer patients receiving erlotinib. Medicine 2015, 94, e887. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.R.; Carroll, N.M.; Sakoda, L.C.; Delate, T.; Hornbrook, M.C.; Jain, R.K.; Kushi, L.H.; Quinn, V.P.; Ritzwoller, D.P. Effect of Angiotensin System Inhibitors on Survival in Patients Receiving Chemotherapy for Advanced Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2017, 18, 189–197.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Chen, W.; Zhou, L.; Wan, H.; Gao, B.; Feng, Y. Impact of Angiotensin I-converting Enzyme Inhibitors and Angiotensin II Type-1 Receptor Blockers on Survival of Patients with NSCLC. Sci Rep. 2016, 6, 21359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y. Telmisartan inhibits NSCLC A549 cell proliferation and migration by regulating the PI3K/AKT signaling pathway. Oncol. Lett. 2018, 15, 5859–5864. [Google Scholar] [CrossRef] [Green Version]
- Surapaneni, S.K.; Nottingham, E.; Mondal, A.; Patel, N.; Arthur, P.; Gebeyehu, A.; Kalvala, A.K.; Rishi, A.K.; Singh, M. Telmisartan Facilitates the Anticancer Effects of CARP-1 Functional Mimetic and Sorafenib in Rociletinib Resistant Non-small Cell Lung Cancer. Anticancer Res. 2021, 41, 4215–4228. [Google Scholar] [CrossRef]
- Halatsch, M.-E.; Kast, R.; Karpel-Massler, G.; Mayer, B.; Zolk, O.; Schmitz, B.; Scheuerle, A.; Maier, L.; Bullinger, L.; Mayer-Steinacker, R.; et al. CTNI-04. RECURRENT GLIOBLASTOMA LONG-TERM SURVIVORS TREATED WITH CUSP9v3. Neuro-Oncology 2021, 23 (Suppl. 6), vi59. [Google Scholar] [CrossRef]



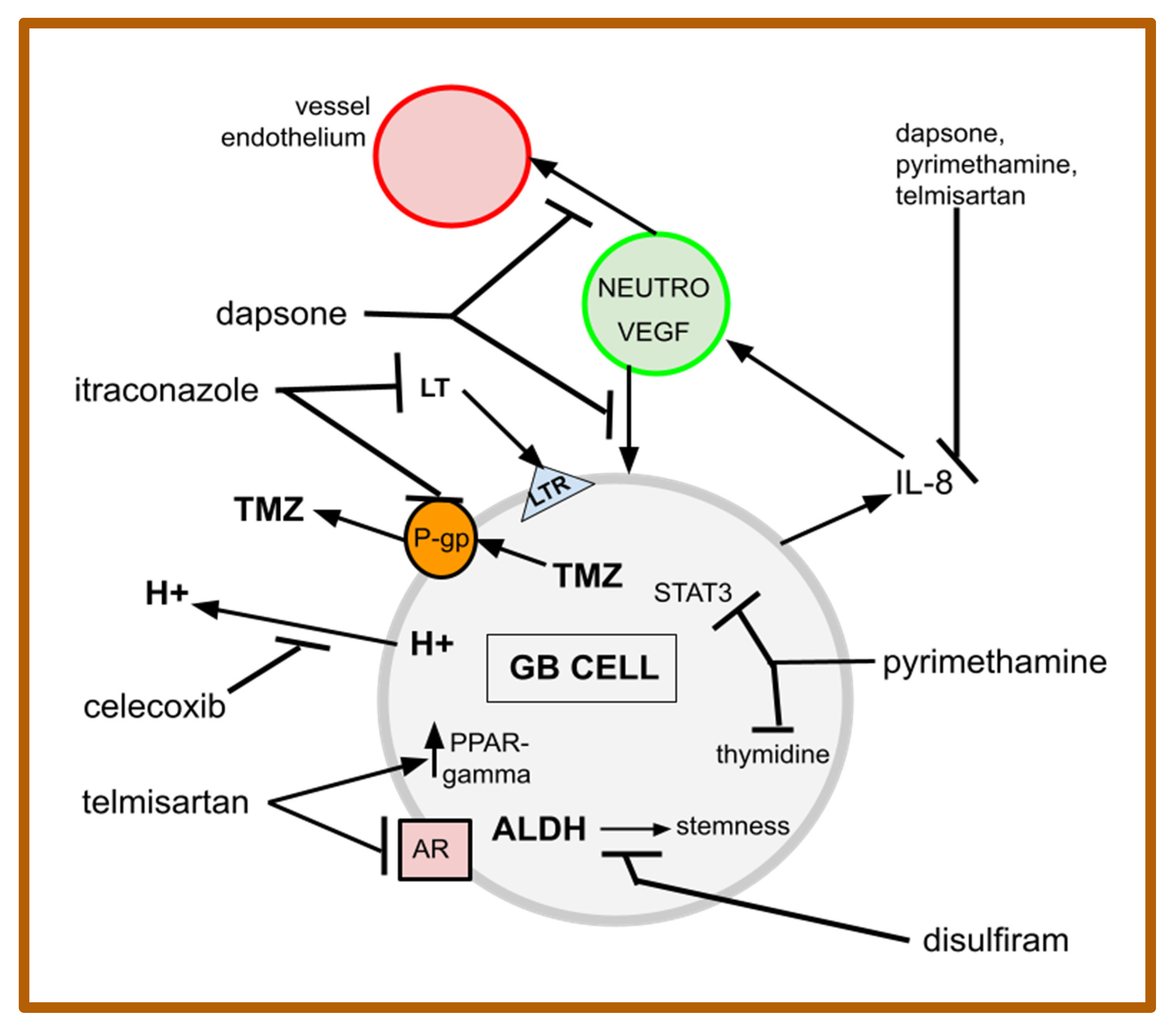{kind=link}
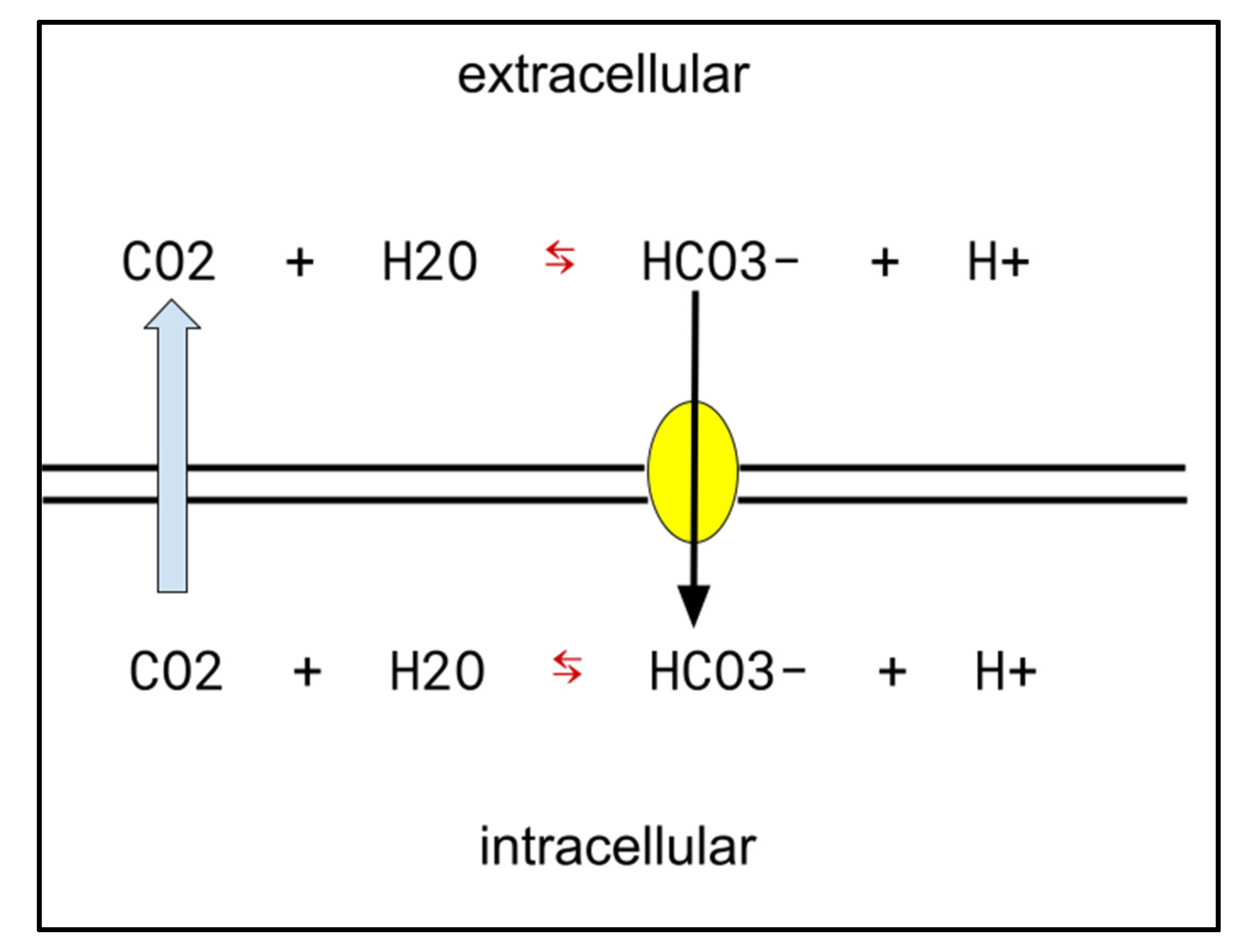{kind=link}
| 1. The principle of breaking more than one link in any chain in a series that leads to undesired outcomes. |
| 2. The principle of Palmer et al., of achieving fractional cell killing with multiple drugs with independent MOAs. |
| 3. The principle of shaping versus decisive operations—both required for a successful cancer treatment, with MDACT regimens being largely shaping operations. |
| 4. A principle adapted from Chow et al. of using multiple simultaneous cytotoxic medicines at low doses. |
| 5. As in CUSP9v3, the principle of using non-oncology drugs from general medical practice, repurposed to block multiple survival paths. |
| 6. The concept borrowed from chess that every move (i.e., medical or other intervention) creates weaknesses and strengths. |
| 7. The principle of mass, where inadequate response is redressed by simply adding more force. |
| 8. The Nile Distributary Problem, where the existence of parallel growth-driving pathways allows signaling flow to proceed when a given pathway is blocked. |
| Drug | Dose | Usual Use—Target Use in gMDACT |
|---|---|---|
| Celecoxib | 600 mg × 2 | Analgesic—COX-2, CA-IX, P-gp |
| Dapsone | 100 mg × 2 | Antibiotic—neutrophils, IL-8, VEGF |
| Disulfiram | 250 mg × 2 | Anti-alcoholism—ALDH, P-gp |
| Itraconazole | 200 mg × 2 | Antifungal—Hh, 5-LO, P-gp |
| Pyrimethamine | 50 mg × 1 | Antibiotic—STAT3, DHFR, IL-8, thymidine phosphorylase |
| Telmisartan | 80 mg × 1 | Anti-hypertensive—PPAR-gamma, ARB, IL-8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kast, R.E.; Alfieri, A.; Assi, H.I.; Burns, T.C.; Elyamany, A.M.; Gonzalez-Cao, M.; Karpel-Massler, G.; Marosi, C.; Salacz, M.E.; Sardi, I.; et al. MDACT: A New Principle of Adjunctive Cancer Treatment Using Combinations of Multiple Repurposed Drugs, with an Example Regimen. Cancers 2022, 14, 2563. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102563
Kast RE, Alfieri A, Assi HI, Burns TC, Elyamany AM, Gonzalez-Cao M, Karpel-Massler G, Marosi C, Salacz ME, Sardi I, et al. MDACT: A New Principle of Adjunctive Cancer Treatment Using Combinations of Multiple Repurposed Drugs, with an Example Regimen. Cancers. 2022; 14(10):2563. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102563
Chicago/Turabian StyleKast, Richard E., Alex Alfieri, Hazem I. Assi, Terry C. Burns, Ashraf M. Elyamany, Maria Gonzalez-Cao, Georg Karpel-Massler, Christine Marosi, Michael E. Salacz, Iacopo Sardi, and et al. 2022. "MDACT: A New Principle of Adjunctive Cancer Treatment Using Combinations of Multiple Repurposed Drugs, with an Example Regimen" Cancers 14, no. 10: 2563. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102563